

Abstract
The total synthesis and biological evaluation of neopeltolide and analogs are reported. The key bond-forming step utilizes a Lewis acid-catalyzed intramolecular macrocyclization that installs the tetrahydropyran ring and macrocycle simultaneously. Independent of each other, neither the macrolide nor the oxazole side chain substituents of neopeltolide can inhibit the growth of cancer cell lines. The biological data of the analogs indicate that alterations to either the ester side chain or the stereochemistry of the macrolide result in a loss of biological activity.
Introduction
Background
Natural products continue to be excellent sources for new drug candidates, especially in the area of anticancer therapeutics.1-3 In many instances, the limited amount of natural material that is typically isolated severely limits the ability of the cancer research community to investigate important lead compounds. Organic synthesis can facilitate the preparation of sufficient quantities of target compounds when natural sources are scarce or no longer available. Additionally, synthetic routes to molecules can be manipulated to create simplified analogs of the target compound that may have the same or enhanced biological activity. Total synthesis can also confirm the structural assignment and absolute stereochemistry of a natural product, which was required for neopeltolide 1.4
In 2007, Wright and co-workers reported the structure of a new, highly cytotoxic, natural product neopeltolide 2, which was isolated from sponges most closely related to the genus Daedalopelta Sollas.5 The key structural features of neopeltolide include a trisubstituted 2,6-cis-tetrahydropyran moiety, which is embedded in a 14-membered macrolactone containing six stereogenic centers. The pyran subunit possesses a highly functionalized oxazole substituent that is identical to the side chain of leucascandrolide A. The molecular formula and tricyclic nature of the molecule were determined by high resolution mass spectrometry (HRMS) and 13C NMR spectroscopy. The planar structure was elucidated by the analysis of one (1D)- and two-dimensional (2D) 1H NMR spectroscopy, including advanced TOCSY and COSY experimentation. The relative stereochemistry of neopeltolide was originally proposed based on 1D and 2D NOESY spectra, which ultimately lead to the incorrect structural assignment. The key protons on the macrolide (H-3, H-7, H-9, H-11, and H-13) were assigned to all be syn in relationship with each other, all adopting a pseudoaxial orientation (Figure 1). The absolute stereochemistry of the molecule could not be initially established due to the lack of available material.
Figure 1. Neopeltolide Structures.

Neopeltolide potently inhibits proliferation of a number of cancer cell lines. Although it exhibits IC50 values in the low nanomolar to subnanomolar range, the efficacy with which neopeltolide inhibits cancer cell growth varies, with some cell lines completely inhibited and others only partially inhibited.5,6 Based on that limited efficacy and data showing an accumulation cells in G1 phase, Wright hypothesized that neopeltolide may be at least partially cytostatic to these cell lines rather than simply cytotoxic. Additionally, neopeltolide is also a potent inhibitor of the fungal pathogen Candida albicans (MIC value of 0.625 μg/mL), which severely and adversely affects the health of AIDS patients, potentially resulting in death. Preliminary investigations into the mechanism of action suggested that it does not interact with either tubulin or actin. Recently, Kozmin, Kron and co-workers have advanced that the cellular target of neopeltolide is the cytochrome bc1 complex and that the molecule inhibits mitochondrial ATP synthesis. However, their investigations seemingly employed racemic material and it is unclear if and how this aspect impacts their biological studies.6
The macrolide ring of neopeltolide has similarities to the macrocycles found in the callipeltosides,7 lyngbyaloside,8,9 lyngbouilloside,10 and the aurisides.11 The key distinction between these natural products is the C-7 hemiketal functionality as opposed to the reduced ether of the tetrahydropyran ring system present in neopeltolide. Additionally, several of these molecules exhibit modest to slight cytotoxicity as opposed to the high biological activity of neopeltolide. Leucascandrolide A is also structurally similar and has comparable activity to neopeltolide.12 Preliminary biological studies of leucascandrolide A have shown that: (i) the macrolide core is essential for tumor cytoxicity, and (ii) the oxazole side chain is important for fungistatic activity. Due to the interesting structural aspects as well as the high biological activity, neopeltolide has garnered the attention of the synthetic community and has resulted in several other total syntheses.6,13-19
Synthesis Plan
An important goal of our synthesis was to develop a highly convergent and flexible synthetic route with the ability to easily prepare analogs. Excision of the oxazole side chain affords macrocyclic core 3 (Scheme 1), which contains an embedded tetrahydropyran that we envisioned forming through our previously developed Lewis acid-catalyzed cyclization.20 This methodology allows for a highly diastereoselective (>20:1 dr) cyclization of β-hydroxy dioxinone 9 with aldehyde 10 in the presence of a catalytic amount of Sc(OTf)3 to furnish bicyclic heterocycle 11 (Scheme 2). Upon heating 11 in the presence of H2O, resulted in the formation of a 2,6-cis-tetrahydropyranone. Notably, our studies have indicated that the use of optically enriched β-hydroxy dioxinone 9 in the cyclization results in complete retention of optical purity. Utilizing this methodology, the tetrahydropyran core could be constructed as a single diastereomer in two different manners: 1) through an intermolecular cyclization followed by a Yamaguchi macrolactonization to create the macrocycle,21 or 2) through a new intramolecular cyclization that would simultaneously form the macrocycle. At the time of our initial publication, we were aware of only a single report in the literature in which a Prins cyclization was employed to provide a pyran-containing macrocycle.22 Since we reported the synthesis of neopeltolide several other research groups have successfully utilized the Prins cyclization strategy as a key step in their syntheses.17,23-26 The precursors to both cyclization pathways are carboxylic acid 7 and alcohol 8.
Scheme 1.

Synthetic Plan
Scheme 2.
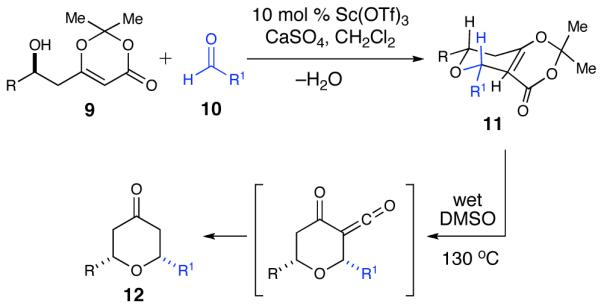
Scandium(III) Triflate Methodology
Results and Discussion
Carboxylic Acid Fragment
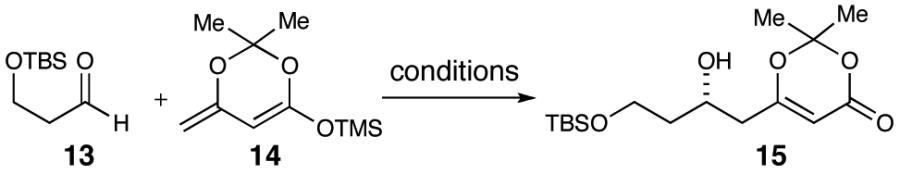
The synthesis began with monoprotection of 1,3-propanediol as a silyl ether followed by subsequent oxidation with TPAP27 to afford aldehyde 13, the precursor to the vinylogous aldol reaction. Using the procedure as reported by Scettri and co-workers,28,29 the aldol reaction with aldehyde 13 and dienoxy silane 1430,31 resulted in moderate yields and enantioselectivities (Table 1, entries 1 and 2). Varying the solvent from THF and trying alternative drying agents did little to improve the yield or enantioselectivity. In the end we observed that 10 mol % Ti(i-OPr)4, 10 mol % (R)-BINOL, flame-dried powdered 4 Å molecular sieves and a higher precomplexation concentration (0.5 M) resulted in our best results (Table 1, entry 11). The resultant β-hydroxy dioxinone was converted to a bis-silyl ether and a selective deprotection of the primary TBS-ether with pPTs was accomplished to furnish primary alcohol 16 (Scheme 3). Oxidation to the carboxylic acid was achieved with PDC in a 97% yield.
Table 1.
Optimization of Aldol Reactiona
 | |||||
|---|---|---|---|---|---|
| entry | solvent | R-BINOL (mol %) | additive | yield (%)b | ee (%)c |
| 1 | THF | 8 | 4 Å MS | 50 | 73d,e |
| 2 | THF | 8 | 4 Å MS | 53 | 71d |
| 3 | THF | 10 | 4 Å MS | 60 | 78d,e |
| 4 | toluene | 10 | 4 Å MS | 30 | 71 |
| 5 | CH2Cl2 | 10 | 4 Å MS | — | — |
| 6 | THF | 10 | 3 Å MSf | 65 | 68 |
| 7 | THF | 10 | 3 Å MS | 66 | 63 |
| 8 | THF | 10 | 3 Å MSf,g | 65 | 81 |
| 9 | THF | 10 | 4 Å MSg | 57 | 74 |
| 10 | THF | 15 | 4 Å MSg | 66 | 80 |
| 11 | THF | 10 | 4 Å MS | 63 | 88 |
| 12 | THF | 15 | 4 Å MS | 64 | 60 |
Reactions performed under N2 at 0.17 M (precomplexation concentration 0.53 M) with Ti(i-OPr)4 and ligand after 60 min, reaction cooled to -78 °C followed by addition of 13 and 14.
Isolated yields after column chromatography.
Determined by chiral HPLC.
precomplexation concentration 0.21 M.
Slowly warmed to 23 °C.
3 Å MS pellets.
Dried in a reduced pressure oven at 145 °C for 2 days.
Scheme 3.
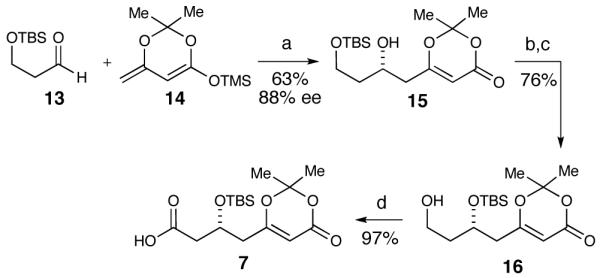
Dioxinone Fragment Synthesisa
a Conditions: (a) Ti(i-OPr)4, (R)-BINOL, 4 Å sieves, THF, 63%, 88% ee. (b) TBSOTf, 2,6-lutidine, CH2Cl2, 91%. (c) pPTs, MeOH, 83%. (d) PDC, DMF, 97%.
Alcohol Fragment
We next turned our attention toward the synthesis of the alcohol fragment, which began with a Noyori reduction of ethyl 3-oxohexanoate 17 (Scheme 4).32,33 Using (R)-tol-BINAP as the chiral ligand, the C-13 stereocenter was set, affording the β-hydroxyester in 94% yield and 97% ee. The ester was then converted to the Weinreb amide followed by PMB protection of the secondary alcohol to give rise to amide 18. Next, iodide 19, which was furnished by using Myers’ pseudoephedrine controlled alkylation methodology,34 underwent lithium-halogen exchange and addition to Weinreb amide 18.35 The yield of this reaction was moderate as a result of a competing elimination of the PMB-alcohol to produce the α,β-unsaturated ketone. Various conditions were attempted to render the reaction less basic (i.e. CeCl3),36 unfortunately the yield remained moderate. The PMB protecting group was removed using DDQ and the resulting β-hydroxy ketone underwent an Evans-Tishchenko anti-reduction with SmI2 and benzaldehyde.37 The anti-relationship was confirmed by Rychnovsky’s 13C NMR spectroscopic analysis.38,39 Methylation of the secondary alcohol and subsequent hydrolysis of the benzyl ester yielded alcohol 8 to complete the second main fragment.
Scheme 4.
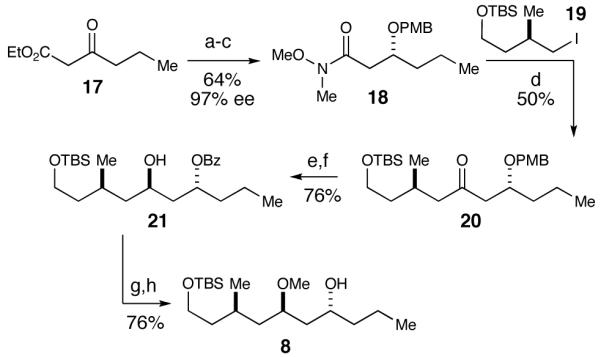
Alcohol Fragment Synthesisa
a Conditions: (a) RuCl2(PhH)2, (R)-tol-BINAP, H2, EtOH, 94%, 97% ee. (b) MeN(H)OMe·HCl, i-PrMgCl, THF, -20 °C, 85%. (c) PMB-imidate, pPTs, cyclohexane, CH2Cl2, 0 °C, 80%. (d) t-BuLi, pentane/Et2O, -78 °C, 50%. (e) DDQ, pH7 buffer, CH2Cl2, 84%. (f) SmI2, PhCHO, THF, 91%. (g) MeOTf, DTBMP, CH2Cl2, 88%. (h) K2CO3, MeOH, 86%.
Oxazole Fragment
The synthesis of oxazole 4 was based upon the routes independently reported by Leighton40 and Kozmin,41 with minor modifications. Carbamate 23 was prepared from the addition of propargylamine 22 to methyl chloroformate, followed by a carboxylation and subsequent reduction with Lindlar’s catalyst (Scheme 5). This was then coupled with L-serine methyl ester hydrochloride and converted to the oxazole.42 The resultant ester was reduced with DIBAL to afford alcohol 24, which underwent subsequent bromination to generate a primary bromide. A Stille coupling was employed to couple vinyltributyltin to the primary bromide.43 Subsequent hydroboration of the terminal olefin followed by a Dess-Martin oxidation afforded aldehyde 26.44 The cis-alkene was then obtained by a Still-Gennari olefination45 and consequent hydrolysis of the methyl ester delivered the oxazole side chain.
Scheme 5.
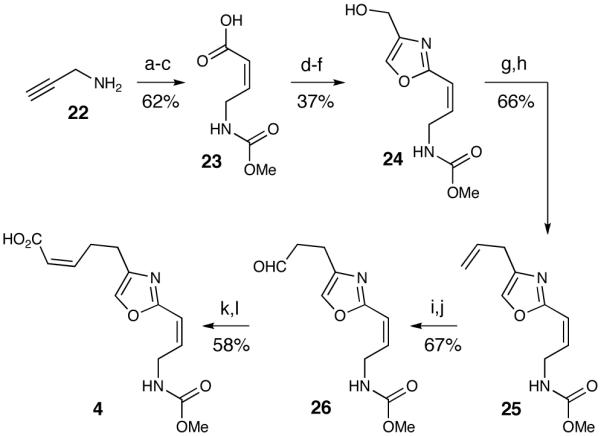
Oxazole Fragment Synthesisa
a Conditions: (a) ClCO2Me, dioxane, sat’d NaHCO3, 89%. (b) n-BuLi, CO2, THF, -78 °C. (c) Lindlar’s catalyst, quinoline, atm H2, EtOAc, 70% over two steps (d) i-BuOCOCl, N-Me-Morpholine, Ser-OMe·HCl, 71%. (e) DAST, CH2Cl2, -20 °C; BrCCl3, DBU, 0 °C, 62%. (f) DIBAL, THF, 0 °C, 85%. (g) CBr4, PPh3, 2,6-lutidine, CH3CN, 80%. (h) n-Bu3SnCH=CH2, Pd2dba3, tri(2-furyl)phosphine, THF, reflux, 83%. (i) 9-BBN, THF, H2O2, 68%. (j) Dess-Martin, CH2Cl2, 98%. (k) (F3CCH2O)2P(O)CH2CO2Me, 18-c-6, KHMDS, THF, 72%. (l) 1.0 M LiOH, THF, 80%.
Coupling the Fragments
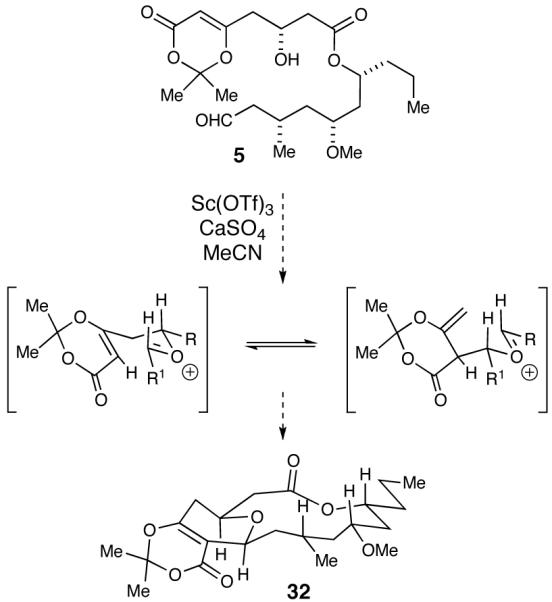
We first attempted to utilize our scandium(III) triflate-promoted cyclization methodology in an intermolecular fashion followed by closure of the macrocycle using a Yamaguchi macrolactonization. Therefore, carboxylic acid 7 was converted into the methyl ester by addition of (trimethylsilyl)diazomethane (Scheme 6 Meanwhile, HF·pyridine was utilized to deprotect 28 and subsequently oxidized to aldehyde 29 with TEMPO.46 Next, subjecting both dioxinone 27 and aldehyde 29 to our cyclization conditions afforded the bicyclic dioxinone as a single diastereomer in a 55% yield, which upon heating in wet DMSO formed pyranone 6. While convinced that this route would lead us to the completion of the proposed structure of neopeltolide, we were also interested in examining the scope of our scandium(III) triflate-catalyzed cyclization for the more elegant and demanding intramolecular macrocyclization route.
Scheme 6.
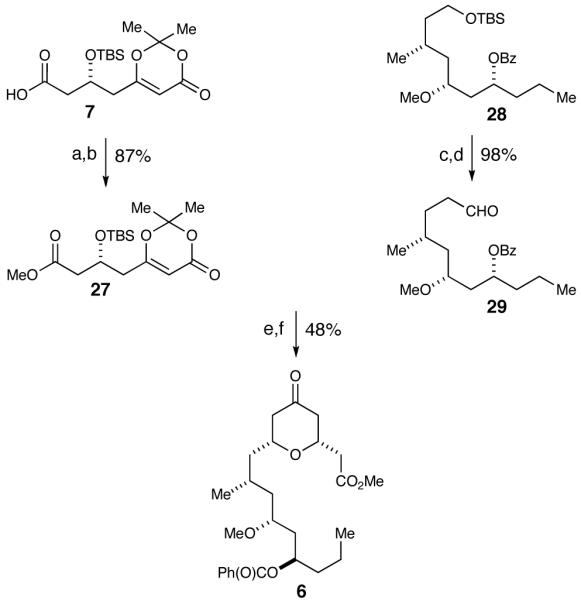
Intermolecular Scandium(III) Triflate Methodologya
a Conditions: (a) TMSCH2N2, CH2Cl2, MeOH, 90%. (b) HF·pyridine, THF, 97%. (c) HF·pyridine, THF, 99%. (d) TEMPO, H5C6I(OAc)2, CH2Cl2, 99%. (e) Sc(OTf)3, CaSO4, CH2Cl2, 55%. (f) DMSO, H2O, 87%.
The two main fragments, 7 and 8, were coupled using Yamaguchi’s esterification protocol (Scheme 7).21 Global deprotection of the silyl ethers proceeded smoothly with the use of HF·pyridine and selective oxidation of the primary alcohol was accomplished with TEMPO to generate acyclic aldehyde 5. In the key step, 10 mol % of scandium(III) triflate promoted the macrocyclization of 5 to furnish the fully elaborated 14-membered tricyclic ring 30 in 25% yield (unoptimized) and greater than 20:1 dr. Formation of the tetrahydropyranone was accomplished by heating 30 in wet DMSO and the pyranone was selectively reduced with NaBH4 to place the requisite alcohol in the equatorial position. Much to our surprise, a Mitsunobu reaction47 of the macrocyclic pyran with oxazole 4 produced a compound (2) whose spectra were similar, but not identical, to the natural product. Throughout the course of the synthesis, extensive NOE experiments on advanced intermediates and the final molecule all indicated that the key protons on the macrolide where syn to each other, as was reported by Wright and co-workers (Figure 2).5
Scheme 7.

Fragment Coupling and Initial Synthesisa
a Conditions: (a) 8, 2,4,6-trichlorobenzoyl chloride, Et3N, DMAP, THF, 80%. (b) HF·pyridine, THF, 93%. (c) TEMPO, H6C5I(OAc)2, CH2Cl2, 92%. (d) Sc(OTf)3, CaSO4, MeCN, 25%. (e) DMSO, H2O, 130 °C, 99%. (f) NaBH4, MeOH, 0 °C, 71%. (g) DIAD, Ph3P, 4, benzene, 80%.
Figure 2.
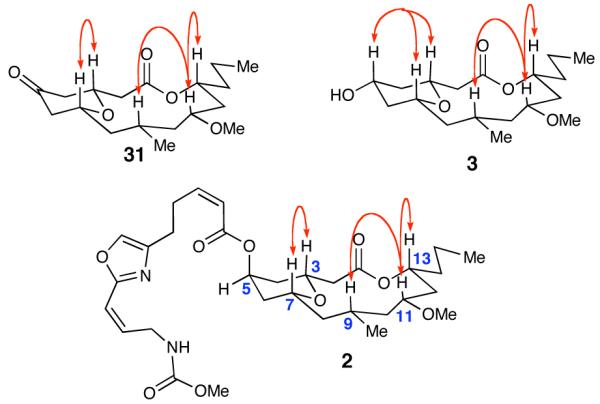
Observed NOEs for Synthetic Compounds
At this point, we hypothesized that two potential scenarios could account for the discrepancies evident in the comparison of our synthetic material and the isolated natural product. First, our macrocyclization, which forms the tricyclic macrocycle, could have potentially proceeded through an oxonia-Cope mechanism48-50 to give the inverted stereochemistry at the C-3 and C-7 positions (Scheme 8). This possibility could be the function of ring strain in the macrocycle that would result in the undesired configuration being the more thermodynamically stable product. Our other hypothesis was that we had successfully synthesized the proposed structure and that this structure differed in some way from the actual configuration of neopeltolide.
Scheme 8.
Oxonia-Cope Pathwaya
Synthesis of a Second Diastereomer (34)
We wanted to examine the viability of the oxonia-Cope pathway by synthesizing the enantiomer of carboxylic acid ent-7, couple this to alcohol 8 and determine if the same tricyclic macrocycle is formed after our cyclization reaction. Toward this end, we synthesized the enantiomer of carboxylic acid fragment ent-7 (Scheme 9). Therefore, utilizing (S)-BINOL for the vinylogous aldol reaction we installed the β-hydroxy substituents with the opposite configuration. With access to both enantiomers, 15 and ent-15, a Mosher ester analysis confirmed that we had correctly assigned the stereochemistry at the hydroxyl position.51 The dioxinone afforded the carboxylic acid ent-7 through the same protection, deprotection and oxidation sequence as described above.
Scheme 9.

Synthesis of Neopeltolide Diastereomer 34a
a Conditions: (a) Ti(i-OPr)4, (S)-BINOL, 4 Å sieves, THF, 63%, 88% ee. (b) TBSOTf, 2,6-lutidine, CH2Cl2, 91%. (c) pPTs, MeOH, 83%. (d) PDC, DMF, 97%. (e) 2,4,6-trichlorobenzoyl chloride, Et3N, DMAP, THF, 76%. (f) HF·pyridine, THF, 80%. (g) TEMPO, H6C5I(OAc)2, CH2Cl2, 98%. (h) Sc(OTf)3, CaSO4, MeCN, 20%. (i) DMSO, H2O, 130 °C, 98%. (j) NaBH4, MeOH, 0 °C, 70%. (k) DIAD, Ph3P, 4, benzene, 82%.
The coupling of fragments ent-7 and 8, following Yamaguchi’s procedure, removal of the silyl protecting groups, and selective oxidation of the primary alcohol furnished aldehyde 33 (Scheme 9). The resultant bicyclic dioxinone was then generated upon subjection of 33 with our scandium(III) triflate-catalyzed cyclization conditions and did not provide matching NMR spectra to that of 30. The material was decarboxylated and extensive NOE experimentation was done on the resulting pyranone indicating that the C-3 and C-7 positions had been inverted (Figure 3). This data indicated that our scandium(III) triflate cyclization reaction did not epimerize the C-3 position and, therefore the stereochemistry about the embedded tetrahydropyran ring was not inverted via the oxonia-Cope pathway in the initial synthesis. Nevertheless, we decided to finish the synthetic route with the inverted pyranone due to the ease of the last two steps. The reduction of pyranone with NaBH4 followed by the Mitsunobu addition of oxazole 4, again did not produce neopeltolide. Additional NOE experimentation performed on 34 confirmed the relative stereochemistry of the protons on the macrolide (Figure 3). We could now exclude the hypothesis that an oxonia-Cope process affected our synthetic route, validating that our initial synthesis afforded the reported structure of neopeltolide.
Figure 3.
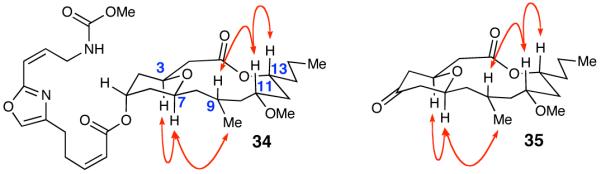
Observed NOEs for Synthetic Compounds
Completion of Neopeltolide
After careful consideration of the original isolation data in conjunction with our synthetic efforts, we postulated that the correct structure for neopeltolide was alcohol diastereomer 36 where the C11 and C13 substituents are inverted (Figure 4). Accessing 36 was accomplished by using (S)-BINAP for the Noyori reduction of ethyl 3-oxahexanoate (Scheme 10). The same reaction sequence was employed on this enantiomer to provide alcohol 36. Additionally, the undesired enantiomer of Weinreb amide 18, from the original synthesis (Scheme 4), could be carried forward due to the availability of the material and it required fewer steps to obtain 36 (Scheme 11). The reduction of 18 and consequent addition of iodide 19 to the resulting aldehyde produced a 1:1 mixture of inseparable diastereomers. Fortunately, upon methylation of the secondary alcohol, the diastereomers were separated and PMB ether 39 was isolated in 34% yield over the two steps as a single diastereomer. Hydrolysis of the benzyl ester followed by inversion of the resultant alcohol via Mitsunobu reaction generated alcohol 36.
Figure 4.
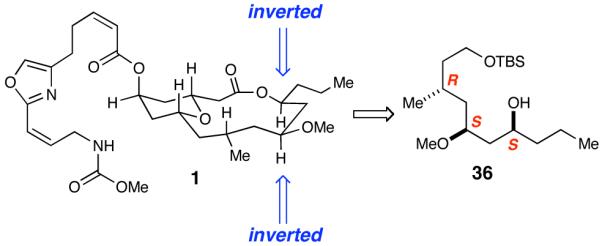
Our Structural Reassignment of Neopeltolide 1
Scheme 10.
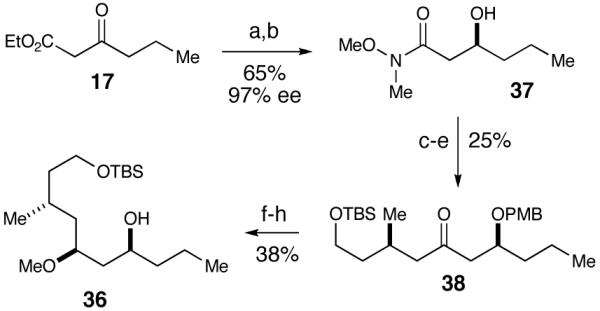
Synthesis of New Alcohol Fragmenta
a Conditions: (a) RuCl2(PhH)2, (S)-tol-BINAP, H2, EtOH, 94%, 96% ee. (b) MeN(H)OMe·HCl, i-PrMgCl, THF, -20 °C, 69%. (c) PMB-imidate, pPTs, cyclohexane, CH2Cl2, 0 °C, 97%. (d) 19, t-BuLi, pentane/Et2O, -78 °C, 28%. (e) DDQ, pH7 buffer, CH2Cl2, 92%. (f) SmI2, PhCHO, THF, 80%. (g) MeOTf, DTBMP, CH2Cl2. (h) K2CO3, MeOH, 48% over two-steps.
Scheme 11.

Completion of the Synthesisa
a Conditions: (a) DIBAL, CH2Cl2, -78 °C, 67%. (b) 19, t-BuLi, pentane/Et2O, -78 °C. (c) MeOTf, DTBMP, CH2Cl2, 34% over two-steps, single diastereomer. (d) DDQ, pH7 buffer, CH2Cl2, 83%. (e) 4-NO2-C6H5, DIAD, PPh3, benzene, 73%. (f) K2CO3, MeOH, 71%.(g) 7, 2,4,6-trichlorobenzoyl chloride, Et3N, DMAP, THF, 82%. (h) HF·pyridine, THF, 93%. (i) TEMPO, H6C5I(OAc)2, CH2Cl2, 99%. (j) Sc(OTf)3, CaSO4, MeCN, 40%. (k) DMSO, H2O, 130 °C, 82%. (l) NaBH4, MeOH, 0 °C, 96%. (m) DIAD, Ph3P, 4, benzene, 76%.
Acyclic aldehyde 40 can be accessed by coupling newly synthesized alcohol 36 and carboxylic acid 7 followed by deprotection and a selective oxidation. Our scandium(III) triflate-catalyzed macrocyclization produced the tricyclic macrocycle in 40% yield. Despite the modest yield, this reaction is remarkable due to the high degree of complexity generated in this single step. Decarboxylation of 41 followed by a subsequent reduction and Mitsunobu reaction with 4, gratifyingly, furnished neopeltolide (1). Our 1H and 13C NMR spectra match those reported in the literature for the isolated natural product. Additionally, the 2D NOESY, HRMS and optical rotation data confirmed a successful synthesis of the natural (+)-enantiomer of neopeltolide (1).
Biological Evaluation
With a highly convergent and flexible route to access the natural product and related compounds, we sought to explore the biological activity and to determine the structure-activity relationships of neopeltolide (1), the two diastereomers (2 and 34), three advanced intermediates (4, 42 and 43), and finally, two analogs of neopeltolide (44 and 45). The two analogs were readily prepared from a Mitsunobu reaction of pyran 43 with the corresponding acid (Scheme 12).
Scheme 12.

First, in order to confirm that synthetic neopeltolide 1 we had created was identical to the natural product itself, we evaluated its biological activity against several tumor cell lines (Figure 5A). Against the human breast adenocarcinoma line MCF-7, our structurally reassigned neopeltolide potently inhibited cell proliferation (as assayed by incorporation of [3H]-thymidine) with an IC50 value of 2.2 ± 0.1 nM (Figure 5A).52 Furthermore, the corrected neopeltolide inhibited proliferation of the murine leukemia line P388 even more potently, with an IC50 of 0.6 ± 0.2 nM. These IC50 values are nearly identical to those reported by Wright and co-workers in their report on the isolation of neopeltolide from Daedalopelta sp., validating our revised structure as the correct one5; conversely, diastereomer 2, which corresponded to the originally proposed structure, was approximately 100-fold less active in both cell lines (Table 2). While neopeltolide completely inhibited the proliferation of P388 cells, there remained a small level of growth (10 - 12%) of the MCF-7 cells that was refractory to neopeltolide. More surprisingly, however, was our finding that despite its potent effect on P388 and MCF-7 cells, neopeltolide was minimally active or inactive against the four other cell lines that we tested (Figure 5A): the human cervical carcinoma line HeLa, the rat adrenal tumor line PC12, the human epidermal carcinoma line KB and the human lung carcinoma line A549. We detected no dose-dependent growth inhibition in A549 or HeLa cells, and minimal inhibition of PC12 and KB cells was observed only at 10 μM neopeltolide. Limited effectiveness of neopeltolide at blocking growth of different cell lines, including A549 cells, has been recently reported5,6 but in these cases there was no rightward shift in IC50 value. Our discovery of entirely resistant cell lines was therefore surprising. Neopeltolide appears to be selective with respect to which cancer cells it inhibits.
Figure 5.

A. Activity of synthetic neopeltolide 1 against cancer cell lines. B. Activity against cultured primary human cells.
Table 2.
Activity of Neopeltolide (1) and Structural Analogs to Inhibit Proliferation of Human Cancer Cells. IC50 values represent the mean ± SEM of three experiments
| analog | MCF-7 cells | P-388 cells | ||
|---|---|---|---|---|
| efficacy (at 10 mM) | potency (IC50) | efficacy (at 10 mM) | potency (IC50) | |
| neopeltolide (1) | 84% inhibition | 2.2 ± 0.1 nM | 100% inhibition | 0.6 ± 0.2 nM |
| diastereomer 1 (2) | 86% inhibition | 219 ± 59.1 nM | 100% inhibition | 42.3 ± 14.4 nM |
| diastereomer 2 (34) | 88% inhibition | 328 ± 28.4 nM | 100% inhibition | 216 ± 62.3 nM |
| benzoyl ester (44) | 78% inhibition | 5.8 ± 4.7 μM | 75% inhibition | 3.3 ± 2.0 nM |
| octanoyl ester (45) | 33% inhibition | > 10 μM | 25% inhibition | > 10 μM |
| oxazole side chain (4) | nominal inhibition | > 10 μM | 10% inhibition | > 10 μM |
| core pyran-alcohol (43) | nominal inhibition | > 10 μM | nominal inhibition | > 10 μM |
| core pyranone (42) | nominal inhibition | > 10 μM | nominal inhibition | > 10 μM |
We have initiated structure-activity studies in order to begin identification of the biologically important structural attributes of neopeltolide. The molecule can be divided structurally into two main components: the core macrolide 43 and the oxazole-containing side chain 4. Our synthetic scheme provided us with variants of both of these components as well as a pair of stereoisomers (2, 34) of the full molecule. In addition, we synthesized a pair of ester variants of the side chain by replacing the oxazole-containing side chain with either a benzoyl (44) or octanoyl group (45). In total, the set of analogs enabled us to evaluate the importance of both the side chain as well as the core macrolide by testing the activity of the analogs in the MCF-7 and P-388 cells (Table 2). The results were similar in both of the cell lines. By themselves, both the oxazole-containing side chain (4) and the core macrolide (with either the pyran 43 or pyranone 42 functionality) are completely without activity and do not inhibit the proliferation of either cell line even at maximum concentrations. As with elimination of the oxazole-containing side chain, substitution of this native portion with the octanoyl (45) or benzoyl (44) groups also abolished the vast majority of activity, with growth inhibition observed at only the maximum concentration: 10 μM of the octanoyl ester inhibits 25% of P-388 cell growth and 33% of MCF-7 cell growth, while the benzyl ester is somewhat more effective at the same concentration and inhibits 75% and 78%, respectively. It has been shown that the olefin in the side chain, which is approximated in the benzoyl ester (44) but not the octanoyl ester (45), plays a significant role in inhibiting proliferation.53 Both the octanoyl and benzoyl side chains are also significantly shorter in length than the natural side chain. Thus tends to support the recent finding that overall side chain length has importance;53 alternatively, the oxazole ring itself may be paramount, although that possibility remains to be firmly established experimentally. Thus, while other pharmacophores may reside apart from the olefin, the importance of the side chain for full activity is clearly demonstrated by the data presented herein and in agreement with the results of others.12,53
This data also reinforce that the core macrolide is necessary for full growth inhibition. As with the side chain, elimination of the macrolide from the molecule also eliminates activity while smaller scale alterations of its structure also markedly reduce potency. Diastereomer 1 (2), in which the stereochemistry of C13 and C11 were inverted, is 84-fold and 100-fold less potent than neopeltolide in P-388 and MCF-7 cells, respectively. The effects of stereocenter inversion appear to be cumulative, as inversion of C11 alone is reported to result in a 10-fold reduction of activity.53 Diastereomer 2 (34), which in addition to the inversions at C13 and C11 also contained inversions at C3, C5 and C7, is an additional 5-fold less potent than diastereomer 1 (2) in P-388 cells but only 50% less potent in MCF-7 cells. The reason why the additional inversions of C3, C5 and C7 have a greater impact on activity in P-388 cells is not known; however, in either cell line the additional reduction in potency is noteworthy. Taken together, the results show that the activity of the molecule is not easily ascribed to any single pharmacophore; rather, full activity is reliant on the concerted contributions of many structural features -- both in the core macrolide and in the side chain -- of which the removal of any one is detrimental to activity to some degree.
Conclusion
We have completed the total synthesis of neopeltolide and related compounds by employing an aggressive intramolecular Sc(III)-catalyzed cyclization between a dioxinone and an in situ generated oxocarbenium ion to furnish the THP and the macrocycle in a single step. The flexible and convergent synthetic route allowed for facile construction of two diastereomers that ultimately lead to the structural revision and absolute stereochemical determination of neopeltolide. The biological evaluation of the synthetic neopeltolide also confirmed that we had correctly reassigned the structure. Neopeltolide has a variable degree of impact on the growth of different cancer cells that may not be useful against all forms of cancer. The basis for this variation inhibition is not clear, but indicates that neopeltolide may interact selectively with specific (cancer) cells and not adversely affect normal cells in the body, which is suggested by its lack of effect on cultured primary cells. Both the core macrolide and the oxazole side chain bound together play a role in mediating the effect of neopeltolide: neither component on its own is active. The substitution of the side chain and the stereochemistry of the macrolide ring carbons, especially C11 and C13, appear to be important for full biological activity. Further investigations of neopeltolide and related analogs to enhance their properties as potential chemotherapeutic agents are ongoing. These studies reinforce the vital role that total synthesis continues to play in determining the actual structures of natural products and the viability of these molecules to be used as lead compounds in a pharmacological setting.
Supplementary Material
Acknowledgements
This work has been supported by the Sloan Foundation, American Cancer Society (Research Scholar Award to K.A.S.), AstraZeneca, GlaxoSmithKline, Boehringer-Ingelheim, and the H-Foundation. C.M.C. gratefully acknowledges the support of the NIH (GM62120). Funding for the NU Integrated Molecular Structure Education and Research Center (IMSERC) has been furnished in part by the NSF (CHE-9871268). We thank FMCLithium, Sigma-Aldrich, Wacker Chemical Company, and Pressure Chemical Company for generous reagent support, Dr. Thad Franczyk for assistance with the details of catalytic hydrogenation, and Professor Regan Thomson for helpful discussions.
References
- (1).Newman DJ, Cragg GM. J. Nat. Prod. 2004;67:1216–1238. doi: 10.1021/np040031y. [DOI] [PubMed] [Google Scholar]
- (2).Newman DJ, Cragg GM. Curr. Med. Chem. 2004;11:1693–1713. doi: 10.2174/0929867043364982. [DOI] [PubMed] [Google Scholar]
- (3).Newman DJ, Cragg GM, Snader KM. J. Nat. Prod. 2003;66:1022–1037. doi: 10.1021/np030096l. [DOI] [PubMed] [Google Scholar]
- (4).Custar DW, Zabawa TP, Scheidt KA. J. Am. Chem. Soc. 2008;130:804–805. doi: 10.1021/ja710080q. [DOI] [PubMed] [Google Scholar]
- (5).Wright AE, Botelho JC, Guzman E, Harmody D, Linley P, McCarthy PJ, Pitts TP, Pomponi SA, Reed JK. J. Nat. Prod. 2007;70:412–416. doi: 10.1021/np060597h. [DOI] [PubMed] [Google Scholar]
- (6).Ulanovskaya OA, Janjic J, Suzuki M, Sabharwal SS, Schumacker PT, Kron SJ, Kozmin SA. Nat. Chem. Biol. 2008;4:418–424. doi: 10.1038/nchembio.94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (7).Zampella A, DAuria MV, Minale L, Debitus C, Roussakis C. J. Am. Chem. Soc. 1996;118:11085–11088. [Google Scholar]
- (8).Luesch H, Yoshida WY, Harrigan GG, Doom JP, Moore RE, Paul VJ. J. Nat. Prod. 2002;65:1945–1948. doi: 10.1021/np0202879. [DOI] [PubMed] [Google Scholar]
- (9).Klein D, Braekman JC, Daloze D, Hoffmann L, Demoulin V. J. Nat. Prod. 1997;60:1057–1059. doi: 10.1021/np9900324. [DOI] [PubMed] [Google Scholar]
- (10).Tan LT, Marquez BL, Gerwick WH. J. Nat. Prod. 2002;65:925–928. doi: 10.1021/np010526c. [DOI] [PubMed] [Google Scholar]
- (11).Sone H, Kigoshi H, Yamada K. J. Org. Chem. 1996;61:8956–8960. doi: 10.1021/jo961302f. [DOI] [PubMed] [Google Scholar]
- (12).DAmbrosio M, Guerriero A, Debitus C, Pietra F. Helv. Chim. Acta. 1996;79:51–60. [Google Scholar]
- (13).Youngsaye W, Lowe JT, Pohlki F, Ralifo P, Panek JS. Angew. Chem. Int. Ed. 2007;46:9211–9214. doi: 10.1002/anie.200704122. [DOI] [PubMed] [Google Scholar]
- (14).Fuwa H, Naito S, Goto T, Sasaki M. Angew. Chem. Int. Ed. 2008;47:4737–4739. doi: 10.1002/anie.200801399. [DOI] [PubMed] [Google Scholar]
- (15).Paterson I, Miller NA. Chem. Commun. 2008:4708–4710. doi: 10.1039/b812914b. [DOI] [PubMed] [Google Scholar]
- (16).Kartika RG,TR, Taylor RE. Org. Lett. 2008;10:5047–5050. doi: 10.1021/ol802254z. [DOI] [PubMed] [Google Scholar]
- (17).Woo SK, Kwon MS, Lee E. Angew. Chem. Int. Ed. 2008;47:3242–3244. doi: 10.1002/anie.200800386. [DOI] [PubMed] [Google Scholar]
- (18).Vintonyak VV, Maier ME. Org. Lett. 2008;10:1239–1242. doi: 10.1021/ol8001255. [DOI] [PubMed] [Google Scholar]
- (19).Tu Wangyang, Floreancig Paul E. Angew. Chem. Int. Ed. 2009;48:4567–4571. doi: 10.1002/anie.200901489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (20).Morris WJ, Custar DW, Scheidt KA. Org. Lett. 2005;7:1113–1116. doi: 10.1021/ol050093v. [DOI] [PubMed] [Google Scholar]
- (21).Inanaga J, Hirata K, Saeki H, Katsuki T, Yamaguchi M. Bull. Chem. Soc. Jpn. 1979;52:1989–1993. [Google Scholar]
- (22).Schulteelte KH, Hauser A, Ohloff G. Helv. Chim. Acta. 1979;62:2673–2680. [Google Scholar]
- (23).Bahnck KB, Rychnovsky SD. J. Am. Chem. Soc. 2008;130:13177–13181. doi: 10.1021/ja805187p. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (24).Boeckman RK, Jr, Potenza JC, Enholm EJ. J. Org. Chem. 1987;52:469–472. [Google Scholar]
- (25).Hoye TR, Danielson ME, May AE, Zhao H. Angew. Chem. Int. Ed. 2008;47:9743–9746. doi: 10.1002/anie.200804049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (26).Wender PA, DeChristopher BA, Schrier AJ. J. Am. Chem. Soc. 2008;130:6658–6659. doi: 10.1021/ja8015632. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (27).Ley SV, Norman J, Griffith WP, Marsden SP. Synthesis. 1994:639–666. [Google Scholar]
- (28).De Rosa M, Acocelli MR, Villano R, Soriente A, Scettri A. Tetrahedron Lett. 2003;44:6087–6090. [Google Scholar]
- (29).De Rosa M, Acocelli MR, Villano R, Soriente A, Scettri A. Tetrahedron: Asymmetry. 2003;14:2499–2502. [Google Scholar]
- (30).Singer RA, Carreira EM. J. Am. Chem. Soc. 1995;117:12360–12361. [Google Scholar]
- (31).Casiraghi G, Zanardi F, Appendino G, Rassu G. Chem. Rev. 2000;100:1929–1972. doi: 10.1021/cr990247i. [DOI] [PubMed] [Google Scholar]
- (32).Noyori R, Ohta M, Hsiao Y, Kitamura M, Ohta T, Takaya H. J. Am. Chem. Soc. 1986;108:7117–7119. [Google Scholar]
- (33).Deng LS, Huang XP, Zhao G. J. Org. Chem. 2006;71:4625–4635. doi: 10.1021/jo0605086. [DOI] [PubMed] [Google Scholar]
- (34).Myers AG, Yang BH, Chen H, McKinstry L, Kopecky DJ, Gleason JL. J. Am. Chem. Soc. 1997;119:6496–6511. [Google Scholar]
- (35).Nahm S, Weinreb SM. Tetrahedron Lett. 1981;22:3815–3818. [Google Scholar]
- (36).Kurosu M, Kishi Y. Tetrahedron Lett. 1998;39:4793–4796. [Google Scholar]
- (37).Evans DA, Hoveyda AH. J. Am. Chem. Soc. 1990;112:6447–6449. [Google Scholar]
- (38).Rychnovsky SD, Rogers B, Yang G. J. Org. Chem. 1993;58:3511–3515. [Google Scholar]
- (39).Rychnovsky SD, Skalitzky DJ. Tetrahedron Lett. 1990;31:945–948. [Google Scholar]
- (40).Hornberger KR, Hamblett CL, Leighton JL. J. Am. Chem. Soc. 2000;122:12894–12895. [Google Scholar]
- (41).Wang Y, Janjic J, Kozmin SA. J. Am. Chem. Soc. 2002;124:13670–13671. doi: 10.1021/ja028428g. [DOI] [PubMed] [Google Scholar]
- (42).Phillips AJ, Uto Y, Wipf P, Reno MJ, Williams DR. Org. Lett. 2000;2:1165–1168. doi: 10.1021/ol005777b. [DOI] [PubMed] [Google Scholar]
- (43).Stille JK. Angew. Chem. Int. Ed. 1986;25:508–523. [Google Scholar]
- (44).Dess DB, Martin JC. J. Am. Chem. Soc. 1991;113:7277–7287. [Google Scholar]
- (45).Still WC, Gennari C. Tetrahedron Lett. 1983;24:4405–4408. [Google Scholar]
- (46).DeMico A, Margarita R, Parlanti L, Vescovi A, Piancatelli G. J. Org. Chem. 1997;62:6974–6977. [Google Scholar]
- (47).Mitsunobu O, Yamada M. Bull. Chem. Soc. Jpn. 1967;40:2380. &. [Google Scholar]
- (48).Rychnovsky SD, Marumoto S, Jaber JJ. Org. Lett. 2001;3:3815–3818. doi: 10.1021/ol0168465. [DOI] [PubMed] [Google Scholar]
- (49).Roush WR, Dilley GJ. Synlett. 2001:955–959. [Google Scholar]
- (50).Jasti R, Rychnovsky SD. J. Am. Chem. Soc. 2006;128:13640–13648. doi: 10.1021/ja064783l. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (51).Dale JA, Mosher HS. J. Am. Chem. Soc. 1968;90:3732–3740. [Google Scholar]
- (52).For all details and procedures for biological evaluations of neopeltolide and analogs, see Supporting Information.
- (53).Vintonyak VV, Kunze B, Sasse F, Maier ME. Chem. Eur. J. 2008;14:11132–11140. doi: 10.1002/chem.200801398. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.