

Abstract
Mitochondrial dysfunction is a prominent feature of Alzheimer's disease (AD) neurons. In this study, we explored the involvement of an abnormal mitochondrial dynamics by investigating the changes in the expression of mitochondrial fission and fusion proteins in AD brain and the potential cause and consequence of these changes in neuronal cells. We found that mitochondria were redistributed away from axons in the pyramidal neurons of AD brain. Immunoblot analysis revealed that levels of DLP1 (also referred to as Drp1), OPA1, Mfn1, and Mfn2 were significantly reduced whereas levels of Fis1 were significantly increased in AD. Despite their differential effects on mitochondrial morphology, manipulations of these mitochondrial fission and fusion proteins in neuronal cells to mimic their expressional changes in AD caused a similar abnormal mitochondrial distribution pattern, such that mitochondrial density was reduced in the cell periphery of M17 cells or neuronal process of primary neurons and correlated with reduced spine density in the neurite. Interestingly, oligomeric amyloid-β-derived diffusible ligands (ADDLs) caused mitochondrial fragmentation and reduced mitochondrial density in neuronal processes. More importantly, ADDL-induced synaptic change (i.e., loss of dendritic spine and postsynaptic density protein 95 puncta) correlated with abnormal mitochondrial distribution. DLP1 overexpression, likely through repopulation of neuronal processes with mitochondria, prevented ADDL-induced synaptic loss, suggesting that abnormal mitochondrial dynamics plays an important role in ADDL-induced synaptic abnormalities. Based on these findings, we suggest that an altered balance in mitochondrial fission and fusion is likely an important mechanism leading to mitochondrial and neuronal dysfunction in AD brain.
Introduction
Characterized by neurofibrillary tangles, senile plaques, and progressive loss of neuronal cells in selective brain regions, Alzheimer's disease (AD) is a fatal degenerative dementia with initial memory impairment that progresses to a total debilitating loss of mental and physical faculties (Smith, 1998). Evidence suggests that memory failure in AD is a result of synaptic loss and, among all the early changes that occur in AD, is a robust correlate of AD-associated cognitive deficits, leading to the notion that synaptic dysfunction plays a critical role in the pathogenesis of AD (DeKosky and Scheff, 1990; Terry et al., 1991; Coleman et al., 2004). A myriad of studies have focused on the central role of amyloid-β (Aβ) in the pathogenesis and progression of AD. Aβ oligomers may bind to dendritic spines, causing synaptic dysfunction (Lacor et al., 2004); however, the molecular mechanism remains elusive.
In mammals, mitochondria are vital organelles known for their essential role in energy metabolism and cell survival (Delettre et al., 2000; Benard et al., 2007). Neurons are particularly vulnerable to mitochondrial dysfunction because of their high metabolic rate dependence and complex morphology (Delettre et al., 2000; Benard et al., 2007). More specifically, mitochondria are of pivotal importance for synaptic development and plasticity, and changes in mitochondria distribution and/or function can result in synaptic dysfunction and loss (Li et al., 2004; Verstreken et al., 2005). Notably, whereas defects in mitochondrial function are a prominent early event in AD (Blass et al., 2000; Swerdlow and Kish, 2002; Zhu et al., 2004; Bubber et al., 2005), whether and how mitochondrial defects contribute to synaptic dysfunction in AD neurons is unclear.
Emerging evidence suggests that mitochondria are dynamic organelles undergoing constant fission and fusion regulated by a machinery involving large dynamin-related GTPases (Chan, 2006). Mitochondrial dynamics not only determines mitochondrial morphology and size, but also regulates mitochondrial distribution and function (Chan, 2006). Given the critical role of mitochondria in neurons, impaired mitochondrial dynamics is increasingly implicated in neurodegenerative diseases including Parkinson and Huntington diseases (Exner et al., 2007; Deng et al., 2008; Mortiboys et al., 2008; Poole et al., 2008; Yang et al., 2008; Park et al., 2009; Wang et al., 2009). In relation to AD, Aβ caused rapid and severe impairment of mitochondrial transport (Rui et al., 2006). More recently, we demonstrated an abnormal mitochondrial morphology and distribution in AD fibroblasts compared with normal healthy fibroblasts from age-matched controls (Wang et al., 2008a) and found that amyloid-β protein precursor (AβPP) overexpression, through Aβ overproduction, caused abnormal mitochondrial dynamics in neurons (Wang et al., 2008b). In this study, we aimed to determine whether an imbalance in mitochondrial fission and fusion occurs in vivo in AD neurons by exploring the expression of mitochondrial fission/fusion proteins [i.e., DLP1 (also referred to as Drp1), Fis1, OPA1 and Mfn1/2] in AD brain and identifying the potential cause and consequence of changes in these proteins related to mitochondrial dynamics and synaptic function.
Materials and Methods
Cell culture and transfection.
The human neuroblastoma cell line, M17, was grown in Opti-MEM medium (Invitrogen), supplemented with 5% or 10% (v/v) fetal bovine serum and 1% penicillin–streptomycin, in 5% CO2 in a humid incubator at 37°C. Cells were transfected using Effectene (Qiagen) according to the manufacturer's instructions. For cotransfection, a 3:1 ratio (indicated plasmid: Mito-DsRed2) was applied. Regular culture medium containing 300 μg/ml Geneticin (Invitrogen), 20 μg/ml Blasticidin (Invitrogen), or 150 μg/ml of hygromycin B (Calbiochem) was used for stable cell line selection. The selective medium was replaced every 3 d until the appearance of foci, each apparently derived from a single stably transfected cell. Stable cell lines were then picked and maintained with 100 μg/ml Geneticin, 5 μg/ml Blasticidin, or 30 μg/ml hygromycin. Primary neurons from embryonic day 18 (E18) rat hippocampus (BrainBits) were seeded at 30,000–40,000 cells per well on eight-well chamber slides coated with poly-d-lysine/laminin (BD Biosciences) in neurobasal medium supplemented with 2% B27 (Invitrogen)/0.5 mm glutamine/25 mm glutamate. One-half of the culture medium was changed every 3 d with neurobasal medium supplemented with 2% B27 (Invitrogen) and 0.5 mm glutamine. All cultures were kept at 37°C in a humidified 5% CO2-containing atmosphere. More than 90% of the cells were neurons after they were cultured for 7–17 d of culture in vitro (DIV), which was verified by positive staining for the neuronal specific markers microtubule-associated protein-2 (MAP2, dendritic marker) and Tau-1 (axonal marker). At DIV 7–12, neurons were transfected using Neurofect (Genlantis) according to the manufacturer's protocol.
Expression vectors and antibodies.
Mito-DsRed2 construct (Clontech), green fluorescent protein (GFP)-tagged wild-type DLP1 (DLP1wt), mutant DLP1 K38A and Fis1 constructs (gifts from Dr. Yisang Yoon, University of Rochester, Rochester, NY), yellow fluorescent protein (YFP)-tagged postsynaptic density protein 95 (PSD-95) (gift from Dr. Ann Marie Craig, University of British Columbia, Vancouver, British Columbia, Canada), and wild-type OPA1 (OPA1wt) and mutant OPA1 K301A constructs (gifts from Dr. Luca Scorrano, Venetian Institute of Molecular Medicine, Venice, Italy) were obtained. The expression plasmids for Myc-tagged wild-type DLP1/DLP1 K38A were constructed based on the pCMV-Tag3 vector (Stratagene), whereas Myc/GFP-tagged wild-type OPA1/OPA1 K301A was based on pCMV-Tag vector (Stratagene) and pcDNA3.1 (Invitrogen), respectively. The micro-RNA (miR) RNA interference (RNAi) vector was generated via the pcDNA 6.2-GW/EmGFP-miR construct (Invitrogen). The miR RNAi sequence targeting the open reading frame region of human or rat mitochondrial fission or fusion proteins are as follows: human OPA1 (ATCACTTGCAAGATAAGCTGG), rat OPA1 (GCGCAGTATTGTTACAGACTT), human Mfn2 (CTCCTTAGCAGACACAAAGAA), rat Mfn2 (GCTGGACAGCTGGATTGATAA), human Mfn1 (ATTTCTTCCCATACCATCCTC), rat Mfn1 (GCAAACAAGGTTTCTTGTGCA), human Fis1 (TTACGGATGTCATCATTGTAC), and rat Fis1 (TGGTGCCTGGTTCGAAGCAAA). miR RNAi construct targeting both human and rat DLP1 was described as previously (Wang et al., 2008a). Primary antibodies used included mouse anti-DLP1/OPA1 (BD Biosciences), mouse anti-Mfn1 (Novus Biologicals), mouse anti-Mfn2/cytochrome c oxidase subunit 1 (COX I) (Sigma), rabbit anti-Fis1 (Imgenex), mouse anti-glyceraldehyde-3-phosphate dehydrogenase (anti-GAPDH) (Chemicon), rabbit anti-α-tubulin (Epitomics), rabbit anti-phospho-DLP1 (Ser616) (Cell Signaling), and mouse anti-α-tubulin/Myc/COX IV (Cell Signaling).
Immunocytochemical procedures.
Hippocampal, frontal cortical, and cerebellar brain tissue obtained postmortem was fixed and consecutive 6-μm-thick sections were prepared essentially as described previously (Zhu et al., 2000). Cases used in this study included AD (n = 5) and control (n = 5) cases. Immunocytochemistry was performed by the peroxidase–antiperoxidase protocol as described before (Zhu et al., 2000). Absorption experiments were performed to verify the specificity of antibody binding. Each case was examined on a Zeiss Axiophot microscope for neurons stained with DLP1, OPA1, Mfn1, Mfn2, Fis1, COX I (Molecular Probes), and tubulin. Positively stained neurons were counted.
Western blot analysis.
Brain samples from gray matter of temporal cortex of AD (n = 13; ages = 60–89 years, postmortem interval = 1–24 h) and age-matched control (n = 12, ages = 58–92 years; postmortem interval = 2–23 h) cases [based on clinical and pathological criteria established by CERAD (the Consortium to Establish a Registry for Alzheimer's Disease) and an National Institute on Aging consensus panel (Khachaturian and Emr, 1984; Mirra, 2000)] or cells were lysed with 1× Cell Lysis Buffer (Cell Signaling), plus 1 mm PMSF (Sigma) and Protease Inhibitor Cocktail (Sigma). Equal amounts of total protein extract (5 μg or 20 μg) were resolved by SDS-PAGE and transferred to Immobilon-P (Millipore). After blocking with 10% nonfat dry milk, primary and secondary antibodies were applied as previously described (Wang et al., 2008a) and the blots developed with Immobilon Western Chemiluminescent HRP Substrate (Millipore).
Electron microscopy.
Hippocampus tissue fixed in 2% paraformaldehyde was embedded in London Resin gold after ethanol dehydration and polymerized with UV exposure. Sections (60–100 nm) were placed on nickel grids and incubated with COX antibody followed by gold-conjugated secondary antibody. Grids were stained with uranyl acetate and lead citrate and viewed in a Zeiss electron microscope at 80 kV (CEM902; Zeiss, Oberkochen, Germany).
Cell treatments and measurements.
Amyloid-β-derived diffusible ligands (ADDLs) were prepared using Aβ1-42 peptide (California Peptide) as described before (Klein, 2002), except that phenol red free neurobasal medium (Invitrogen) was used instead of phenol red free F12 medium. Primary hippocampal cells were usually treated at DIV 7–16. The negative control Aβ42-1 peptide (Anaspec) was subject to the same preparation procedure as ADDLs were. Cytotoxicity was measured by use of a cytotoxicity detection kit (LDH; Roche). For differentiation of M17 cells, serum content was reduced to 2%, and 1 μm retinoic acid (Sigma-Aldrich) was included in the culture medium.
To measure mitochondrial reactive oxygen species (ROS), the fluorescent probe MitoSOX (Invitrogen) was used according to the manufacturer's protocol. Briefly, neurons were incubated with 2.5 μm MitoSOX in culture medium for 10 min, followed by three washes with prewarmed Hanks' balanced salt solution (Invitrogen). Cells were fixed in 4% paraformaldehyde for 30 min and then stained and observed using a Zeiss LSM 510 inverted fluorescence microscope.
Mitochondria were harvested from human brain samples or cells by use of a mitochondrial isolation kit (Pierce) following the manufacturer's protocol. The cytosolic extracts were also used to determine DLP1 levels. The biotin-switch assay was performed using an S-nitrosylated protein detection assay kit (Cayman) following the manufacturer's protocol. Biotinylated DLP1 was immunoprecipitated with anti-DLP1 antibody in radioimmunoassay precipitation buffer using Protein G-agarose beads (Roche Diagnostics) according to the manufacturer's protocol and analyzed by Western blot using avidin–HRP (Cayman).
Immunofluorescence.
Cells were cultured on chamber slides. After treatment, cells were fixed and stained as described previously (Wang et al., 2008a). All fluorescence images were captured with a Zeiss LSM 510 inverted fluorescence microscope or a Zeiss LSM 510 inverted laser-scanning confocal fluorescence microscope. Confocal images of far-red fluorescence were collected using 633 nm excitation light from a HeNe laser and a 650 nm long-pass filter; images of Mito-DsRed2 red fluorescence were collected using 543 nm excitation light from an argon laser and a 560 nm long-pass filter; and green fluorescence images were collected using 488 nm excitation light from an argon laser and a 500–550 nm bandpass barrier filter. Image analysis was performed with open-source image analysis program Wright Cell Imaging Facility (WCIF) ImageJ (developed by W. Rasband; NIH).
Time-lapse imaging.
Neurons were seeded in glass-bottom dishes (MatTek) and then transfected with Mito-Dendra2. Forty-eight hours after transfection, cells were placed in a well-equipped live imaging station (Zeiss CTI-Controller 3700) with controlled CO2 content, humidity, and temperature of stage, objective, and the air. Images were captured with a Zeiss LSM 510 inverted laser-scanning confocal fluorescence microscope. Images of red signal were collected using 543 nm excitation light from an argon laser and a 560 nm long-pass filter; those of green fluorescence were collected using 488 nm excitation light from an argon laser and a 500–550 nm bandpass barrier filter. During time-lapse imaging, frames were captured every 10 s for at least 1 h without apparent phototoxicity or photobleaching. Image analysis was also performed with open-source image-analysis programs WCIF ImageJ (developed by W. Rasband; NIH).
Results
Changes in levels and distribution of mitochondrial fission/fusion proteins in neurons from AD hippocampus
We investigated the expression pattern of mitochondrial fission proteins (i.e., DLP1 and Fis1) and fusion proteins (i.e., OPA1, Mfn1, and Mfn2) in hippocampal tissues from 13 AD patients and 12 age-matched normal subjects. Immunoblot analysis revealed significant changes in the levels of all five proteins: DLP1 levels were dramatically reduced by 74.3% in AD brains, and OPA1, Mfn1, and Mfn2 levels were also reduced by 61.4%, 27.8%, and 33.6%, respectively. Interestingly, unlike other fission/fusion proteins, Fis1 levels were significantly increased 4.8-fold in AD brains compared with age-matched control brains (Fig. 1A, B). No significant changes in overall mitochondrial contents were noted between AD and control samples, as evidenced by the constant expression levels of COX IV, a mitochondrial marker (Fig. 1A). Real-time PCR studies of five control and five AD cases confirmed a 60% increase in Fis1 mRNA levels in AD but no changes in mRNA levels of other mitochondrial fission/fusion proteins (not shown). Immunocytochemical analysis of these proteins revealed extensive immunoreactivity of each of these proteins in pyramidal neurons in hippocampal tissues from age-matched control patients (Fig. 1C). Interestingly, only background neuronal immunoreactivity of DLP1 was noted in the hippocampus from AD patients (Fig. 1C), consistent with the greatly reduced levels revealed by immunoblot analysis. In contrast, extensive neuronal immunoreactivity was noted for the other four mitochondrial fission/fusion proteins in hippocampal tissues from AD patients; however, they displayed a distinctive distribution pattern compared with that of control hippocampus (Fig. 1C). Although all these proteins demonstrate a uniform distribution throughout the soma and neurites of pyramidal neurons in age-matched control hippocampus, in AD hippocampal neurons, they accumulated in the soma and were depleted in the neurite (Fig. 1C). Positively stained neurons were counted and categorized into three groups: neurons with only soma staining, neurons with only axon staining, and neurons with both axon and soma staining. Indeed, all of these four proteins (i.e., Fis1, OPA1, Mfn1, and Mfn2) demonstrated only soma staining in >80% of pyramidal neurons in AD hippocampus, significantly different from what was seen for control hippocampal neurons (Fig. 1D). Antibodies against each of these fission/fusion proteins from two different sources were used in both immunocytochemical and immunoblot studies, and similar results were obtained (not shown). The specificity of each antibody was confirmed by preabsorption of primary antibodies with their immunizing peptides. Since Fis1, Mfn1, and Mfn2 are mitochondrial outer membrane proteins and OPA1 is a mitochondrial inner membrane protein, the consistent pattern of their redistribution to soma in AD pyramidal neurons implies that mitochondria are redistributed in these neurons.
Figure 1.

Mitochondrial fission and fusion protein expression and distribution in AD hippocampus. A, B, Representative immunoblot (A) and quantification (B) analysis revealed that in AD hippocampus (n = 13), levels of DLP1/OPA1/Mfn1/Mfn2 were reduced significantly, whereas levels of Fis1 were increased significantly compared with age-matched controls (n = 12) (*p < 0.05, Student's t test). Equal protein amounts (30 μg) were loaded and confirmed with actin staining (A). C, Representative immunocytochemistry of DLP1, OPA1, Mfn1, Mfn2, and Fis1 in hippocampus from AD (right) and age-matched controls (left). Scale bars, 50 μm. D, Positively stained neurons were categorized into three groups: neurons with soma staining only, neurons with axon staining only, and neurons with both axon and soma staining. Quantification in five AD brains and five age-matched controls indicated that Fis1, OPA1, Mfn1, and Mfn2 demonstrated soma staining in >80% of pyramidal neurons in AD hippocampus, significantly different from what was seen for control hippocampal neurons.
Mitochondria are redistributed in AD pyramidal neurons
To confirm the altered distribution of mitochondria in AD neurons, we used a widely used mitochondrial marker, COX I, which also demonstrated exclusive neuronal staining in both AD and age-matched control cases (Fig. 2A). The mitochondrial localization of COX I in pyramidal neurons was confirmed by electron microscopy (Fig. 2B). Notably, immunocytochemical analysis also revealed striking changes in the distributions of COX I immunoreactivity within neurons between AD and control cases: localization of COX I in control cases is seen throughout the soma and processes of most pyramidal neurons (>85%) of the hippocampus, whereas in AD cases, the immunoreactivity is essentially limited to the soma with no more than 20% neurons demonstrating positively stained processes. Since some neurons may lose processes in sections as a result of the angle of cutting in both AD and control cases, which may mask the difference, to further clarify the changes in COX I distribution, immunofluorescence analysis of tubulin and COX I double staining of AD and age-matched control hippocampal tissues was performed (Fig. 2C). Quantitative analysis of tubulin staining revealed no significant difference in the percentages of neurons with soma staining only, neurite staining only, and both soma and processes staining between AD and age-matched control sections (Fig. 2D). However, in control cases, the majority (>90%) of those neurons with long processes contain COX I throughout the soma and processes, whereas in AD cases, only a minority of those neurons with long processes contain COX I in the soma and processes (<20%) and the majority of those neurons with long processes contain COX I in the soma only (Fig. 2C, D). Since COX I is a mitochondrial inner membrane protein widely used as a mitochondrial marker, this finding confirms that mitochondria accumulate in the soma and are reduced in neuronal processes in AD pyramidal neurons.
Figure 2.

Altered distribution of mitochondria in AD. A, Representative immunocytochemistry of COX I confirmed that in AD hippocampus, COX I staining of neuronal processes was significantly reduced compared with age-matched controls (*p < 0.05, Student's t test). B, A representative immunoelectron micrograph of COX I gold labeling of mitochondria is shown. C, A representative immunofluorescence picture of COX I staining (green) and tubulin (red) in AD hippocampus (bottom) and age-matched control (top). D, Quantification revealed that mitochondria were evenly distributed in the control (n = 5), whereas they were significantly constricted in the soma in AD hippocampus (n = 5) (*p < 0.05, Student's t test).
Changes in subcellular localization and modification of DLP1 in AD brain
Previously we reported shorter mitochondria in AD neurons (Wang et al., 2008b), suggestive of enhanced mitochondrial fission. DLP1 is known to play a critical role in the fission process; however, our results indicated reduced levels of DLP1 in AD neurons. Given that the majority of DLP1 in mammalian cells is cytosolic and it is the mitochondrial DLP1 that participates in mitochondrial fission (Smirnova et al., 2001), we hypothesized that there may be comparable or even increased levels of mitochondrial DLP1 in AD compared with control brain samples. To test this hypothesis, we conducted subcellular fractionation experiments from AD and control brain homogenates, and the resultant fractions were analyzed by probing immunoblots with anti-DLP1 antibody as well as antibodies for GAPDH to track cytosolic fractions, and with COX IV to track mitochondrial fractions. We found slightly increased levels of mitochondrial DLP1 (not significant) in AD brain samples (Fig. 3A). The immunoblot analysis also confirmed significantly reduced levels of cytosolic DLP1 in AD samples (Fig. 3A). Phosphorylation of DLP1 at several sites regulates its mitochondrial fission activity: whereas there is still debate over whether phosphorylation of DLP1 at Ser637 enhances or inhibits mitochondrial fission activity (Cereghetti et al., 2008; Han et al., 2008), it appears that phosphorylation of DLP1 at Ser 616 activates mitochondrial fission activity (Taguchi et al., 2007). Given the known imbalance of kinase and phosphatase activities such that increased phosphorylation of multiple proteins was identified in AD brain, we determined the levels of DLP1 phosphorylated at Ser616 in AD brain and found significantly increased phosphorylated DLP1 at this site in both mitochondrial and cytosolic fractions from AD brains compared with control brains (Fig. 3A). A recent study suggested that S-nitrosylation of DLP1 activates GTPase activity and mitochondrial fission and reported increased S-nitrosylation of DLP1 in AD brain tissues (Cho et al., 2009); we also determined DLP1 nitrosylation in our studies by biotin-switch assay. Indeed, we confirmed significantly increased S-nitrosylation of DLP1 in AD brain tissues (Fig. 3B). These data suggest that, despite the reduction in overall levels of DLP1, there were comparable levels of mitochondrial DLP1 in AD neurons, which likely contributes to enhanced mitochondrial fission.
Figure 3.

Changes in subcellular localization and modification of DLP1 in AD brain. A, Representative immunoblot and quantification analysis of DLP1 and phospho-DLP1 (Ser616) (p-DLP1) in the mitochondrial and cytosolic fraction from AD brains (n = 8) and age-matched control brains (n = 8) indicated that, although relative mitochondrial DLP1 level (DLP1/COX IV) did not change significantly, the relative ratio of p-DLP1 to DLP1 increased significantly in both mitochondrial and cytosolic fractions from AD brain samples. All samples were also immunoblotted with antibodies to detect mitochondrial (COX IV) and cytoplasmic (GAPDH) markers. The absence of GAPDH in the mitochondrial fraction confirms the purity of preparations from brain tissues. B, Representative immunoblot and quantification analysis of S-nitrosylated DLP1 (SNO-DLP1) by biotin-switch assay of AD brains (n = 8) and age-matched control brains (n = 8) showed that relative SNO-DLP1 formation increased significantly in AD. All experiments were repeated three times (*p < 0.05, Student's t test).
Modulations of mitochondrial fission/fusion proteins, mimicking changes in AD neurons, cause abnormal mitochondrial distribution in M17 cells
Since mitochondrial fission/fusion proteins not only control mitochondrial morphology but also regulate mitochondrial distribution, we hypothesized that changes in mitochondrial fission/fusion proteins may also change mitochondrial morphology and distribution in AD neurons. To test this hypothesis, we evaluated whether changes in these proteins, mimicking the changes found in AD neurons, affected mitochondrial morphology and distribution in the M17 human neuroblastoma cell line. Stable M17 cell lines were generated with reduced expression of DLP1, OPA1, Mfn1, and Mfn2, i.e., mitochondrial fission/fusion proteins that demonstrated reduced levels in AD neurons, and overexpression of Fis1, i.e., the mitochondrial fission/fusion protein that demonstrated increased levels in AD neurons. The overexpression or reduced expression of mitochondrial fission/fusion proteins was confirmed by Western blotting (Fig. 4A). There was no increase in basal cell death in any of the cell lines compared with control cells (i.e., untransfected or empty vector-transfected cells) (data not shown). To visualize mitochondria, stable M17 cell lines were transiently transfected with Mito-DsRed2. Forty-eight hours after transfection, cells were fixed, stained, and evaluated using laser confocal microscopy (Fig. 4B). Detailed analysis of mitochondrial length revealed that these manipulations, mimicking changes in AD neurons, caused differential effects on mitochondrial morphology: the majority of the M17 cells in control lines demonstrated short tubular-form mitochondria, while almost 100% of stable M17 cells with reduced DLP1 expression demonstrated elongated mitochondria, and the majority of M17 cells with reduced OPA1, Mfn1, or Mfn2 expression or increased Fis1 expression demonstrated fragmented mitochondria (72.6 ± 8.1%, 77.4 ± 7.3%, 63.2 ± 4.2%, or 84.7 ± 6.4%, respectively) (Fig. 4C). Most notably, despite their differential effects on morphology, all manipulations caused a similar abnormal effect on mitochondrial distribution when compared with untransfected or empty vector-transfected control cells: in control cells, mitochondria were distributed evenly throughout the cytoplasm (>95% cells) (Fig. 4D). However, in stable M17 cells with reduced DLP1 expression, almost 100% of the cells demonstrated an abnormal mitochondrial distribution with mitochondria aggregating in the perinuclear area, whereas more remote cytoplasmic areas were devoid of mitochondria; 63.5 ± 3.2%, 87.8 ± 3.2%, 78.3 ± 5.3%, or 82.5 ± 7.3% of cells in the stable M17 cell lines with reduced OPA1, Mfn1, or Mfn2 expression or increased Fis1 expression, respectively, demonstrated similar abnormal mitochondrial distributions (Fig. 4D).
Figure 4.

Modulations of mitochondrial fission/fusion proteins, mimicking changes in AD neurons, cause abnormal mitochondrial distribution in M17 cells. A, Representative immunoblot analysis of M17 cells stably knocking down or overexpressing mitochondrial fission and fusion proteins. Equal protein amounts (15 μg) were loaded. Tubulin immunoblot was used as an internal loading control. B, Representative confocal pictures of mitochondria in M17 cells either stably overexpressing (OE) or knocking down (RNAi) mitochondrial fission/fusion proteins. Cells were transfected with Mito-DsRed2 to label mitochondria. For knockdown experiments, GFP was tagged to the micro-RNAi construct. For overexpression experiments, tubulin was immunostained as green to label the cell shape. C, D, Quantifications of mitochondrial morphology (C) and abnormal distribution (D) in M17 cell either stably overexpressing or knocking down mitochondrial fission/fusion proteins revealed differential effects on mitochondrial morphology but similar effects on abnormal mitochondrial distribution for those manipulations mimicking changes in AD. Scale bars, 20 μm. At least 500 cells were analyzed in triplicate for each cell line (*p < 0.05, Student's t test).
We also generated stable M17 cell lines overexpressing dominant negative DLP1 or OPA1 mutants. As expected, they caused effects similar to those of DLP1 or OPA1 knockdown on mitochondrial morphology and distribution (Fig. 4C, D): overexpression of the DLP1 K38A mutant caused elongated mitochondria, whereas overexpression of OPA1 K301A mutant caused fragmented mitochondria (Fig. 4C), but both caused abnormal mitochondrial distribution with mitochondria accumulating in the perinuclear area (Fig. 4D).
In addition, we further generated stable M17 cells lines overexpressing DLP1 or OPA1 or with reduced expression of Fis1, i.e., changes opposite to those observed in AD neurons (Fig. 4C, D). The analysis of mitochondrial morphology and distribution in these cell lines revealed that DLP1 overexpression caused primarily fragmented mitochondria, and Fis1 knockdown caused primarily elongated mitochondria, whereas overexpression of OPA1 in cells resulted in a heterogeneous population of mitochondria that varied in shape (Fig. 4C). However, neither of these manipulations caused abnormal perinuclear accumulation of mitochondria (Fig. 4D).
Modulations of mitochondrial fission/fusion proteins, mimicking changes in AD, cause abnormal mitochondrial distribution in differentiated primary neuronal cells
To study the effect of modulating this mitochondrial fission/fusion expression in differentiated cells, primary rat hippocampal neurons (DIV 7–12) were transfected with GFP-tagged miR RNAi expression vectors targeting DLP1, OPA1, Mfn1, or Mfn2 or a Myc-tagged Fis1 expression vector. Cells were fixed and stained 3 or 4 d after transfection. Positively transfected cells can be identified by GFP fluorescence or Myc immunostaining. In nontransfected control hippocampal neurons, mitochondria were abundant and overlapping in soma and proximal neurites but were separated from each other and evenly distributed in distal neurites and usually demonstrated short tubular forms with an average length of 2.1 ± 0.08 μm (Fig. 5A, B). In DLP1 knockdown neurons, many mitochondria in neurites became extremely long (>10 μm) and the average mitochondrial length increased significantly (4.1 ± 0.56 μm) (Fig. 5B). However, mitochondrial number was significantly reduced in neuronal processes (Fig. 5C), leaving large lengths of neurites devoid of mitochondria (Fig. 5E). Indeed, the neurite mitochondrial index (total mitochondrial length/neurite length) in neuronal processes 200 μm in length beginning from the cell body, used as an index for mitochondrial coverage in neuronal processes, was significantly decreased (Fig. 5D). OPA1, Mfn1, and Mfn2 knockdown as well as Fis1 overexpression caused mitochondria to become globular and significantly shorter in positively transfected neurons (Fig. 5A, B). Mitochondrial aggregates were often observed in the neuronal processes of Mfn1 knockdown or Fis1-overexpressing neurons. Most importantly, despite their differential effect on mitochondrial morphology compared with DLP1 knockdown, manipulations of OPA1, Mfn1, Mfn2, or Fis1 all caused significantly reduced neuritic mitochondrial density (Fig. 5C), decreased neurite mitochondrial index (Fig. 5D), and increased length of neurites devoid of mitochondria in the neuronal processes (Fig. 5E) in positively transfected hippocampal neurons, a phenomenon also observed in DLP1 knockdown neurons, consistent with the redistribution pattern of mitochondria in M17 cells.
Figure 5.

Modulations of mitochondrial fission/fusion proteins, mimicking changes in AD, cause similar abnormalities in differentiated primary neuronal cells. A, Representative pictures of mitochondria in primary rat E18 hippocampal neurons (DIV 7–12) transiently transfected with GFP-tagged miR RNAi expression vector targeting DLP1, OPA1, Mfn1, or Mfn2 (DLP1, OPA1, Mfn1, or Mfn2 RNAi) or a Myc-tagged Fis1 expression vector (Fis1 OE) and Mito-DsRed2 (Red) to label mitochondria. For Myc-tagged Fis1 overexpression, a GFP-expressing vector was also cotransfected to show neurites and soma. Positively transfected cells were identified by GFP fluorescence (green) or Myc immunostaining (white). B–E, Quantification of mitochondrial length (B), number (C), neurite mitochondrial index (total mitochondrial length/neurite length) (D), and axial length of neurites devoid of mitochondria (E) in a segment of neuronal process 400 μm in length beginning from the cell body of neurons either overexpressing or knocking down mitochondrial fission/fusion proteins (*p < 0.05, Student's t test). Scale bars, 20 μm. At least 20 cells were analyzed in each experiment, and experiments were repeated three times.
Differentiated hippocampal neurons were also transiently transfected with dominant negative DLP1 or OPA1 mutants and essentially caused effects similar to DLP1 or OPA1 knockdown on mitochondrial morphology and distribution (Fig. 5B–E). In contrast, overexpression of DLP1 caused fragmented mitochondria but increased mitochondrial number in neuronal processes with an overall increase in neurite mitochondrial index (Fig. 5B–E). OPA1 overexpression led to a heterogeneous effect on mitochondrial morphology and Fis1 knockdown caused mitochondria elongation, but neither changed mitochondrial density in neuronal processes or neurite mitochondrial index in rat primary neurons (Fig. 5B–E).
Effect of mitochondrial fission/fusion proteins on dendritic spines
As one approach to determine whether alterations in mitochondrial dynamics correlate with neuronal changes, we compared the number of dendritic spines, an important index of synaptic plasticity. Rat E18 primary hippocampal neurons (DIV 9) were transfected with GFP-tagged miR RNAi expression vectors targeting DLP1, OPA1, Mfn1, or Mfn2 or a Myc-tagged Fis1 expression vector together with Mito-DsRed2 to label mitochondria. Three days later, neurons were fixed and stained. Positively transfected neurons were identified by GFP fluorescence or Myc immunostaining. For each neuron, dendritic segments with 100–200 μm in length beginning 100 μm from the cell body were selected. In dendrites with large diameters, mitochondria were overlapping and abundant in the proximal area but separated from each other in the distal area similarly to what was seen for dendrites with small diameter. Protrusions of 0.5–5 μm in length that had a clear neck and expanded mushroom-like heads or a stubby shape were defined as dendritic spines. At DIV 12, dendrites of control neurons were extensively covered with dendritic spines (Fig. 6A). However, neurons with DLP1 downregulation showed a markedly reduced spine density (1.1 ± 0.35/10 μm) compared with nontransfected or control RNAi-transfected neurons (4.2 ± 0.46/10 μm). Similarly OPA1, Mfn1, or Mfn2 knockdown as well as Fis1 overexpression also caused markedly reduced spine density (Fig. 6A, B). To determine whether these changes correlated with a loss of mitochondria in the vicinity of the spine, the frequency of spine occupancy by mitochondria was measured. It was found that the number of spines containing at least one mitochondrion in a 1-μm-diameter region centered in the middle of the spine was significantly reduced in all neurons with DLP1/OPA1/Mfn1/Mfn2 knockdown or Fis1 overexpression (Fig. 6C).
Figure 6.
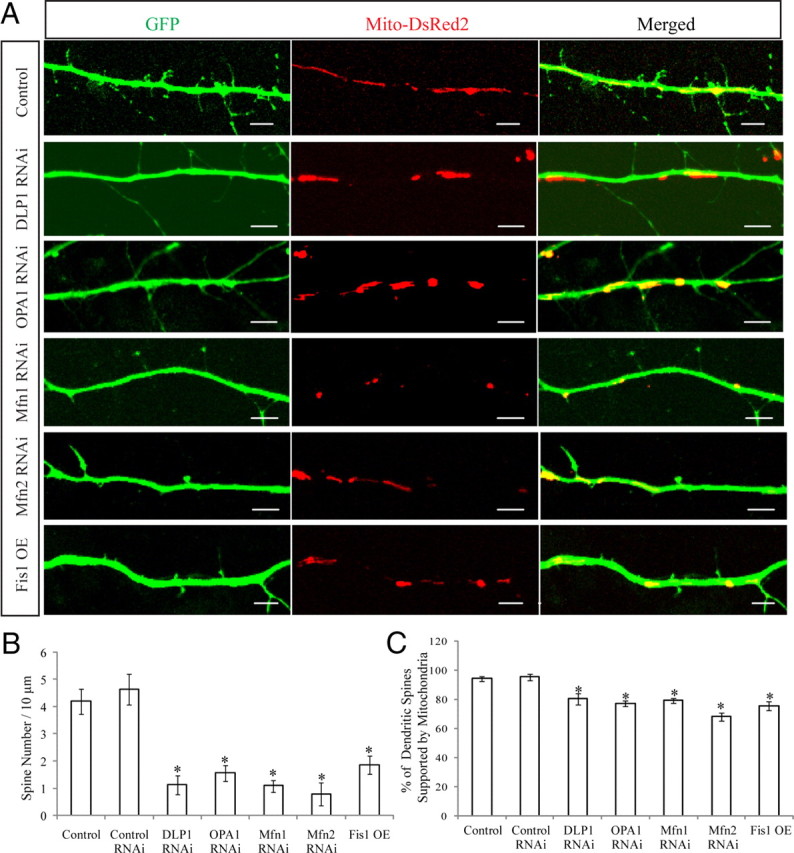
Effect of mitochondrial fission/fusion proteins on dendritic spines. Primary rat E18 primary hippocampal neurons were transfected at DIV 9 with the indicated plasmids. For each neuron, dendritic segments of 100–200 μm in length beginning 100 μm from the cell body were selected. A, Representative pictures of positively transfected neurons are shown. B, C, Quantification of spine number and the percentage of spines supported by mitochondria (*p < 0.05, Student's t test). Scale bars, 5 μm. At least 20 cells were analyzed in each experiment, and experiments were repeated three times.
Effects of Aβ on mitochondrial dynamics in primary neurons
It is believed that soluble Aβ oligomers are involved in the pathogenesis of AD. We previously demonstrated that AβPP overexpression, likely through Aβ overproduction, induces mitochondrial dysfunction through altered mitochondrial dynamics (Wang et al., 2008b). It was reported that Aβ25-35 induced mitochondrial fragmentation before cell death. To directly examine the potential effect of Aβ oligomers on mitochondria, rat hippocampal neurons (DIV 7–9) were cotransfected with a GFP expression vector to label cell shape and Mito-DsRed2 to highlight mitochondria. Two days after transfection, neurons were incubated with 800 nm oligomeric ADDLs. As a control, in parallel experiments, neurons were treated with 10 μm Aβ42-1; the reverse peptide was subjected to the same preparation steps as ADDLs. After treatment for 24 h, cells were fixed, stained, and imaged by laser confocal microscopy. Treatment with ADDLs led to significantly reduced mitochondrial length in neurites, suggestive of enhanced mitochondrial fragmentation (Fig. 7A, B). More importantly, unlike the vehicle- or Aβ42-1-treated controls, large segments of neurites were devoid of mitochondria (Fig. 7E). In fact, the neuritic mitochondrial density and the neurite mitochondrial index were also significantly reduced in neuronal processes of ADDL-treated primary neurons (Fig. 7C, D). The negative control, Aβ42-1, had no effect on any of these mitochondrial parameters.
Figure 7.

Effects of ADDLs on mitochondrial morphology and distribution in primary neurons. Primary rat E18 hippocampal neurons (DIV 9–12) transfected with GFP and Mito-DsRed2 were treated with 800 nm ADDLs for 24 h. Twenty-four hours of treatment of Aβ42-1, subject to the same procedure that produces ADDLs, was used as a control. A, Representative pictures of positively transfected neurons. Red, DsRed; green, GFP; white: tubulin staining. B–E, Quantification of mitochondrial length (B), density (C), neurite mitochondrial index (D), and axial length of neurites devoid of mitochondria (E) in a segment of neuronal process 400 μm in length beginning from the cell body of neurons (*p < 0.05, Student's t test). At least 20 cells were analyzed in each experiment, experiments were repeated three times. F, Demonstration of the effect of ADDLs on mitochondrial fission and fusion events. Rat E18 hippocampal neurons (DIV 9) were transfected with Mito-DsRed2. Twenty-four hours after incubation with or without 800 nm ADDLs at DIV 11, neurons were imaged in time lapse (10 s interval, 20 min). Representative thresholded time-lapse pictures showed active mitochondrial fission and fusion in the segment of axon ∼100 μm in length beginning 300 μm from the cell body of control or ADDL-treated neurons. Active mitochondrial fission (filled arrows) and fusion (empty arrows) and fast-moving mitochondria (asterisks) are marked. G, H, Both fusion and fission were impaired significantly by ADDLs (G), and mitochondria spent significantly less time in the post-fusion fused state than in the post-fission single state (H). At least 20 neurons were analyzed in three independent experiments (*p < 0.05, Student's t test). I–L, Immunoblot and quantitative analysis of DLP1 and OPA1 levels in neurons treated at the indicated dosages of ADDLs for 24 h (I, J) or at 800 nm for the indicated periods of time (K, L) revealed that ADDLs induced a dose- and time-dependent decrease in DLP1 and OPA1 levels (*p < 0 0.05, Student's t test). C, Control, UNT, untreated. M, Unlike ADDLs, 10 μm Aβ42-1 had no effect on the expression of DLP1 and OPA1. Equal protein amounts (15 μg) were loaded. Actin immunoblot was used as an internal loading control.
The readily discernible individual mitochondrion in distal neurites makes it possible to measure the occurrence of mitochondrial fusion and fission events. A fusion event was defined as two separate mitochondria moving toward each other, forming a physical connection into one intact organelle and moving together. A fission event was recognized by the division of a single mitochondrion into two distinct mitochondria. Both fission and fusion events were counted in the segment of axon ∼100 μm in length beginning 300 μm from the cell body of control or ADDL-treated neurons during 20 min time-lapse recordings. Mitochondria rapidly underwent constant fission and fusion in control neurons (0.0263 ± 0.0102 events/μm in 20 min for fusion and 0.0399 ± 0.0098 events/μm in 20 min for fusion) (Fig. 7F, G). Similar rates were seen in neurons treated with the reverse peptide, Aβ42-1. In contrast, mitochondrial fission and fusion occurred at a much lower frequency in neurons treated with ADDLs (0.0036 ± 0.0019 events/μm in 20 min for fusion and 0.0048 ± 0.002 events/μm in 20 min for fusion), suggesting that both mitochondrial fission and fusion were impaired in ADDL-treated primary neurons (Fig. 7F, G). Further, the ratios of the total time each mitochondrion involved in fission/fusion spent in the post-fission “single state” (defined as the time interval from fission to fusion) and post-fusion “fused state” (defined as the time interval from fusion to fission) during the 20 min recording time were determined. In both control neurons and those treated with Aβ42-1, the total times each mitochondrion spent in the post-fusion state and post-fission state were similar (Fig. 7H). However, in the ADDL-treated neurons, mitochondria spent significantly less time in the post-fusion state compared with the post-fission state (Fig. 7H).
We next sought to determine whether Aβ oligomers induced changes in the expression of these mitochondrial fission and fusion proteins in rat hippocampal neurons. Primary neurons (DIV 7∼12) were incubated with different doses of ADDLs (50 nm–10 μm) for 24 h or with 800 nm ADDLs for various period of time (0–24 h) and cell lysates were prepared for immunoblot analysis of mitochondrial fission and fusion proteins. As a negative control, neurons were also treated with 10 μm Aβ42-1 for 24 h. No apparent cell death under these conditions with either ADDLs or Aβ42-1 treatment was observed, as determined by LDH assay (data not shown). Interestingly, ADDL treatment caused a significant decrease in DLP1 and OPA1 levels in a dose-dependent manner (Fig. 7I, J), whereas there was no significant change in Mfn1, Mfn2, or Fis1 level (data not shown). In fact, 800 nm ADDL treatment induced a significant reduction in DLP1 or OPA1 levels as early as 2 h, and the effect lasted for at least 24 h (Fig. 7K, L). In contrast, Aβ42-1 treatment did not change mitochondrial fission and fusion protein levels compared with what was seen for untreated or vehicle-treated neurons (Fig. 7M).
We further determined the levels of DLP1 in the mitochondrial fraction and found that Aβ oligomers induced significantly increased levels of mitochondrial DLP1 (Fig. 8A). Similar to what was seen for AD brain, increased mitochondrial DLP1 was accompanied by Aβ oligomer-induced increased levels of phosphorylation (Fig. 8A) and S-nitrosylation (Fig. 8B) of DLP1.
Figure 8.

Effects of ADDLs on subcellular localization and modification of DLP1 in primary rat E18 hippocampal neurons. A, Representative immunoblot and quantification analysis showed that both relative mitochondrial DLP1 level (DLP1/COX IV) and the relative ratio of phospho-DLP1 (p-DLP1) to DLP1 increased significantly in primary rat E18 hippocampal neurons (DIV 12) treated with 800 nm ADDLs for 24 h. Unlike ADDLs, 10 μm Aβ42-1 had no effect on either relative mitochondrial DLP1 level (DLP1/COX IV) or the relative ratio of p-DLP1/DLP1 (not shown). All samples were also immunoblotted with antibodies to detect mitochondrial (COX IV) and cytoplasmic (GAPDH) markers. The absence of GAPDH in the mitochondrial fraction confirms the purity of preparations from cell lysates. B, Representative immunoblot and quantification analysis further revealed that S-nitrosylated DLP1 (SNO-DLP1) formation was also enhanced in primary rat E18 hippocampal neurons (DIV 12) treated with 800 nm ADDLs for 24 h. As a control, 10 μm Aβ42-1 did not affect SNO-DLP1 formation. All experiments were repeated three times (*p < 0.05, Student's t test).
Effects of DLP1 and OPA1 on Aβ-induced alterations in mitochondrial dynamics
To determine the role of DLP1 and/or OPA1 in Aβ-induced abnormal mitochondrial morphology and distribution, rat hippocampal neurons (DIV 9) were transiently transfected with Myc-tagged DLP1wt or GFP-tagged OPA1wt. Mito-DsRed2 was cotransfected to label mitochondria. Two days after transfection, neurons were treated with 800 nm ADDLs for 24 h, fixed, and stained. Positively transfected cells were selected based on DsRed fluorescence signal and Myc immunoreactivity (or GFP fluorescence signal). As shown before, in the absence of ADDL treatment, mitochondria became fragmented in neurons overexpressing DLP1 but were more heterogeneous in shape, with an increase in average mitochondrial length in neurons overexpressing OPA1 (Fig. 9C). Mitochondrial number and coverage in the neuronal processes were increased in neurons overexpressing DLP1 but unchanged in neurons overexpressing OPA1 (Fig. 9A, B). In control primary neurons expressing only vector, ADDLs induced mitochondrial fragmentation, reduced mitochondrial number and neuritic mitochondrial index, and increased the average length of neuritic segments devoid of mitochondria in the neuronal processes (Fig. 9B–E). Interestingly, in the presence of ADDLs, OPA1 overexpression restored mitochondrial length to a level comparable with what was seen for untreated controls (Fig. 9C). However, OPA1 overexpression had no effects on ADDL-induced decrease in mitochondrial number and neurite mitochondrial index or increase in average length of neuritic segments devoid of mitochondria in the neuronal processes in positively transfected primary neurons (Fig. 9A–E). On the other hand, in contrast to OPA1-overexpressing neurons, in the presence of ADDLs, DLP1 overexpression did not change mitochondrial length but restored mitochondrial number, the neurite mitochondrial index, and the average length of neuritic segments devoid of mitochondria to a level comparable with untreated controls in positively transfected neurons (Fig. 9A–E).
Figure 9.
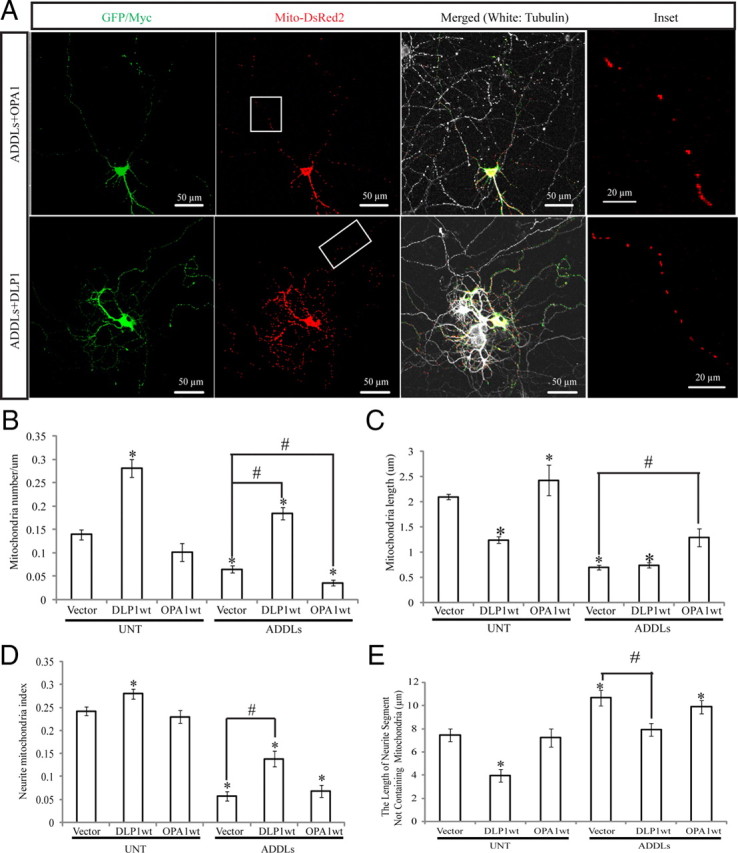
Effects of DLP1 and OPA1 on ADDL-induced mitochondrial dynamics changes. A, Representative pictures of neurons (DIV 12) cotransfected with GFP-, Mito-DsRed2-, and Myc-tagged wild-type DLP1 or wild-type OPA1 after 24 h treatment with 800 nm ADDLs. Areas enclosed in white boxes are shown at higher magnification in Inset panels (right) to allow better appreciation of changes in mitochondrial morphology and density. Red, DsRed; green, Myc; white, Tubulin staining. B–E, Quantification of mitochondrial number (B), length (C), neurite mitochondrial index (D), and axial length of neurites not covered by mitochondria (E) in neurons with indicated treatment or manipulation (*p < 0.05, when compared with the nontransfected or empty vector-transfected normal control cells; #p < 0.05, when compared with control or empty vector-transfected cells with ADDL treatment; Student's t test). At least 20 neurons were analyzed in three independent experiments. UNT, Untreated.
Effects of DLP1 and OPA1 on Aβ-induced functional changes
Mitochondria serve as the primary source of endogenous ROS. By using a fluorescence red dye, MitoSOX, to measure mitochondrial ROS production, we were able to demonstrate that treatment of rat hippocampal neurons with ADDLs resulted in a large increase of mitochondrial ROS levels in rat hippocampal neurons (Fig. 10A, B). In the absence of ADDL treatment, OPA1 overexpression led to a significantly decreased basal level of mitochondrial ROS, as evidenced by the lower fluorescence signal in positively transfected cells compared with those of neighboring nontransfected cells and GFP vector-transfected control cells (Fig. 10A, B). However, DLP1 overexpression did not significantly change the basal mitochondrial ROS levels. Similarly, in the presence of ADDL treatment, OPA1 overexpression significantly lowered mitochondrial ROS levels to a level comparable with that of untreated controls, whereas DLP1 overexpression had no effects (Fig. 10A, B).
Figure 10.

Effects of DLP1 and OPA1 on ADDL-induced changes in mitochondrial function and neuronal function. A, B, Representative fluorescent pictures and quantification of mitochondrial ROS in neurons (DIV 12) transfected with GFP-tagged wild-type DLP1 or wild-type OPA1 with or without ADDL treatment. Mitochondrial ROS was labeled by MitoSOX; Positive transfected cells were selected by GFP signal. C, D, Representative pictures and quantification of dendritic spine in neurons with or without ADDL treatment. Neurons (DIV 9) were cotransfected with GFP- and Myc-tagged wild-type DLP1 or OPA1. Positive transfected cells were selected on the basis of GFP and Myc staining. E, F, To study the effect of Aβ on PSD-95, neurons (DIV 9) were cotransfected with YFP-tagged PSD-95 to label excitatory synapses, Mito-DsRed2 to label mitochondria, and Myc-tagged DLP1 or OPA1 constructs. Shown are representative pictures and quantification of PSD-95 puncta in neurons with or without ADDL treatment and manipulation. Red, DsRed; green, YFP; blue, MAP2A. Scale bars, 5 μm. At least 20 neurons were analyzed in three independent experiments (*p < 0.05, when compared with the nontransfected or empty vector-transfected normal control cells; #p < 0.05, when compared with control or empty vector-transfected cells with ADDL treatment; Student's t test).
Consistent with previous findings demonstrating that ADDL treatment negatively impacted synaptic function (Lacor et al., 2004), we found that 24 h treatment with 800 nm ADDLs also induced a significantly reduced dendritic spine number in rat hippocampal neurons (DIV 12) (Fig. 10C, D). Notably, in the presence of ADDL treatment, DLP1 overexpression significantly alleviated ADDL-induced decreases in dendritic spine number (Fig. 10C, D). In contrast, OPA1 overexpression had no effects on dendritic spine number with or without ADDL treatment. We further studied the effects of ADDL treatment on excitatory synapses by measuring the expression of PSD-95 in primary neurons (Fig. 10E, F). We transfected primary neurons (DIV 9) with YFP-tagged PSD-95 to label excitatory synapses and Mito-DsRed2 to label mitochondria together with or without Myc-tagged DLP1wt or OPA1wt. At DIV 11, neurons were treated with 800 nm ADDLs for 24 h and then fixed. Positively transfected cells were selected based on YFP/DsRed fluorescence signal and Myc immunoreactivity. In the absence of ADDL treatment, DLP1 overexpression led to an increased number of PSD-95 puncta, whereas OPA1 overexpression had no effect on puncta number (Fig. 10E, F). As expected, ADDL treatment significantly decreased the number of PSD-95 puncta (Fig. 10E, F). Interestingly, ADDL-induced reduction in the number of PSD-95 puncta was effectively prevented by DLP1 overexpression but not by OPA1 overexpression. Overall, these data suggested that DLP1-regulated mitochondrial distribution played an important role in ADDL-induced change of dendritic spine and synapse plasticity.
Discussion
In this study, we demonstrated significant alterations in the expression pattern of mitochondrial fission/fusion proteins in hippocampal tissues from AD compared with age-matched control brain. We found reduced levels of DLP1/OPA1/Mfn1/Mfn2 and increased levels of Fis1 in AD hippocampus, consistent with the notion that mitochondrial dynamics are altered in AD. More importantly, all of these proteins accumulated in the soma and were depleted in neuronal processes in AD neurons. Given that OPA1, Mfn1/2, and Fis1 are mitochondrial membrane proteins, these data suggest a potential redistribution of mitochondria in AD neurons. In this regard, we also demonstrated that COX I, a widely used mitochondrial marker, was redistributed in a similar pattern, confirming that mitochondria are redistributed in AD neurons. To explore the consequence of changes in the expression levels of mitochondrial fission/fusion proteins, the expression of these proteins was manipulated in M17 cells and primary hippocampal neurons in a way that mimicked their expression changes in AD. Although these manipulations caused different effects on mitochondrial morphology, they all caused reduced mitochondrial density in the cell periphery (M17 cells) or neuronal processes (primary neurons) which correlated with reduced spine numbers (primary neurons), suggesting that the altered expression of these proteins may play an important role in mitochondrial redistribution and synaptic dysfunction in AD neurons. In an attempt to address the potential cause of abnormal mitochondrial dynamics, we found that ADDL treatment led not only to mitochondrial fragmentation, consistent with previous studies (Barsoum et al., 2006), but also to mitochondrial depletion from neuronal processes. ADDLs likely caused abnormal mitochondrial dynamics directly through adverse effects on DLP1 and OPA1 expression, since DLP1 overexpression rescued ADDL-induced abnormal mitochondrial distribution, whereas OPA1 overexpression restored ADDL-induced mitochondrial fragmentation. Most importantly, we found that ADDL-induced mitochondrial depletion from dendrites correlated with reduced dendritic spine density and PSD-95 puncta number, which could be effectively reversed by DLP1 overexpression through repopulation of neuronal processes with mitochondria, suggesting that abnormal mitochondrial dynamics plays an important role in ADDL-induced synaptic dysfunction and loss.
Our previous finding of reduced mitochondrial length in the soma of AD neurons compared with controls suggested that an abnormal mitochondrial dynamics toward enhanced fission may be involved in neuronal dysfunction in AD (Wang et al., 2008b). The major finding of the current study is an altered expression pattern of mitochondrial fission and fusion proteins in vivo in AD brain, confirming that the balance of mitochondrial fission and fusion is indeed tipped in AD neurons, which thus likely contributes to mitochondrial dysfunction and neurodegeneration in vivo. However, although OPA1/Mfn1/Mfn2 reduction or Fis1 overexpression could lead to mitochondrial fragmentation, DLP1 reduction could cause mitochondrial elongation and thus may make the net effect on mitochondrial morphology uncertain (Chan, 2006). Interestingly, despite the overall reduction in DLP1 levels, we found mitochondrial DLP1 levels remained similar between AD and control samples and were increased in ADDL-treated neurons. In this regard, it is of interest to note that DLP1 phosphorylation at Ser616 and S-nitrosylation activate its mitochondrial fission activity (Taguchi et al., 2007; Cho et al., 2009), and we found increased DLP1 phosphorylation and S-nitrosylation in AD and ADDL-treated neurons. Given that the majority of DLP1 in mammalian cells is cytosolic and it is the mitochondrial DLP1 that participates in mitochondrial fission (Smirnova et al., 2001), the comparable or increased mitochondrial DLP1 levels in AD or ADDL-treated samples, coupled with increased DLP1 S-nitrosylation, suggested that changes in DLP1 in AD and ADDL-treated neurons, along with changes in other fission/fusion proteins, likely contributes to enhanced mitochondrial fission. Such a notion is obviously supported by the net outcome of mitochondrial fragmentation in ADDL-treated neurons.
Interestingly, a detailed measurement of mitochondrial fission and fusion events in ADDL-treated primary neurons revealed that although neuronal mitochondria are still capable of fusing and dividing, they fuse and divide at a significantly slower rate, suggesting that both fission and fusion are impaired, consistent with altered expression of both fission and fusion proteins. It is unclear whether a slower but still balanced mitochondrial fission and fusion process has any effect on mitochondrial function. Notably, although both fission and fusion slowed down at similar rates, we found that mitochondria switched from spending similar amounts of time in the post-fusion fused state and post-fission single state in control cells to spending the majority of their time in the post-fission single state in ADDL-treated cells, which is likely the reason that a net outcome of decreased mitochondrial length was observed in these cells. Such a switch of steady-state morphology is likely attributable to the reduced expression of OPA1 caused by ADDL treatment, because recent studies demonstrated that nonfusing mitochondria are characterized by reduced OPA1 expression and that OPA1 depletion in individual mitochondrion may preclude its fusion even if surrounded by fusion-competent mitochondria (Twig et al., 2008).
Our results also showed that the distribution of mitochondrial membrane proteins OPA1/Mfn1/Mfn2/Fis1 was altered in AD neurons such that neuronal processes were devoid of these proteins, a mitochondrial redistribution in AD neurons that was also confirmed with a more widely used mitochondrial marker, COX I. Intracellular mitochondria distribution is of critical importance to neurons since the great morphological complexity and dependency on mitochondria for energy at multiple selective sites make neurons particularly sensitive to perturbations in mitochondrial distribution (Kann and Kovacs, 2007). The loss of mitochondria from axon terminals leads to synaptic dysfunction in flies (Stowers et al., 2002; Melov, 2004; Guo et al., 2005). Most interestingly, in relation to mitochondrial distribution, we demonstrated in M17 neuroblastoma cells that knockdown of DLP1/OPA1/Mfn1/Mfn2 or overexpression of Fis1, conditions that mimic changes in AD neurons, all caused perinuclear accumulation of mitochondria, leaving more remote areas of the cell devoid of mitochondria. Similarly, all of these manipulations also resulted in a significant reduction of mitochondrial density and uneven mitochondrial coverage in neuronal processes in differentiated primary hippocampal neurons with significantly greater axial neurite length devoid of mitochondria. Our data suggest that changes in the expression of these mitochondrial fission and fusion proteins likely underlie abnormal mitochondrial distribution in AD neurons.
Many in the field believe that Aβ plays a central role in the pathogenesis of AD (Kamenetz et al., 2003). Early changes in the AD brain include loss of synapses and synaptic loss is the most robust correlate of AD-associated cognitive deficits. Aβ is a homeostatic regulator of synaptic strength (Kamenetz et al., 2003), suggesting that perturbations in soluble Aβ levels might be linked to the learning and memory deficits in AD patients (Masliah et al., 2001). Indeed, AD mouse models with elevated Aβ levels exhibit decreased neuronal synaptophysin and PSD-95 staining as well as dendritic spine loss (Mucke et al., 2000; Lanz et al., 2003; Almeida et al., 2005; Spires et al., 2005) along with learning deficits well before the formation of senile plaques. Application of Aβ or soluble oligomers (e.g., ADDLs), either in vitro or in vivo, adversely affects LTP and synaptic transmission (Lambert et al., 1998; Walsh et al., 2002; Wang et al., 2002; Cleary et al., 2005). Along these lines, Aβ overexpression decreases spine density (Hsieh et al., 2006) and ADDLs directly bind to dendritic spines (Lacor et al., 2004) and induce abnormalities in spine composition, shape, and abundance (Lacor et al., 2007). However, it remains unclear how these Aβ effects are transduced to synaptic dysfunction. In this study, we found that ADDLs induced mitochondrial fragmentation accompanied by a decrease in mitochondrial coverage in neurites. At the same time, ADDL treatment also caused significant decreases in the levels of both DLP1 and OPA1. Functionally, ADDL treatment increased mitochondrial ROS levels and decreased spine density and PSD 95-positive puncta. It was reported that number of dendritic mitochondria affected the number and plasticity of spines and synapses and, indeed, there is a correlation between dendritic spine morphogenesis and recruitment of nearby mitochondria (Li et al., 2004). In our study, we found that DLP1 overexpression could not prevent mitochondrial ROS overproduction induced by ADDLs but could efficiently alleviate synaptic loss or dysfunction caused by ADDLs, suggesting that mitochondrial distribution regulated by DLP1 probably accounted for the synaptic dysfunction induced by ADDLs. On the other hand, we showed that OPA1 could alleviate ADDL-induced ROS overproduction by mitochondria. These data suggest that OPA1 plays an important role in ADDL-induced mitochondrial dysfunction, and yet one cannot rule out the possibility that OPA1 is also involved in ADDL-induced synaptic abnormalities through its effect on mitochondrial function.
Together, our data demonstrate significant changes in the expression and distribution of mitochondrial fission and fusion proteins in vivo in AD and suggest altered mitochondrial dynamics likely contribute to mitochondrial and neuronal dysfunction in disease pathogenesis. We further found that Aβ-induced abnormal mitochondrial dynamics plays an important role in Aβ-induced mitochondrial and synaptic dysfunction. Modulating the expression of mitochondrial fission and fusion proteins likely represents a potential novel therapeutic strategy for treating AD.
Footnotes
This study is supported by the National Institutes of Health (AG031852) and the Alzheimer's Association (IIRG-07-60196).
References
- Almeida CG, Tampellini D, Takahashi RH, Greengard P, Lin MT, Snyder EM, Gouras GK. Beta-amyloid accumulation in APP mutant neurons reduces PSD-95 and GluR1 in synapses. Neurobiol Dis. 2005;20:187–198. doi: 10.1016/j.nbd.2005.02.008. [DOI] [PubMed] [Google Scholar]
- Barsoum MJ, Yuan H, Gerencser AA, Liot G, Kushnareva Y, Gräber S, Kovacs I, Lee WD, Waggoner J, Cui J, White AD, Bossy B, Martinou JC, Youle RJ, Lipton SA, Ellisman MH, Perkins GA, Bossy-Wetzel E. Nitric oxide-induced mitochondrial fission is regulated by dynamin-related GTPases in neurons. EMBO J. 2006;25:3900–3911. doi: 10.1038/sj.emboj.7601253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Benard G, Bellance N, James D, Parrone P, Fernandez H, Letellier T, Rossignol R. Mitochondrial bioenergetics and structural network organization. J Cell Sci. 2007;120:838–848. doi: 10.1242/jcs.03381. [DOI] [PubMed] [Google Scholar]
- Blass JP, Sheu RK, Gibson GE. Inherent abnormalities in energy metabolism in Alzheimer disease. Interaction with cerebrovascular compromise. Ann N Y Acad Sci. 2000;903:204–221. doi: 10.1111/j.1749-6632.2000.tb06370.x. [DOI] [PubMed] [Google Scholar]
- Bubber P, Haroutunian V, Fisch G, Blass JP, Gibson GE. Mitochondrial abnormalities in Alzheimer brain: mechanistic implications. Ann Neurol. 2005;57:695–703. doi: 10.1002/ana.20474. [DOI] [PubMed] [Google Scholar]
- Cereghetti GM, Stangherlin A, Martins de Brito O, Chang CR, Blackstone C, Bernardi P, Scorrano L. Dephosphorylation by calcineurin regulates translocation of Drp1 to mitochondria. Proc Natl Acad Sci U S A. 2008;105:15803–15808. doi: 10.1073/pnas.0808249105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chan DC. Mitochondrial fusion and fission in mammals. Annu Rev Cell Dev Biol. 2006;22:79–99. doi: 10.1146/annurev.cellbio.22.010305.104638. [DOI] [PubMed] [Google Scholar]
- Cho DH, Nakamura T, Fang J, Cieplak P, Godzik A, Gu Z, Lipton SA. S-nitrosylation of Drp1 mediates beta-amyloid-related mitochondrial fission and neuronal injury. Science. 2009;324:102–105. doi: 10.1126/science.1171091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cleary JP, Walsh DM, Hofmeister JJ, Shankar GM, Kuskowski MA, Selkoe DJ, Ashe KH. Natural oligomers of the amyloid-beta protein specifically disrupt cognitive function. Nat Neurosci. 2005;8:79–84. doi: 10.1038/nn1372. [DOI] [PubMed] [Google Scholar]
- Coleman P, Federoff H, Kurlan R. A focus on the synapse for neuroprotection in Alzheimer disease and other dementias. Neurology. 2004;63:1155–1162. doi: 10.1212/01.wnl.0000140626.48118.0a. [DOI] [PubMed] [Google Scholar]
- DeKosky ST, Scheff SW. Synapse loss in frontal cortex biopsies in Alzheimer's disease: correlation with cognitive severity. Ann Neurol. 1990;27:457–464. doi: 10.1002/ana.410270502. [DOI] [PubMed] [Google Scholar]
- Delettre C, Lenaers G, Griffoin JM, Gigarel N, Lorenzo C, Belenguer P, Pelloquin L, Grosgeorge J, Turc-Carel C, Perret E, Astarie-Dequeker C, Lasquellec L, Arnaud B, Ducommun B, Kaplan J, Hamel CP. Nuclear gene OPA1, encoding a mitochondrial dynamin-related protein, is mutated in dominant optic atrophy. Nat Genet. 2000;26:207–210. doi: 10.1038/79936. [DOI] [PubMed] [Google Scholar]
- Deng H, Dodson MW, Huang H, Guo M. The Parkinson's disease genes pink1 and parkin promote mitochondrial fission and/or inhibit fusion in Drosophila. Proc Natl Acad Sci U S A. 2008;105:14503–14508. doi: 10.1073/pnas.0803998105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Exner N, Treske B, Paquet D, Holmström K, Schiesling C, Gispert S, Carballo-Carbajal I, Berg D, Hoepken HH, Gasser T, Krüger R, Winklhofer KF, Vogel F, Reichert AS, Auburger G, Kahle PJ, Schmid B, Haass C. Loss-of-function of human PINK1 results in mitochondrial pathology and can be rescued by parkin. J Neurosci. 2007;27:12413–12418. doi: 10.1523/JNEUROSCI.0719-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guo X, Macleod GT, Wellington A, Hu F, Panchumarthi S, Schoenfield M, Marin L, Charlton MP, Atwood HL, Zinsmaier KE. The GTPase dMiro is required for axonal transport of mitochondria to Drosophila synapses. Neuron. 2005;47:379–393. doi: 10.1016/j.neuron.2005.06.027. [DOI] [PubMed] [Google Scholar]
- Han XJ, Lu YF, Li SA, Kaitsuka T, Sato Y, Tomizawa K, Nairn AC, Takei K, Matsui H, Matsushita M. CaM kinase I alpha-induced phosphorylation of Drp1 regulates mitochondrial morphology. J Cell Biol. 2008;182:573–585. doi: 10.1083/jcb.200802164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hsieh H, Boehm J, Sato C, Iwatsubo T, Tomita T, Sisodia S, Malinow R. AMPAR removal underlies Abeta-induced synaptic depression and dendritic spine loss. Neuron. 2006;52:831–843. doi: 10.1016/j.neuron.2006.10.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kamenetz F, Tomita T, Hsieh H, Seabrook G, Borchelt D, Iwatsubo T, Sisodia S, Malinow R. APP processing and synaptic function. Neuron. 2003;37:925–937. doi: 10.1016/s0896-6273(03)00124-7. [DOI] [PubMed] [Google Scholar]
- Kann O, Kovacs R. Mitochondria and neuronal activity. Am J Physiol. 2007;292:C641–C657. doi: 10.1152/ajpcell.00222.2006. [DOI] [PubMed] [Google Scholar]
- Khachaturian ZS, Emr M. Research workshop on the diagnosis of Alzheimer-disease. Neurobiol Aging. 1984;5:161–162. [Google Scholar]
- Klein WL. A beta toxicity in Alzheimer's disease: globular oligomers (ADDLs) as new vaccine and drug targets. Neurochem Int. 2002;41:345–352. doi: 10.1016/s0197-0186(02)00050-5. [DOI] [PubMed] [Google Scholar]
- Lacor PN, Buniel MC, Chang L, Fernandez SJ, Gong Y, Viola KL, Lambert MP, Velasco PT, Bigio EH, Finch CE, Krafft GA, Klein WL. Synaptic targeting by Alzheimer's-related amyloid beta oligomers. J Neurosci. 2004;24:10191–10200. doi: 10.1523/JNEUROSCI.3432-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lacor PN, Buniel MC, Furlow PW, Clemente AS, Velasco PT, Wood M, Viola KL, Klein WL. Aβ oligomer-induced aberrations in synapse composition, shape, and density provide a molecular basis for loss of connectivity in Alzheimer's disease. J Neurosci. 2007;27:796–807. doi: 10.1523/JNEUROSCI.3501-06.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lambert MP, Barlow AK, Chromy BA, Edwards C, Freed R, Liosatos M, Morgan TE, Rozovsky I, Trommer B, Viola KL, Wals P, Zhang C, Finch CE, Krafft GA, Klein WL. Diffusible, nonfibrillar ligands derived from Abeta1–42 are potent central nervous system neurotoxins. Proc Natl Acad Sci U S A. 1998;95:6448–6453. doi: 10.1073/pnas.95.11.6448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lanz TA, Carter DB, Merchant KM. Dendritic spine loss in the hippocampus of young PDAPP and Tg2576 mice and its prevention by the ApoE2 genotype. Neurobiol Dis. 2003;13:246–253. doi: 10.1016/s0969-9961(03)00079-2. [DOI] [PubMed] [Google Scholar]
- Li Z, Okamoto K, Hayashi Y, Sheng M. The importance of dendritic mitochondria in the morphogenesis and plasticity of spines and synapses. Cell. 2004;119:873–887. doi: 10.1016/j.cell.2004.11.003. [DOI] [PubMed] [Google Scholar]
- Masliah E, Mallory M, Alford M, DeTeresa R, Hansen LA, McKeel DW, Jr, Morris JC. Altered expression of synaptic proteins occurs early during progression of Alzheimer's disease. Neurology. 2001;56:127–129. doi: 10.1212/wnl.56.1.127. [DOI] [PubMed] [Google Scholar]
- Melov S. Modeling mitochondrial function in aging neurons. Trends Neurosci. 2004;27:601–606. doi: 10.1016/j.tins.2004.08.004. [DOI] [PubMed] [Google Scholar]
- Mirra S. Vascular lesions and the Consortium to Establish a Registry for Alzheimer's Disease (CERAD) Brain Pathol. 2000;10:591. [Google Scholar]
- Mortiboys H, Thomas KJ, Koopman WJ, Klaffke S, Abou-Sleiman P, Olpin S, Wood NW, Willems PH, Smeitink JA, Cookson MR, Bandmann O. Mitochondrial function and morphology are impaired in parkin-mutant fibroblasts. Ann Neurol. 2008;64:555–565. doi: 10.1002/ana.21492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mucke L, Masliah E, Yu GQ, Mallory M, Rockenstein EM, Tatsuno G, Hu K, Kholodenko D, Johnson-Wood K, McConlogue L. High-level neuronal expression of Aβ1–42 in wild-type human amyloid protein precursor transgenic mice: synaptotoxicity without plaque formation. J Neurosci. 2000;20:4050–4058. doi: 10.1523/JNEUROSCI.20-11-04050.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park J, Lee G, Chung J. The PINK1-Parkin pathway is involved in the regulation of mitochondrial remodeling process. Biochem Biophys Res Commun. 2009;378:518–523. doi: 10.1016/j.bbrc.2008.11.086. [DOI] [PubMed] [Google Scholar]
- Poole AC, Thomas RE, Andrews LA, McBride HM, Whitworth AJ, Pallanck LJ. The PINK1/Parkin pathway regulates mitochondrial morphology. Proc Natl Acad Sci U S A. 2008;105:1638–1643. doi: 10.1073/pnas.0709336105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rui Y, Tiwari P, Xie Z, Zheng JQ. Acute impairment of mitochondrial trafficking by β-amyloid peptides in hippocampal neurons. J Neurosci. 2006;26:10480–10487. doi: 10.1523/JNEUROSCI.3231-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smirnova E, Griparic L, Shurland DL, van der Bliek AM. Dynamin-related protein Drp1 is required for mitochondrial division in mammalian cells. Mol Biol Cell. 2001;12:2245–2256. doi: 10.1091/mbc.12.8.2245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith MA. Alzheimer disease. Int Rev Neurobiol. 1998;42:1–54. doi: 10.1016/s0074-7742(08)60607-8. [DOI] [PubMed] [Google Scholar]
- Spires TL, Meyer-Luehmann M, Stern EA, McLean PJ, Skoch J, Nguyen PT, Bacskai BJ, Hyman BT. Dendritic spine abnormalities in amyloid precursor protein transgenic mice demonstrated by gene transfer and intravital multiphoton microscopy. J Neurosci. 2005;25:7278–7287. doi: 10.1523/JNEUROSCI.1879-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stowers RS, Megeath LJ, Górska-Andrzejak J, Meinertzhagen IA, Schwarz TL. Axonal transport of mitochondria to synapses depends on milton, a novel Drosophila protein. Neuron. 2002;36:1063–1077. doi: 10.1016/s0896-6273(02)01094-2. [DOI] [PubMed] [Google Scholar]
- Swerdlow RH, Kish SJ. Mitochondria in Alzheimer's disease. Int Rev Neurobiol. 2002;53:341–385. doi: 10.1016/s0074-7742(02)53013-0. [DOI] [PubMed] [Google Scholar]
- Taguchi N, Ishihara N, Jofuku A, Oka T, Mihara K. Mitotic phosphorylation of dynamin-related GTPase Drp1 participates in mitochondrial fission. J Biol Chem. 2007;282:11521–11529. doi: 10.1074/jbc.M607279200. [DOI] [PubMed] [Google Scholar]
- Terry RD, Masliah E, Salmon DP, Butters N, DeTeresa R, Hill R, Hansen LA, Katzman R. Physical basis of cognitive alterations in Alzheimer's disease: synapse loss is the major correlate of cognitive impairment. Ann Neurol. 1991;30:572–580. doi: 10.1002/ana.410300410. [DOI] [PubMed] [Google Scholar]
- Twig G, Elorza A, Molina AJ, Mohamed H, Wikstrom JD, Walzer G, Stiles L, Haigh SE, Katz S, Las G, Alroy J, Wu M, Py BF, Yuan J, Deeney JT, Corkey BE, Shirihai OS. Fission and selective fusion govern mitochondrial segregation and elimination by autophagy. EMBO J. 2008;27:433–446. doi: 10.1038/sj.emboj.7601963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Verstreken P, Ly CV, Venken KJ, Koh TW, Zhou Y, Bellen HJ. Synaptic mitochondria are critical for mobilization of reserve pool vesicles at Drosophila neuromuscular junctions. Neuron. 2005;47:365–378. doi: 10.1016/j.neuron.2005.06.018. [DOI] [PubMed] [Google Scholar]
- Walsh DM, Klyubin I, Fadeeva JV, Cullen WK, Anwyl R, Wolfe MS, Rowan MJ, Selkoe DJ. Naturally secreted oligomers of amyloid beta protein potently inhibit hippocampal long-term potentiation in vivo. Nature. 2002;416:535–539. doi: 10.1038/416535a. [DOI] [PubMed] [Google Scholar]
- Wang H, Lim PJ, Karbowski M, Monteiro MJ. Effects of overexpression of huntingtin proteins on mitochondrial integrity. Hum Mol Genet. 2009;18:737–752. doi: 10.1093/hmg/ddn404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang HW, Pasternak JF, Kuo H, Ristic H, Lambert MP, Chromy B, Viola KL, Klein WL, Stine WB, Krafft GA, Trommer BL. Soluble oligomers of beta amyloid (1-42) inhibit long-term potentiation but not long-term depression in rat dentate gyrus. Brain Res. 2002;924:133–140. doi: 10.1016/s0006-8993(01)03058-x. [DOI] [PubMed] [Google Scholar]
- Wang X, Su B, Fujioka H, Zhu X. Dynamin-like protein 1 reduction underlies mitochondrial morphology and distribution abnormalities in fibroblasts from sporadic Alzheimer's disease patients. Am J Pathol. 2008a;173:470–482. doi: 10.2353/ajpath.2008.071208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang X, Su B, Siedlak SL, Moreira PI, Fujioka H, Wang Y, Casadesus G, Zhu X. Amyloid-beta overproduction causes abnormal mitochondrial dynamics via differential modulation of mitochondrial fission/fusion proteins. Proc Natl Acad Sci U S A. 2008b;105:19318–19323. doi: 10.1073/pnas.0804871105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang Y, Ouyang Y, Yang L, Beal MF, McQuibban A, Vogel H, Lu B. Pink1 regulates mitochondrial dynamics through interaction with the fission/fusion machinery. Proc Natl Acad Sci U S A. 2008;105:7070–7075. doi: 10.1073/pnas.0711845105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu X, Rottkamp CA, Boux H, Takeda A, Perry G, Smith MA. Activation of p38 kinase links tau phosphorylation, oxidative stress, and cell cycle-related events in Alzheimer disease. J Neuropathol Exp Neurol. 2000;59:880–888. doi: 10.1093/jnen/59.10.880. [DOI] [PubMed] [Google Scholar]
- Zhu X, Smith MA, Perry G, Aliev G. Mitochondrial failures in Alzheimer's disease. Am J Alzheimers Dis Other Demen. 2004;19:345–352. doi: 10.1177/153331750401900611. [DOI] [PMC free article] [PubMed] [Google Scholar]