

Abstract
Signal transducer and activator of transcription 3 (Stat3) is involved in aberrant growth and survival signals in malignant tumor cells and is a validated target for anti-cancer drug design. We are targeting its SH2 domain to prevent docking to cytokine and growth factor receptors and subsequent signaling. The amino acids of our lead phosphopeptide, Ac-pTyr-Leu-Pro-Gln-Thr-Val-NH2, were replaced with conformationally constrained mimics. Structure-affinity studies led to the peptidomimetic, pCinn-Haic-Gln-NHBn (21) which had an IC50 of 162 nM (fluorescence polarization), as compared to 290 nM for the lead phosphopeptide (pCinn = 4-phosphoryloxycinnamate, Haic = (2S,5S)-5-amino-1,2,4,5,6,7-hexahydro-4-oxo-azepino[3,2,1-hi]indole-2-carboxylic acid). pCinn-Haic-Gln-OH was docked to the SH2 domain (AUTODOCK) and the two highest populated clusters were subjected to molecular dynamics simulations. Both converged to a common peptide conformation. The complex exhibits unique hydrogen bonding between Haic and Gln and Stat3 as well as hydrophobic interactions between the protein and pCinn and Haic.
Introduction
Signal transducer and activator of transcription 3 (Stat3) is a latent, cytosolic transcription factor that transmits signals directly from cell surface receptors to the nucleus (Reviewed in references 1-3). On stimulation by IL-6 family of cytokines, or growth factors such as EGF or PDGF, Stat3 is recruited to phosphotyrosine residues on the cell surface receptor via its SH2 domain and becomes phosphorylated on Tyr705. The protein then forms a dimerized complex in which the SH2 domain of one protein binds to the phosphotyrosine of the other and vice versa. The Stat3-dimer then translocates to the nucleus where it initiates transcription of cell cycling genes such and cyclin D1 and p21waf, antiapoptotic genes such as survivin and Bcl-XL, and angiogenesis factors such as vascular endothelial growth factor. Stat3 is constitutively activated in cancers of the breast, prostate, lung, head and neck, multiple myeloma, leukemia, and others (reviewed in references 4-8). Studies on cell lines have showed that inhibiting Stat3 activity with the use of antisense oligonucleotides, dominant negative constructs,4-8 and decoy oligonucleotides9-14 reduces the growth of cancer cells and induces apoptosis. Thus Stat3 is a target for antitumor drug design.
We are developing Stat3 inhibitors by targeting the SH2 domain with peptidomimetics derived from phosphopeptides. Such compounds are expected to prevent binding to cytokine or growth factor receptors thus preventing phosphorylation of Tyr705 and subsequent dimer formation. Therefore, translocation to the nucleus and binding to Stat3 response elements on the promotors of the above-mentioned cell cycling, survival and angiogenesis genes will be inhibited resulting in reduced tumor growth. Peptidomimetic development requires information gained from structure activity relationship (SAR) and structural studies to design or otherwise find non-peptidic scaffolds to present the pharmacophores of a lead peptide in the correct spatial arrangement for optimal binding to the target. To find a lead peptide for inhibitor development, we screened several receptor docking sites for Stat3 and found that the sequence from gp130 904 – 909, Ac-pTyr-Leu-Pro-Gln-Thr-Val-NH2 (1), is a high affinity ligand for the Stat3 SH2 domain.15 We recently reported SAR studies on the peptide which showed that the pY-1 acetyl group could be replaced by a hydrocinnamoyl group, the optimum residue at pY+1 is Leu, cis-3,4-methanoProline at pY+2 results in enhanced interaction, Gln is optimal at pY+3 and the side chain CONH2 is involved in important hydrogen bonds, and that the C-terminal Thr-Val dipeptide can be replaced with a benzyl group.16 This study provided important information on the pharmacophores of the lead peptide but gave little information on the conformation of the bound peptide.
NMR or X-ray crystal structures of phosphopeptides bound to SH2 domains have provided the basis for the development of inhibitors of Grb2 and Src-family kinases (Reviewed in references 17-21). Information on intermolecular interaction and on the bioactive conformation has led to the development of highly potent, non-peptide inhibitors of Src22,23, Lck,24 and Grb2.25-28 The crystal structure of Stat3β dimer in complex with DNA29 reveals the interactions between the phosphopeptide segment, Tyr(PO3H2)705-Leu-Lys-Thr-Lys-Phe, of one protein molecule and the SH2 domain of the other. The pTyr-Leu dipeptide makes contacts in an analogous manner to the pY and pY+1 residues in other SH2-phosphopeptide complexes.26,30-38 However, because of the structural difference between Lys-Thr-Lys-Phe of Stat3 and Pro-Gln-Thr-Val of our lead peptide, no direct information on the conformation or intermolecular interaction of the pY+2 — pY+4 residues of the latter can be obtained from this model. In this communication, we describe the incorporation of conformational constraints into our lead peptide that provided information on the bound conformation. The result was a high affinity, peptidomimetic inhibitor with enhanced affinity for Stat3, compound 21.
Having found conformational constraints that improve the affinity of our lead peptide, we studied potential inhibitor-protein interactions using AUTODOCK39 and selected two complexes for further study. Extended molecular dynamics simulations resulted in a converged conformation of the inhibitor and provided insight into the basis of affinity of the peptidomimetic.
Chemistry
Peptide Synthesis
Peptides were synthesized using manual solid phase techniques employing the Fmoc protection scheme and either Rink resin or Wang resin. Couplings were mediated with either diisopropylcarbodiimide (DIC)/1-hydroxybenzotriazole (HOBt) or PyBOP/HOBt/DIEA. Fmoc removal was accomplished with 2 treatments of 20% piperidine in DMF for 5 min each. Peptides incorporating a C-terminal glutamine α-benzyl amide were synthesized by coupling Fmoc-Glu-NHBn,16 via the side chain, to Rink resin. Peptides were cleaved from the solid support with TFA:triethylsilane:H2O (95:2.5:2.5)40 and were purified by reverse phase HPLC.
Synthesis of inhibitors incorporating constrained tyrosine mimics
4-Phosphoryloxycinnamic acid (pCinn) was introduced onto peptide chains using the protected building block, 4-(di-tert-butoxyphosphoryloxy) cinnamic acid (39, Scheme 1). This was prepared by methodology initially described by J. Perich et al. for the synthesis of phosphotyrosine derivatives.41,42 The carboxyl group of 4-hydroxycinnamic acid (36) was selectively protected as a TBDMS ester and the hydroxyl group was phosphitylated with di-tert-butyl-N,N-diisopropyl phosphoramidite in the presence of tetrazole. The phosphorous was oxidized with tert-butylhydroperoxide and the TBDMS ester was removed with aqueous bicarbonate to afford 4-(di-tert-butoxyphosphoryloxy) cinnamic acid (39). This agent was coupled to peptides using PyBOP/HOBt/DIPEA. The other constrained phosphotyrosine mimics, 5-phosphoryloxyindole-2-carboxylic acid (pInd), N-acetyl-4-phosphoryloxy-1,2,3,4-tetrahydro-3-isoquinolinecarboxylic acid (Ac-Tic), 2-phosphoryloxy-7-carboxyl-naphthalene, and 6-phosphoryloxyindole-3-acetic acid, were prepared by global phosphorylation of the corresponding hydroxy compounds on solid supports using di-benzyl-N,N-diisopropyl phosphoramidite/tetrazole and oxidation with tert-butylhydroperoxide after coupling to their peptide chains (reviewed in reference 43). Peptides were cleaved and purified as above.
Scheme I.

Synthesis of 4-(di-tert-butyl-phosphoryloxy)-cinnamic acid
Synthesis of analogues of Fmoc-Haic, (2S,5S)-5-(9-fluorenyloxycarbonyl)amino-1,2,4,5,6,7-hexahydro-4-oxo-azepino[3,2,1-hi]indole-2-carboxylic acid
Fmoc-Haic-OH (44) was obtained commercially or was synthesized by the method of De Lombaert et al.44,45 (Scheme 2). Friedel-Crafts cyclization of 41 results in approximately 10% of the 2R,5S diastereoisomer (43) which was isolated by fractional crystallization of the crude material in Et2O. It can be converted to the 2S,5S ketone 42 by treatment with DBU and taken on to 44. To prepare the 2R,5S diastereoisomer for use in solid phase synthesis (45), compound 43 was hydrogenated, deprotected with LiOH and the amino group capped with an Fmoc group. To prepare 6,7-dehydro Haic, partial reduction of ketone 42 was accomplished by hydrogenation in THF to give alcohol 46 which was dehydrated using trifluoracetic anhydride and TFA. The amino and carboxyl protecting groups were removed with LiOH and Fmoc protection was applied to the amino group providing compound 47. Stereoisomers of Fmoc-2-amino-azabicyclo[4.3.0]nonane-9-carboxylate (ABN) were synthesized as described.46
Scheme II.

Synthesis of Fmoc-Haic-OH and its analogues
Synthesis of inhibitors containing glutamine analogues
Inhibitors 27, 29, and 30 (Table 5) were prepared by coupling Fmoc-Glu-OMe (50a), Fmoc-Glu-NHMe (50b), and Fmoc-Glu-NHiPr (50c), respectively to Rink resin via their side chains using DIPCDI/HOBt followed by assembly of the remainder of the compound using standard peptide synthesis protocols. Glutamine analogues 50a-c were synthesized by derivatization of Fmoc-Glu(OtBu)-OH (48) with MeOH or the appropriate amine with PyBOP/DIPEA followed by cleavage of the side chain tert-butyl group with TFA (Scheme 3). Fmoc-pyrrolidine-3-acetic acid, used in the synthesis of 25, was prepared as described by Coleman et al.16
Table 5.
Contributions of Gln-X to the binding of pCinn-Haic-based peptidomimetics to Stat3
| Compound | IC50 (nM) | |
|---|---|---|
| 26 | pCinn-Haic-Gln-OH | 359 ± 30 |
| 27 | pCinn-Haic-Gln-OMe | 141 ± 14 |
| 28 | pCinn-Haic-Gln-NH2 | 233 ± 27 |
| 29 | pCinn-Haic-Gln-NHMe | 269 ± 22 |
| 30 | pCinn-Haic-Gln-NHiPr | 305 ± 46 |
| 31 | pCinn-Haic-NH2 | 35,600 ± 5,010 |
| 32 | pCinn-Haic-Ala-NH2 | 25,060 ± 2,750 |
| 33 | pCinn-Haic-Nle-NH2 | 19,200 ± 1,300 |
| 34 | pCinn-Haic-Met(O)-NH2 | 7,600 ± 10 |
| 35 | pCinn-Haic-Met(O2)-NH2 | 7,030 ± 1,300 |
Scheme III.

Synthesis of α-carboxy analogues of Fmoc-glutamine
Results and Discussion
In a previous study the lead peptide, 1, was truncated to pentapeptide 2 and tetrapeptide 3.16 The latter two peptides showed reduced affinity for Stat3 suggesting that hydrophobic contacts exist between the C-terminal Val and Stat3 (Table 1). Replacement of the Thr-Val dipeptide unit with a benzyl group brought the IC50 to 409 nM, thus confirming the presence of these contacts.16 Peptides 2 – 4 served as leads to which the effects of the constrained amino acid substitutions described below were compared.
Table 1.
IC50 values of peptide inhibitors of Stat3.a
| Peptide | IC50 (nM) | |
|---|---|---|
| 1 | Ac-Tyr(PO3H2)-Leu-Pro-Gln-Thr-Val-NH2 | 290 ± 63 |
| 2 | Ac-Tyr(PO3H2)-Leu-Pro-Gln-Thr-NH2 | 739 ± 31 |
| 3 | Ac-Tyr(PO3H2)-Leu-Pro-Gln-NH2 | 856 ± 41 |
| 4 | Ac-Tyr(PO3H2)-Leu-Pro-Gln-NHBn | 409 ± 15 |
IC50 values from Coleman et al.16.
Phosphotyrosine constraints
The X-ray crystal structure of dimerized Stat3β bound to DNA29 shows interactions between the phosphate group of pTyr705 and the opposing SH2 domain which are analogous to those observed in NMR and crystal structures of Src,30-33 Grb2,26,34,35 Lck,36 and p85 PI3K.37,38 In the Stat3 structure,29 the dihedral angle of the C, Cα, Cβ, and Cγ atoms of pTyr is 174°, also typical of those found in phosphopeptide ligands of other SH2 domains26,30-38 4-Phosphoryloxycinnamate (pCinn)47 and 5-phosphoryloxyindole-2-carboxylate (pInd)48 both constrain this dihedral angle to 180°. Substitution of the pTyr of peptide 2 with these two mimics enhanced the activity of our peptide 4-5 fold, decreasing the IC50 from 739 nM to 136 nM and 186 nM, respectively (5 and 6, Table 2). The use of pCinn as a phosphotyrosine replacement in inhibitors of the Src SH2 domain was reported by Shahripour et al.47 In this paper the activity of the lead was reduced 11-fold by replacing pTyr with 4-phosphoryloxycinnamide. Researchers from Tularik, Inc. used pCinn in inhibitors of Stat649,50 although no comparison with phosphotyrosine was provided in their patents. Vu et al.48 reported that the use of pInd in development of inhibitors of the SH2 domain of Zap70 reduced activity of their lead 7-fold. In the case of Stat3, constraining the C,Cα,Cβ,Cγ dihedral angle to 180° appears to have locked the aromatic phosphate in a favorable orientation for contact with the protein which likely reduced the entropy penalty on binding. Substitution of pTyr with Ac-pTic, 2-phosphoryloxy-7-carboxyl-naphthalene, or 6-phosphoryoxyindole-3-acetic acid resulted in inactive compounds (7 – 9). Movement of the phosphate group to position 3 of the aromatic ring of cinnamic acid also abrogated activity (peptide 10).
Table 2.
Effect of pTyr mimics on the affinity of XXX-Leu-Pro-Gln-Thr-NH2
| XXX | IC50 (nM) | |
|---|---|---|
| 5 | 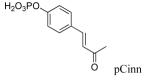 |
136 ± 23 |
| 6 | 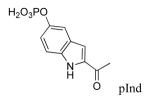 |
186 ± 23 |
| 7 | >1.00×105 | |
| 8 | >1.00×105 | |
| 9 | 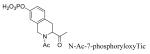 |
>1.00×105 |
| 10 | 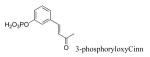 |
>1.00×105 |
Replacement of pY+1 — pY+2 dipeptide unit
We recently reported that substitution of proline of peptide 2 with the pseudo-proline (ΨPro) analogues 2,2-dimethyl-1,3-oxazolidine-5-carboxylate and 2,2,4-trimethyl-1,3-oxazolidine-5-carboxylate resulted in reduced affinity for Stat3.51 That the lower activity correlated with the increased extent of cis isomerization of the Leu-ΨPro peptide bond suggests that the corresponding peptide bond is trans in our lead peptides.51 Azabicyclo[4.3.0]nonane-9-carboxylate (ABN) has been studied in the context of β-turn mimetics and has been used to good effect as a conformational constraint.52-59 Replacement of the Leu-Pro unit with ABN would hold the peptide bond in the trans conformation and would also constrain the Φ dihedral angle of Leu, thus reducing the amount of conformational space available to the peptide and the entropy penalty on binding. Incorporation of (3S,6S,9S) ABN into peptide 2 increased the IC50 from 740 nM to 3,530 nM, an approximately 5-fold loss in affinity (11, Table 3). Incorporation of the (3S,6R,9R) isomer was even more deleterious (compound 12, IC50 = 24,700 nM). In compound 12 the configuration of Cα of the 5-membered ring (C9) is R, analogous to D-Pro, which we reported to greatly reduce affinity for Stat3.16
Table 3.
Effect of Leu-Pro mimics on the affinity of Ac-pTyr-XXX-Gln-Thr-NH2
| XXX | IC50 (nM) | |
|---|---|---|
| 2 | 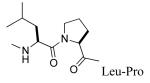 |
739 ± 31 |
| 11 | 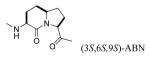 |
3,530 ± 270 |
| 12 | 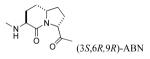 |
24,700 ± 2,400 |
| 13 | 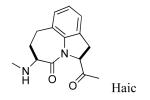 |
231 ± 22 |
| 14 | 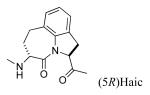 |
1,650 ± 99 |
| 15 | 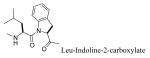 |
4,640 ± 45 |
| 16 | 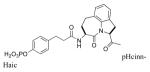 |
3,240 ± 169 |
Replacing Leu-Pro with Haic (5-[(S)-amino]-1,2,4,5,6,7-hexahydroazepino[3,2,1-hi]indole-4-one-2-(S)-carboxylate) produced an inhibitor with IC50 of 231 nM (compound 13, Table 3). The synthesis of Haic by the method of DeLombaert et al.44,45 produces roughly 90% of the 2S,5S diastereomer and 10% of the 2S,5R diastereomer during the annelation of the 7-membered ring. We completed the synthesis of Fmoc-(2S,5R)-Haic (45) and used this to prepare 14. The IC50 of the peptide containing this diastereomer was 1650 nM, a 7-fold reduction in activity compared to the (2S,5S) stereoisomer. Compound 14 was only 2-fold lower in affinity than the parent peptide, 2. The 7-membered ring of Haic displays noticeable flexibility.44 In spite of the opposite configuration at C5, it appears that the (2S,5R) stereoisomer can indeed fold to present the phenylphosphate and carboxamide pharmacophores for interaction with the SH2 domain. The tricyclic structure of Haic is important. Uncoupling of the azepine ring of Haic by substituting with leucinyl-(S)-imidazoline-2-carboxylate caused a 20-fold loss in affinity (15, IC50 = 4,640 nM). Replacing pTyr of compound 13 with the 4-phosphoryloxydihydrocinnamate resulted in a 14-fold loss in affinity (16, IC50 = 3240 nM). The acetamido group appears to add to the affinity of 13.
In a previous publication16 Gln was substituted with a wide variety of flexible and constrained ω-amino acid amides. None were as effective as glutamine. We also found that substituting a benzylamide on the C-terminus (4) produced a slight enhancement in affinity over the Thr-NH2 in peptide 2.94 To create peptidomimetics, pCinn was added to the ABN and Haic units and the Thr-NH2 was replaced with a benzylamide. Again, the ABN unit was not as favorable as Haic (Table 4). Incorporation of (3S,6S,9S) ABN (18) resulted in an IC50 of 604 nM, 4-fold lower affinity than the Leu-Pro analogue, 17 (IC50 = 138 nM). The (3S,6R,9S) and (3S,6R,9R) diastereomers of ABN caused large decreases in affinity (compounds 19 and 20, IC50 = 45,700 nM and 59,800 nM, respectively). Incorporation of Haic resulted in an IC50 value of 162 nM (21), nearly identical to the Leu-Pro analogue, 17. In an attempt to constrain the conformation of the 7-membered ring of the Haic unit we synthesized Fmoc-6,7-dehydroHaic (47) and incorporated it into the mimetic to give compound 22. The IC50 of 195 nM is somewhat lower than that of the saturated compound, 21 (162 nM). Incorporating pInd onto the Haic-Gln-NHBn framework (22) resulted in an IC50 of 190 nM. This slight reduction compared to pCinn (20) parallels that found in the more peptidic compounds, 6 vs 5. Haic is a known dipeptide mimetic that has been used in protease inhibitor development,60,61 bradykinin antagonists,62 major histocompatibility complex antagonist studies,63 and opioid receptor agonists.64 This is the first reported use in an SH2 domain inhibitor.
Table 4.
Affinities of derivatives of 4, Ac-pTyr-Leu-Pro-Gln-NHBn
| Compound | IC50 (nM) | |
|---|---|---|
| 17 | pCinn-Leu-Pro-Gln-NHBn | 138 ± 8a |
| 18 | pCinn-(3S,6S,9S)-ABN-Gln-NHBn | 604 ± 68 |
| 19 | pCinn-(3S,6R,9S)-ABN-Gln-NHBn | 45,700 ± 7,710 |
| 20 | pCinn-(3S,6R,9R)-ABN-Gln-NHBn | 59,800 ± 7,580 |
| 21 | pCinn-Haic-Gln-NHBn | 162 ± 19 |
| 22 | pCinn-ΔHaic-Gln-NHBn | 195 ± 20 |
| 23 | pInd-Haic-Gln-NHBn | 190b |
| 24 | pCinn-Haic-Gaba | 815 ± 84 |
| 25 | pCinn-Haic-pyrrolidine acetamide | 595 ± 92 |
From reference 69
A single assay was run.
Incorporation of glutamine mimics
Glutamine is essentially 4-carboxy-4-aminobutyramide, an analogue of Gaba. Replacement of Gln-NHBn of compound 21 with Gaba (24) resulted in an IC50 value of 815 nM, a 5-fold loss in affinity (Table 4). Constraining the conformation of Gaba by tethering the nitrogen with C3, i.e. using pyrrolidine-3-acetamide (compound 25, IC50 = 815 nM) did not have an appreciable effect on affinity. Thus, Gln-NHBn produced the greatest affinity, which is in keeping with earlier work in which Gaba and pyrrolidine-3-acetamide provided less affinity than Gln-NHBn in the peptide Ac-pTyr-Leu-Pro-XXX.16
To ascertain the contribution of the aromatic ring of the C-terminal α-benzylamide in the pCinn-Haic-Gln-NHBn, a series of substitutions was made on the main chain carboxyl group of Gln of compound 21. Thus, carboxyl, methyl ester, and a set of amides were assayed (compounds 26-30, Table 5). The difference in affinity of a negatively charged carboxyl group (26, IC50 = 359 nM and the benzyl group (21, IC50 = 162 nM) is only 2-fold, and the other substitutions fall within that range. Thus the contribution of the C-terminal benzene ring of 21 is relatively small. However, the contribution of the alkyl carboxamide side chain is large. pCinn-Haic-NH2 (31), has an IC50 value of 35 μM, 42-fold weaker than Gaba containing compound 24 (819 nM, Table 2) and 150-fold weaker than compound 28 containing Gln-NH2 (IC50 = 233 nM). Replacement of glutamine in 28 with alanine (32) or norleucine (33), neither of which possess a side chain CONH2 group, also resulted in compounds with IC50 values in the micromolar range.
Docking of pCinn-Haic-Gln-OH to the SH2 domain of Stat3
To determine possible inhibitor conformations and to study intermolecular interactions, pCinn-Haic-Gln-OH, 26, was docked to the SH2 domain of Stat3 and then subjected to minimization and molecular dynamics simulations. Stat3, like all STATs, is a multidomain protein consisting of an amino terminal domain, a coiled coil domain, a DNA binding domain, a linker domain, an SH2 domain, and the C-terminal transactivation domain. The Stat3β isoform lacks the amino terminal domain and the 55 residues of the transactivation domain, which are replaced by 7 unique amino acids (Schaefer et al., 1995). The coordinates from the crystal structure of Stat3β,29 PDB code 1BG1, were used for the simulations. For the initial docking experiments, residues 136-688 were used and the ligand was docked into the SH2 domain. This fragment of Stat3 does not contain amino acids C-terminal to the SH2 domain. For the molecular dynamics simulations, residues 585-688, consisting of the linker and SH2 domains, were employed. The smaller protein fragment was used to reduce computation time and inclusion of the linker domain ensured the conformation of the distal side of the SH2 domain would not change during the trajectories. In the discussion below, amino acids from the protein will be designated by the one-letter code followed by the residue number, e.g., E638. Features from the inhibitor will be designated by fragment, e.g., Gln side chain. Rigid-protein/flexible-ligand docking of 25 to Stat3 was modeled in an automated manner using AUTODOCK 4.0.39 Two poses, in which 25 was in an extended conformation (Pose A, Figure 1A) and a bent conformation (Pose B, Figure 1B), were selected from the docking study for subsequent molecular modeling explorations. Pose A was chosen because it was a member of the largest cluster of docking poses. Pose B was selected because of proximity of the glutamine side chain amide to residues E638, P639, and Y640 of Stat3, which were hypothesized to be close in peptide complexes with the SH2 domain of Stat3.65
Figure 1.

Structures of the poses of docking compound 26 to the SH2 domain of Stat3 from the initial AUTODOCK study. A. Pose A, a representative from the most populated cluster from the AUTODOCK study exhibiting the extended-conformation. B. Pose B, a representative from the cluster possessing the bent-conformation. These figures represent complexes that were energy minimized with implicit solvent followed by minimization with explicit solvent. Water molecules were removed for clarity. Compound 26 is depicted as sticks: green, carbon; blue, nitrogen; red, oxygen. Hydrogens were removed for clarity. Stat3 is represented as a surface rendition. The following residues are highlighted: Phosphate binding pocket (blue); S636 and P639 (purple); E638 (red); Y640 (yellow); Y657 (orange) M660 (brown). The images were generated with PyMol.70
After extensive energy minimization with implicit solvent followed by explicit solvent, the two poses were separately heated, equilibrated, and subjected to 3 ns molecular dynamics simulations containing explicit water. After about 1.5 ns both simulations exhibited a decrease in main-chain RMSD of both ligand and protein and convergence to a similar inhibitor conformation and inter-molecular hydrogen bonding features. Both trajectories were separately clustered based on similarity of the RMSD of the inhibitor.
The negatively charged phosphate of pCinn is located in the electropositive phosphate binding pocket formed partly by K591 and R609 throughout the entirety of both simulations. These ionic interactions are analogous to those observed in the crystal structure of the Stat3 dimer29 and are typical of phosphopeptide-SH2 domain complexes.26,30-38 The phosphate-R609 hydrogen bond exists for 100.0% and 99.9% of the total 3 ns MD trajectories of Poses A and B, respectively. Similarly the phosphate-K591 hydrogen bond exists for 98.0% and 97.9% of the 3 ns MD trajectories of Poses A and B, respectively. Other hydrogen bonds observed between Stat3 and the phosphopeptide phosphate in both the Stat3 crystal structure and the molecular dynamics simulations are as follows. The interaction between the backbone nitrogen of E612 and one of the phosphate oxygens exists for 93.0% and 84.9% of the 3 ns trajectories of Poses A and B, respectively; and the hydrogen bond between the side-chain hydroxyl group of Stat3 S613 and one of the phosphate oxygens exists for 62.4% and 10.3% in the trajectories of Poses A and B, respectively, which differs from the crystal structure of Stat3.29 The placement of the phosphate moiety in this pocket is consistent with site-directed mutations which demonstrated that K591 and R609 are essential for binding 4-6 residue phosphopeptides with pYXXQ consensus sequences.66 Based on fidelity of the hydrogen bonds observed in the Stat3 crystal structure29 and consistency with experimental evidence, it can be concluded that the molecular dynamics simulations reasonably predict the interaction between Stat3 and the pCinn unit of 26.
Hydrogen bonds between the pY+1 main chain amide nitrogen and the backbone carbonyl oxygen of the βD4 residue exist in most known complexes between SH2 domains and phosphopeptide ligands26,30-38 and the STATs are no exception.29,67,68 In crystal structures of Stat3 and Stat1 the backbone oxygen of Stat3, S636 (Stat1:A630) makes a hydrogen bond with the pY+1 backbone nitrogen of the opposing protein29,67 or a phosphopeptide ligand68. A structure-activity relationship study demonstrated through leucine N-methylation that this hydrogen bond is essential for high affinity recognition of phosphopeptides by Stat3.16 However, hydrogen bonds between the analogous exocyclic amide nitrogen of Haic and the backbone oxygen of S636 were observed for only 22-25% of the time in the two trajectories (Figure 2). A hydrogen bond between the endocyclic carbonyl oxygen of Haic and the backbone nitrogen of E638 exists for 60% in the MD trajectory of the Pose A as compared to only 23% in that of Pose B (Figure 2). Compound 26 makes a hydrogen bond with either S636 or E638 for 80% of the trajectory starting with Pose A and 50% of that starting with Pose B. The data suggest that the inhibitor may toggle back and forth in position between these two interaction sites.
Figure 2.

Percentage of time that specific hydrogen bonds exist between 26 and Stat3 in the final 1.5 ns of the MD simulations starting with Pose A and Pose B.
A hydrogen bond exists between the side chain amide oxygen of Gln and the side-chain hydroxyl group of Y640 approximately 70% of the final 1.5 ns of both MD simulations (Figure 2). The difference in the percent of the simulation of this hydrogen bond between the two conformations is only 1.9%. A hydrogen bond also exists between the exocyclic carbonyl oxygen of Haic and the phenolic hydroxyl group of Y657 side-chain hydroxyl at least 74% of the time for both conformations with only a 4.8% difference between the two simulations. The persistence of hydrogen bonds between 25 and the side-chains of Y640 and Y657 in both the simulations suggests these interactions are important for stabilizing the inhibitor-protein complex. An analogous hydrogen bond between the main chain C=O of the Pro of phosphopeptide ligand Ac-pTyr-Asp-Lys-Pro-His-NH2 and the side chain hydroxyl group of Y651 (analogous to Y657 of Stat3) was observed in the recently reported crystal structure of Stat1.68
In both MD trajectories, at least one water molecule bridges the side chain NH2 of Gln to the main chain carbonyl oxygens of either E638 or P639. For example, in the most populated cluster of the Pose A simulation (61% of the snapshots) a water molecule coordinates a hydrogen bond between the backbone oxygen of E638 and the side-chain amide nitrogen of Gln. In the second cluster of the Pose A simulation (26% of the snapshots), two water molecules are involved in a hydrogen bond network between the following atoms: the backbone oxygens of E638 and P639, the side-chain oxygen of E638, and the side-chain amide nitrogen of 26.
Methionine sulfoxide, Met(O), is a glutamine isostere in which the side chain carbonyl group is replaced by a sulfoxide and the NH2 is replaced by a methyl group. The sulfonyl group could serve as a hydrogen bond acceptor from Y640 but the terminal methyl group is not a hydrogen bond donor. To test for the presence of hydrogen bonding between the side chain NH2 of Gln and Stat3, Met(O) and the dioxygenated analogue, methionine sulphone (Met(O2)), were incorporated into compound 28 (chosen for the ease of synthesis). The modified analogues 34 (containing Met(O)) and 35 (containing Met(O2)) resulted in about a 10-fold loss in affinity, which is consistent with loss of hydrogen bonding between Gln and Stat3.
In addition to the similarity of intermolecular hydrogen bonding behavior during the final 1.5 ns of both MD trajectories, the contributions to the binding free energies of each residue of Stat3 are identical within standard deviation with the exception of M660. In the complex starting with Pose A, this residue contributes -3.7 kcal/mol to the total molecular mechanical/Generalized Born (GB) binding free energy. Interestingly, there is negligible contribution in the structure resulting from Pose B. Decomposition of the contribution of M660 into its individual energetic components reveals a significant van der Waals interaction (-3.2 kcal/mol) and hydrophobic interaction (-0.4 kcal/mol) in the complex starting with Pose A.
The discrepancy in the contribution of M660 to the binding free-energy between the two simulations correlates with the distance between this amino acid and the hydrophobic ring-system of Haic. In the MD simulation starting with Pose B, the distance between M660 Cβ and the fused-ring system of Haic was 5.1 Å - 12.8 Å in the final 1.5 ns of the trajectory. The closest distance of 5.1 Å occurs in only one of the 1500 snapshots and in this instance there are three water molecules between M660 Cβ and rings of Haic, which would preclude hydrophobic interactions. The existence of water molecules between M660 and Haic persisted throughout the entire final 1.5 ns of the 3 ns trajectory.
In contrast, in the MD trajectory of Pose A, the distance between M660 Cβ and Haic ranged from 3.3 Å to 6.3 Å throughout the final 1.5 ns. Moreover, it is within 5.1 Å for 1433 ps out of 1500 ps (95.5%). There are no water molecules between the M660 Cβ and Haic in this simulation, which, in conjunction with the GB free-energy calculations, suggest significant hydrophobic interactions.
In the simulation of Pose A, a hydrogen bond exists between the main chain NH of M660 and the side chain hydroxyl group of Y657 6% of the final 1.5 ns. This pair is <3.5 Å in the remaining simulation indicating high potential for hydrogen bonding.
M660 resides in a loop of about 10 residues from K658 to S668. RMSD analysis of the backbone atoms of residues within this loop reveals significantly more deviation in the Pose A trajectory as compared to that of Pose B. The data seem counterintuitive considering that M660 is “tethered” to Haic by hydrophobic and indirect hydrogen bond interactions in trajectory A but exhibits no interaction at all in trajectory B. Visual analysis of the latter trajectory shows that the loop maintains its β-sheet secondary structure throughout the simulation and remains in place relative to the remainder of the protein. This is likely due to water molecules that packed the space between the loop and the inhibitor when we hydrated the molecular system. In trajectory A the loop does not retain its intraloop hydrogen bonds and it moves relative to the remainder of the protein. No waters were able to fit between this loop and the inhibitor because in the starting complex of Pose A the Gln side chain covered the loop during the solvent soaking step prior to the molecular dynamics simulation. The lack of water molecules allowed movement of the loop resulting in hydrophobic contact between M660 and Haic and hydrogen bonding to Y657.
It is unknown whether interaction with M660 contributes to the binding of compound 15, in which Haic has been “split” into leucine and the bicyclic proline analogue, 2-carboxyindoline. The reduction in affinity may be due to increased entropy penalty resulting from rotation of the Leu Ψ bond which is not present in Haic. Alternatively, collisions between the side chain of Leu and the closest aromatic proton of 2-carboxyindoline may result in conformations of peptide 15 which are unfavorable for binding.
Both E638 and P639 contribute to the free energy of binding in the MD simulations of both poses, through van der Waals and hydrophobic interactions. The side chains of these residues contact the β and aromatic carbons of pCinn. These interactions were observed in the crystal structure of the Stat3 dimer29 and the model of Ac-pTyr-Leu-Pro-Gln-NHBn bound to the SH2 domain of Stat3.65
Integration of the experimental evidence with the molecular dynamics data is consistent with the predicted central/representative structure of the largest cluster (61% of the snapshots) from the extended-conformation MD trajectory (Figure 3). The hydrogen bonds of all but the phosphate are depicted in Figure 3B.
Figure 3.

Proposed model for pCinn-Haic-Gln-OH bound to the SH2 domain of Stat3. The central representative structure from the largest cluster of the MD simulation starting with Pose A is shown. A. Compound 26 is depicted as sticks: green, carbon; blue, nitrogen; red, oxygen. Hydrogens were removed for clarity. Stat3 is shown as a surface representation with the following residues highlighted: Phosphate binding pocket (blue); S636 and P639 (purple); E638 (red); Y640 (yellow); Y657 (orange) M660 (brown). B. Hydrogen bonds between 26 and Stat3. The side chain of E638 was removed for clarity. Stat3 amino acids are depicted in the light blue coloring scheme: light blue, carbon; blue, nitrogen; red, oxygen. The images were generated with PyMol.70
Conclusions
Compared to the original lead hexapeptide, 1, we have significantly reduced peptide character and by judicious incorporation of conformational constraints have increased the affinity nearly 2-fold. Constraining the C-Cα-Cβ-Cγ dihedral angle of the phosphotyrosine 180° led to a major increase in affinity, likely the result of reduced entropy penalty of binding. The affinities of compounds incorporating Haic provide further evidence that the Leu-Pro peptide bond in peptide inhibitors 1 – 4 is in the trans conformation. Modeling of pCinn-Haic-Gln-OH suggests that the basis of affinity of the Haic molecules is, in addition to the ionic interactions with the phosphate, mediated by hydrogen bonds between the inhibitor and various groups on the protein and to hydrophobic interactions with the aromatic ring of the dipeptide mimic. Loop658-668 of Stat3 moved in relation to the bulk of the protein. This movement placed M660 in proximity to the inhibitor allowing a hydrophobic contact with the aromatic groups of Haic, which may be one reason why this Leu-Pro mimic was more effective than the ABN groups. Compound 21, pCinn-Haic-Gln-NHBn, is a peptidomimetic containing only one natural amino acid, Gln.
Currently two models have been published on the nature of phosphopeptide binding to the SH2 domain of Stat3. Shao et al.66 proposed that on docking to the protein, peptides form β-turns and that, in addition to the phosphotyrosine interactions, the side chain carbonyl group of Gln at position pY+3 participates in hydrogen bonding with the main chain NH of E638. McMurray65 hypothesized in a model of peptide 4 that the side chain of Gln participates in an extensive hydrogen bonding network in which the NH2 interacts with the C=O’s of E638 and P639 and carbonyl oxygen participates in a hydrogen bond with the side chain of Q644 and a water mediated hydrogen-bond with the side chain of E638. The models generated here suggest that the side chain of Gln does indeed participate in hydrogen bonds but to residues that are different than the previous models. It seems reasonable that the constrained nature of Haic does not allow the Gln to insert itself as deeply into the pocket as does the more flexible Leu-Pro central dipeptide in McMurray’s model. However, Haic provides interactions with M660 that the peptide does not. These, in addition to the entropic gain of restricted rotation of the pCinn and Leu Ψ dihedral angle, provide binding energy that peptides such as 4 do not exhibit. Thus, the IC50 values of the Haic peptides are low. The interactions suggested by the modeling have generated ideas for modification of Haic and Gln, which will be reported on completion of those studies.
Several authors have reported the development of Stat3 inhibitors that incorporate phopshotyrosine or phosphotyrosine mimic.71-76 These compounds did not contain glutamine or a mimic and displayed low affinity for the target, as judged by electrophoretic mobility shift assays or fluorescence polarization. Modifications to the structure of glutamine reported previously15,16,69 and here point to the importance of the alkyl carboxamide group for affinity. Our previous model65 and the models generated here suggest that the side chain amide group participates in hydrogen bonding interactions with Stat3 that have a major impact on binding energy.
Experimental procedures
Synthesis of 4-(di-tert-butoxyphosphoryloxy)-cinnamic acid, 39
To a suspension of 4-hydroxy cinnamic acid (1.0 g, 6.1 mmol) and tert-butyldimethylsilyl chloride (0.92g, 6.1mmol) in 10 mL of dry THF, N-ethylmorpholine (0.6 mL, 5.5 mmol) was added at room temperature. After 10 min, 1H-tetrazole (1.5 g, 21.3 mmol) was added in one portion followed by the addition of di-tert-butyl N,N-diethylphosphoramidite (4.2 g, 15.2 mmol) in 10 mL of dry THF. After stirring for 2 h, 70% aq. tert-butylhydroperoxide (2.5 mL, 18.3 mmol) was added at 0°C and the mixture was stirred for 1 h more. The reaction was quenched with slow addition of an aqueous solution of 10% Na2S2O5 and stirred for 20 min. It was then diluted with ether (50 mL) and extracted with an additional amount of ether (2 × 50 mL). The combined organic parts were washed with water followed by brine and dried (MgSO4). Solvent was removed and the crude product was triturated with hexane-ether. The product was collected by filtration and dried to give 0.65 g of a white solid. To obtain more product, the filtrate was concentrated and extracted with 5% NaHCO3 (3 × 10 mL). The combined extracts were carefully acidified to pH 3.5-4.0 with slow addition of solid citric acid and were extracted with DCM (3 × 25 mL). The combined organic parts were washed with brine, dried (MgSO4) and evaporated to dryness to give the product as white foam (0.21g). Compound 39 was used in solid phase synthesis with no further purification. 1H NMR (CDCl3, 500 MHz) δ1.5 (s, 18H), 6.36 (d, J = 16.0 Hz, 1H), 7.25 (d, J = 8.0 Hz, 2H), 7.5 (d, J = 8.0 Hz, 2H), 7.7 (d, J = 16.0 Hz, 1H).
Synthesis of Fmoc-(5-[(R)-amino]-1,2,4,5,6,7-hexahydroazepino[3,2,1-hi]indole-4-one-2-(S)-carboxylate), 45
A solution of (R)-4,7-dioxo-5-(2,2,2-trifluoroacetylamino)-1,2,4,5,6,7-hexahydroazepino[3,2,1-hi]indole-2-carboxylic acid methyl ester (43)44,45 (850 mg, 2.3 mmol) was suspended in AcOH (65 mL) and hydrogenated with 10% Pd/C (850 mg) at 50 psi and ambient temperature for 24hrs. The catalyst was removed by filtration through Celite and the solvent removed under reduced pressure. The residue was dissolved in EtOAc (50 mL) and the solution washed with saturated NaHCO3 (2 × 50 mL) and saturated brine (50 mL) before drying (MgSO4). Filtering and removal of the solvent under reduced pressure gave a white solid (744 mg, 87%) which was used without further purification. 1H NMR (CDCl3, 300 MHz) δ 2.0 (m,1H), 2.5 (m,1H), 3.1 (m,2H), 3.5 (m,2H), 3.8 (s,3H), 4.5 (dd,1H), 5.1 (m,1H), 7.1 (m,3H), 7.8 (s,1H).
The intermediate (520 mg, 1.46 mmol) was dissolved in THF (15 mL) and LiOH (210 mg, 8.76 mmol) in H2O (7.5 mL) was added. The mixture was stirred vigorously for 2 h, acidified with 2N HCl (6 mL) and the solvents removed under reduced pressure. Sodium carbonate (950 mg, 8.93 mmol) dissolved in water (18 mL) was added to the solid residue and the mixture stirred for 5 minutes. Fmoc-OSu (492 mg, 1.46 mmol) dissolved in dioxane (18mL) was added and the stirring continued for 20 h. The solvent was removed under reduced pressure, water (150 mL) was added to the solid residue and the mixture acidified with 2N HCl (12 mL). The aqueous suspension was extracted with EtOAc (2×100 mL). The extract was washed with water (3×100 mL) and saturated brine (100 mL) before drying over anhydrous magnesium sulfate. Removal of the solvent gave a solid residue (649 mg) of which 600 mg were purified by silica gel chromatography: EtOAc (300 mL), EtOAc/AcOH 99.7:0.3 (650 mL). Yield: 353mg, 56%. HRMS (M+H), 469.1742 (Theory, 469.1763). 1H NMR (DMSO-d6, 300 MHz) δ 2.0 (m, 2H), 3.0 (m, 3H), 3.4 (m, 1H), 4.2 (m, 4H), 4.9 (dd, 1H), 7.0 (m, 3H), 7.3 (m, 4H), 7.8 (m, 5H), 12.7 (s, 1H). 13C NMR (DMSO-d6) δ 27.6, 30.8, 31.1, 47.2, 56.6, 61.1, 66.1, 120.6, 123.8, 125.7, 126.1, 127.6, 128.1, 129.7, 131.7, 138.9, 141.2, 144.3, 144.4, 156.3, 169.4, 172.9.
Synthesis of (S)-7-hydroxy-4-oxo-5-(2,2,2-trifluoro-acetylamino)-1,2,4,5,6,7-hexahydroazepino[3,2,1-hi]indole-2-carboxylic acid methyl ester, 46
A solution of 4244,45 (500 mg, 1.35 mmol) and AcOH (1 mL) in THF (10 mL) was hydrogenated with 10% Pd/C (21 mg) at 49 psi and ambient temperature for 4 h. The catalyst was removed by filtration through Celite and the filtrate washed with saturated aqueous NaHCO3 (2 × 30 mL) and saturated brine (20 mL) before drying (MgSO4). Filtration and removal of the solvent under reduced pressure gave a white solid (470 mg). Purification was by silica gel chromatography: EtOAc/hexane 1:4 (400 mL), EtOAc/hexane 3:7 (180 mL) 30 mg (1), EtOAc/hexane 3:7 (680 mL) 200 mg (3), EtOAc/hexane 4:6 (410 mL) gave 430 mg of 46 (86%): 1H NMR (CDCl3, 300 MHz) δ 2.4 (m,1H), 2.7 (m,1H), 3.2 (dd,1H), 3.5 (m,1H), 3.8 (s,3H), 4.5 (m,1H), 5.25 (m,1H), 5.35 (m,1H), 7.2 (m,2H), 7.5 (m,1H), 7.8 (d,1H) ppm.
Synthesis of Fmoc-(5-[(S)-amino]-1,2,4,5-tetrahydroazepino[3,2,1-hi]indole-4-one-2-(S)-carboxylate), 47
Compound 46 (780 mg, 2.1 mmol) was dissolved in trifluoroacetic acid (10 mL) and stirred at room temperature for 5 h. The solvent was removed under reduced pressure and the residue dissolved in EtOAc (100 mL). This solution was washed with 5% NaHCO3 (2 × 75 mL) and saturated brine (50 mL) before drying over MgSO4. Filtration and removal of the solvent gave a solid (760 mg) which was purified by silica gel chromatography: EtOAc/hexane 1:4 (400mL) 680 mg, 92% (4), mp 120-124 °C: 1H NMR (CDCl3, 300 MHz) δ 3.3 (dd,1H), 3.65 (m,1H), 3.7 (s,3H), 4.7 (m,1H), 5.4 (dd,1H), 5.7 (dd,1H),6.8 (dd,1H), 7.2 (m,3H), 7.9 (s,1H).
The unsaturated intermediate (520 mg, 1.49 mmol) was dissolved in THF (18 mL) and LiOH (212 mg, 8.80 mmol) in H2O (9 mL) was added. The mixture was stirred vigorously for 2 h, acidified with HCl and the solvent removed under reduced pressure. Sodium carbonate (933 mg, 8.80 mmol) dissolved in water (15 mL) was added to the solid residue and the mixture stirred for 5 minutes. Fmoc-OSu (562 mg, 1.49 mmol) dissolved in dioxane (15 mL) was added and the stirring continued for 20 h The solvent was removed under reduced pressure, water (70 mL) was added to the solid residue and the mixture acidified with 2N HCl (6 mL). The aqueous suspension was extracted with EtOAc (3 × 70 mL). The extract was washed with water (4 × 70 mL) and saturated brine (70 mL) before drying (MgSO4). Removal of the solvent gave a solid residue (734 mg) which was purified by silica gel chromatography: EtOAc/hexane 1:1 (250 mL), EtOAc/AcOH 99.7:0.3 (410 mL). Yield, 317 mg, 46%. HRMS, 467z.1587 (Theory, 467.1607) 1H NMR (DMSO, 300 MHz) δ 3.1 (d,1H), 3.7 (m,1H), 4.3 (m,4H), 5.1 (d,1H), 5.7 (dd,1H), 6.8 (dd,1H), 7.1 (t,1H), 7.2 (d,1H), 7.3 (m,3H), 7.4 (t,2H), 7.7 (t,2H), 7.9 (d,2H), 8.1 (d,1H), 12.9 (s,1H) ppm. 13C NMR (DMSO, 300MHz) δ 32.6, 47.1, 54.6, 61.8, 66.3, 120.6, 124.0, 125.1, 125.7, 125.8, 126.3, 127.6, 128.1, 128.3, 132.8, 139.2, 141.2, 144.2, 144.3, 156.5, 166.4, 172.5.
Synthesis of Fmoc-Glu(OtBu)-OMe, 49a
To a stirred solution of Fmoc-Glu(OtBu)-OH (1.0 g, 2.35 mmol), DIPEA (0.9 mL, 4.7 mmol) in 10 mL of dry CH2Cl2 and 2mL of dry MeOH, at 0 °C, a solution of PyBOP (1.4 g, 2.6mmol) in 5 mL of dry CH2Cl2 was added dropwise. The mixture was stirred overnightand was transferred to a separatory funnel, with an additional amount of CH2Cl2 (30 mL). The organic layer then washed with 10% HCl, 10% NaHCO3, brine and dried (MgSO4). Solvent was removed, the crude then purified by silica gel column chromatography elution with 25% EtOAc-hexane to yield yielded 0.90gm of pure Fmoc-Glu(OtBu)-OMe. 1H NMR (DMSO-d6, 500 MHz) δ 1.4 (s, 9H), 1.8 (m, 1H), 1.95 (m, 1H), 2.3 (m, 2H), 3.64 (s, 3H), 4.1 (m, 1H), 4.24 (m, 1H), 4.32 (d, J= 7.0 Hz, 2H), 7.34 (t, J = 7.0Hz, 2H), 7.43 (t, J= 7.0Hz, 2H), 7.72 (m, 2H), 7.78 (d, J = 8.0Hz, 1H), 7.91 (d, J = 7.5 Hz, 2H). 13C NMR (DMSO-d6, 125.0 MHz) δ 26.5, 28.2, 31.5, 47.1, 52.4, 53.4, 66.1, 80.3, 120.6, 125.7, 127.5, 128.1, 141.2, 144.3, 156.5, 171.9, 173.0. Anal (C25H29NO6) C, H, N: Cald: C, 68.32; H, 6.65; N, 3.19. Found: C, 68.50; H, 6.70; N, 3.24.
Synthesis of Fmoc-Glu-OMe, 50a
Fmoc-Glu(OtBu)-OMe (0.50 g) was treated with neat TFA (5 mL) for 1h. The TFA was removed under vacuum and residual acid was removed by adding and evaporating tolune (2 × 5 mL). The product was recrystallised from ether —hexane to yield 0.40 g of a white powder. 1H NMR (DMSO-d6, 500 MHz) δ 1.82 (m, 1H), 1.98 (m, 1H), 2.34 (m, 2H), 3.64 (s, 3H), 4.1 (m, 1H), 4.24 (m, 1H), 4.32 (m, 2H), 7.35 (t, J = 7.5 Hz, 2H), 7.43 (t, J = 7.5 Hz, 2H), 7.73 (d, J = 7.5 Hz, 2H), 7.8 (d, J = 8.0 Hz, 1H), 7.9 (d, J = 7.5 Hz, 2H), 12.2 (s, 1H). 13C NMR (DMSO-d6, 125.0 MHz) δ 26.4, 30.4, 47.1, 52.4, 53.5, 66.1, 120.6, 125.7, 127.5, 128.1, 141.2, 144.2, 156.6, 173.0, 174.1. Anal (C21H21NO6) C, H, N: Cald: C, 65.79; H, 5.52; N, 3.65. Found: C, 64.99; H, 5.53; N, 3.66.
Synthesis of Fmoc-Glu(OtBu)-NHMe, 49b
Prepared from Fmoc-Glu(OtBu)-OH (1g, 2.35 mmol) using identical conditions as for 49a except that MeNH2·HCl (0.16g, 2.35 mmol) was used in place of the MeOH. Yield, 0.93 gm of 49b. 1H NMR (DMSO-d6, 500 MHz) δ 1.4 (s, 9H), 1.75 (m, 1H), 1.9 (m, 1H), 2.2 (m, 2H), 2.6 (d, J = 4.5Hz, 3H), 3.96 (m, 1H), 4.2-4.3 (m, 2H), 4.32 (m, 1H), 7.33 (t, J = 7.0 Hz, 2H), 7.42 (t, J = 7.0 Hz, 2H), 7.5 (d, J = 8.0 Hz, 1H), 7.74 (t, J = 7.0 Hz, 2H), 7.8 (d, J = 4.5 Hz), 7.9 (d, J = 7.5 Hz, 2H). 13C NMR (DMSO-d6, 125.0 MHz) δ26.1, 27.7, 28.2, 31.9, 47.2, 54.4, 66.1, 80.1, 120.6, 125.8, 127.5, 128.1, 141.2, 144.2, 144.4, 156.4, 172.1. Anal (C25H30N2O5) C, H, N: Cald: C, 68.47; H, 6.90; N, 6.39. Found: C, 68.45; H, 6.90; N, 6.37.
Synthesis of Fmoc-Glu-NHMe, 50b
Prepared from 49b (0.50 g) using identical conditions as for 50a. Yield: 0.38 g of a white powder. 1H NMR (DMSO-d6, 500 MHz) δ 1.77(m, 1H), 1.95(m, 1H), 2.25 (m, 2H), 2.6 (s, 3H), 4.0(m, 1H), 4.2-4.33 (m, 3H), 7.33 (t, J = 7.0 Hz, 2H), 7.42 (t, J = 7.0 Hz, 2H), 7.5 (d, J = 8.0 Hz, 1H), 7.74 (m, 2H), 7.8 (d, J = 4.5 Hz, 1H), 7.9 (d, J = 7.5 Hz, 2H), 12.1 (s, 1H). 13C NMR (DMSO-d6, 125.0 MHz) δ26.1, 27.7, 30.7, 47.2, 54.5 66.1, 120.6, 125.8, 127.5, 128.1, 141.2, 144.2, 144.4, 156.4, 172.2, 174.3. Anal (C21H22N2O5) C, H, N: Cald: C, 65.96; H, 5.80; N, 7.33. Found: C, 66.11; H, 5.95; N, 7.23.
Synthesis of Fmoc-Glu (OtBu)-NHiPr, 49c
Prepared from Fmoc-Glu(OtBu)-OH (1g, 2.35 mmol) using identical conditions as in 49a except that isopropylamine (0.2mL, 2.6 mmol) in 5 mL of dry CH2Cl was used instead of MeOH. Yield: 0.90 gm of pure Fmoc-Glu(OtBu)-NHCH(Me)2. 1H NMR (DMSO-d6, 500 MHz) δ1.07 (m, 6H), 1.4 (s, 9H), 1.78 (m, 1H), 1.9 (m, 1H), 2.23 (m, 2H), 3.86 (m, 1H), 4.0 (m, 1H), 4.22-4.3 (m, 3H), 7.33 (m, 2H), 7.40-7.46 (m, 3H), 7.65-7.8 (m, 3H), 7.9 (d, J = 7.0Hz, 2H). 13C NMR (DMSO-d6, 125.0 MHz) δ 22.7, 22.8, 28.2, 31.9, 47.2, 54.4, 66.1, 80.1, 120.5, 125.8, 127.5, 128.1, 141.2, 144.2, 144.4, 156.3, 170.7, 172.1. Anal (C27H34N2O5) C, H, N: Calcd: C, 69.50; H, 7.35; N, 6.00. Found: C, 69.11; H, 7.32; N, 5.91.
Synthesis of Fmoc-Glu-NHiPr, 50c
Prepared from 49c (0.50 g) using identical conditions as for 50a. Yield: 0.40 g of a white powder. 1H NMR (DMSO-d6, 500 MHz) δ 1.06 (m, 6H), 1.75(m, 1H), 1.88(m, 1H), 2.26 (m, 2H), 3.83 (m, 1H), 4.0(m, 1H), 4.2-4.3 (m, 3H), 7.33 (m, 2H), 7.4-7.46 (m, 3H), 7.74 (m, 3H), 7.9 (d, J = 7.5 Hz, 2H), 12.1 (s, 1H). 13C NMR (DMSO-d6, 125.0 MHz) δ 22.7, 22.8, 28.1, 30.8, 47.2, 54.4, 66.1, 120.5, 125.8, 127.5, 128.1, 141.2, 144.2, 144.4, 156.3, 170.8, 174.4. Anal (C23H26N2O5) C, H, N: Calcd: C, 67.30; H, 6.38; N, 6.82. Found: C, 66.38; H, 6.39; N, 6.68.
Molecular Modeling of the interactions of 26 and the SH2 domain of Stat3
The coordinates for Stat3β (PDB code 1bg1)29 were obtained from the Research Collaboratory for Structural Bioinformatics (RCSB) Brookhaven Protein Data Bank (PDB; http://www.rcsb.org/pdb).77 Compound 26 and fragments thereof were computationally constructed using the Builder module of INSIGHTII 98.0.78 The coordinates of Haic from a peptidomimetic inhibitor bound to the MHC class II HLA-DR (PDB code 1d5x)79 were used as a template for this dipeptide mimic.
Preparation of ligands
Fragments pCinn-NH2 and H-Haic-Gln-OH were assigned consistent valence force-field (CVFF) partial charges with INSIGHTII 98.0/FDISCOVER 2.98.78 Two of the oxygen atoms of the pCinn-NH2 phosphate group were manually assigned a formal charge of -1, bringing the total formal charge of the fragment to -2. One of the oxygen atoms of the carboxyl group of H-Haic-Gln-OH was manually assigned a formal charge of -1 bringing the total formal charge of the fragment to -1. Formal charges were initially assigned with the CVFF in INSIGHTII 98.0 so that the bond order in the output structure would be correctly set for subsequent processing in the ANTECHAMBER module of the ASSISTED MODEL BUILDING WITH ENERGY REFINEMENT 8 (AMBER 8) molecular simulation programs.80 pCinn-NH2 and H-Haic-Gln-OH were processed separately because the complete compound 26 (68 atoms) was too large for the ANTECHAMBER module. Atomic partial charges, originally assigned through CVFF, were adjusted using the generalized Amber force-field (GAFF). Partial atomic charges were assigned using the AM1 — bond charge correction (AM1-BCC) method.81 GAFF was also used to assign bond and angle force-field potentials.82 To create the complete compound 26, the NH2 group of pCinn-NH2 was deleted in INSIGHTII 98.078 and the acyl group was appended to the exocyclic amino group of N-Haic-Gln-OH in the LEAP module of AMBER8. To compensate for the abrupt change in charge distribution caused by joining these fragments, a third fragment comprised of pCinn, up to but excluding the phosphate and the phenyl groups, and Haic, was generated and processed in ANTECHAMBER. The overlapping charges assigned to the third fragment were used to replace the corresponding charges in the complete 26 construct so as to better approximate a true charge distribution.
Energy Minimization
The Amber99 force-field83 was converted by the AMBERFFC 1.3 program84 to a format which could be read by INSIGHTII 98.0/CDISCOVER 3.78 To reduce the number of torsions during the automated rigid-protein/flexible-ligand docking, 26 was separated in INSIGHTII 98.078 into pCinn-NH2 and pCinn-Haic-OH fragments. The pCinn-NH2 atoms, excluding the phosphophenyl group, were subjected to 40 steps of steepest-descent (SD) followed by 200 steps of conjugate-gradient (CG) energy minimizations in INSIGHTII 98.0/CDISCOVER 3,78at which point the change in total energy (non-bonded and internal) was negligible and the derivative of the gradient was less than 0.01. Energy minimizations were executed using a distance-dependent dielectric constant to implicitly represent continuum solvent, and without non-bonded (electrostatic and van der Waals) cutoffs. The coordinates for the atoms of the phosphophenyl group were held fixed during the minimizations.
Automated Rigid-Protein/Flexible-Ligand Docking
Hydrogen atoms were added to the Stat3 crystal structure at pH 7.4 in INSIGHTII 98.0.78 The charges and Lennard-Jones potentials for all the atoms, including the added hydrogens, were subsequently adjusted with Amber99 force-field parameters.83 All ligands (26 and its fragments) were docked to Stat3 with AUTODOCK 4.039 in an automated rigid-protein/flexible-ligand manner, using a free-energy and charge-based desolvation scoring function.85 Stat3 (residues 136 to 688) served as the rigid-protein, and all atomic partial-charges assigned by the Amber99 force-field83 in INSIGHTII 98.078 were accepted. Atomic partial-charges assigned by ANTECHAMBER to 26 and its fragments were accepted. All ligands were docked to the Stat3 SH2 domain using the Lamarckian genetic algorithm (LGA) method86 using a distance-dependent dielectric. A maximum of 256 LGA runs were executed for each docking experiment. The torsional free-energies (internal 1-4 interactions) and internal electrostatics of all ligands were included in the free-energy calculations. The ligand structures resulting from each docking study were clustered with an RMSD of 1.0 Å. All dockings utilized ligand-centered grid maps of 0.375 Å spacing for each atom type of the ligand and for the electrostatic (e) and desolvation (d) maps.
Docking of pCinn-NH2
The pCinn-NH2 fragment was docked to the Stat3 SH2 domain first. The C, N, P, A, OA, HD, d, and e centered grid maps, with 36, 30, and 32 grid points in the X, Y, and Z directions, respectively, were calculated to create the final affinity grid. LGA was used to generate an initial population of 300 different structures. The structure with the lowest (most favorable) free-energy of binding (FEB) survived to seed the next population of 300 structures. A maximum of 27,000 generations of populations of 300 structures were seeded, and a maximum of 25 million energy calculations were evaluated. AUTODOCK 4.0 identified four torsional degrees of freedom (torsdof) in pCinn-NH2 to be used in the FEB calculation, and all four torsions (ndihe) were active and allowed to rotate in 30° increments during the docking. The pCinn-NH2 fragment was allowed to translate in 1 Å increments, and rotate about its axis in 30° increments. All other parameters, including the LGA parameters, were assigned the AUTODOCK 4.0 default values.
Docking of pCinn-Haic-OH
After the pCinn-NH2 fragment was docked to the Stat3 SH2 domain, one structure was selected as a precursor to the pCinn-Haic-OH starting structure. The same set of docking conditions used for pCinn-NH2 was used for pCinn-Haic-OH except that 40, 40, and 54 grid points in the X, Y, and Z directions respectively, were calculated to create the final affinity grid. Also, a maximum of 2.5 million energy calculations were evaluated, and only three of six torsdof were used in the FEB calculation.
Docking of pCinn-Haic-Gln-OH, 26
One structure from the pCinn-Haic docking study was selected as a precursor for the starting structure for the complete inhibitor 26. The same set of pCinn-Haic docking conditions were used for 26 except 42, 40, and 42 grid points in the X, Y, and Z directions respectively, were calculated to create the final affinity grid, and only three of six torsdof were used in the FEB calculation. After 26 was docked to the Stat3 SH2 domain, one structure was selected as a precursor to another starting structure. A second docking experiment was executed with identical parameters to that of the first 26. Two structures from the second docking experiment were selected as final docking poses, one of which was a member of the largest docking cluster (extended-conformation, Pose A) and the other belonging to a cluster that placed the glutamine side-chain in the pocket formed by Stat3 residues E638, P639, and Y640 (bent-conformation, Pose B).
Implicit-Solvent Energy Minimization
The two poses were assigned Amber99 and GAFF force-field parameters77 in INSIGHTII 98.0/CDISCOVER 3,78 respectively. The energy landscapes of both systems were searched for local energy minima to remove high energy atomic “clashes” that may have arisen between Stat3 and 26. Energy minimizations were executed in a step-wise manner using a distance-dependent dielectric constant to implicitly represent continuum solvent, and without non-bonded (van der Waals and electrostatic) cutoffs. Each minimization was terminated when the change in total energy of the complex was negligible and the derivative of the gradient was less than 0.01.
Pose A
The Stat3/26 complex of pose A was subjected to the following 24,500 step energy minimization procedure: 1) 1,000 SD followed by 1,500 CG on compound 26 hydrogens; 2) 1,000 SD followed by 1,000 CG on 26 and Stat3 hydrogens; 3) 2,000 SD followed by 3,000 CG on the 26 hydrogens and the Stat3 side-chains; 4) 2,000 SD followed by 3,000 CG on 26, and the Stat3 side-chains including the alpha carbons; 5) 2,000 SD followed by 8,000 CG on all atoms of both 26 and Stat3.
Pose B
The Stat3/26 complex of pose B was subjected to the following 13,700 step energy minimization procedure: 1) 600 SD followed by 600 CG on compound 26 hydrogens; 2) 500 CG on the Stat3 hydrogens; 3) 500 SD followed by 500 CG on the Stat3 side-chains excluding the alpha carbons; 4) 1,000 SD followed by 1,000 CG on 26 and the Stat3 side-chains excluding the alpha carbons; 5) 1,000 SD followed by 8,000 CG on all atoms of both 26 and Stat3.
Explicit-solvent Molecular Dynamics
Both docking poses were solvated in a 15 Å truncated octahedral box with explicit water molecules using the XLEAP module of AMBER8. The total charge on the Stat3 protein, which was protonated with the CVFF at pH 7.4, was +1 and the total charge on 21 was -3, bringing the total charge on the poses to -2. The charges on each simulation system was neutralized by replacing two of the explicit waters in the truncated octahedral boxes with two sodium ions (Na+) at a distance of greater than 3.5 Å from the protein/ligand complexes. This rendered the net charge on the simulation system zero which is a requirement of the method that we used for treating long range electrostatic effects (see below). Water molecules were modeled using the TIP3P force-field parameters.87
Systems Setup
The heating, equilibration, and production phases of all molecular dynamics (MD) simulations were executed in NAMD using a fixed number of particles, fixed pressure, and fixed temperature (NPT) ensemble. The pressures of the systems were fixed to 1.01325 bar by coupling to a Berendsen pressure bath,88 and were assigned a compressibility of 4.57 × 10-5 bar. After heating and during equilibration, the temperatures of the systems were fixed to 310K by coupling to a temperature bath with temperature reassignment every 1 ps. Atomic positions, trajectories, forces, and other system parameters were sampled in 2 fs time intervals by fixing hydrogen covalent bond lengths using the SHAKE algorithm89 with a 1.0 x 10-8 Å tolerance. A 12 Å interaction cutoff, which gradually approached zero beginning at a distance of 8 Å, was used to truncate the non-bonded and long-range electrostatics of the systems. Any atom pairs that vacillated beyond and within the 12 Å cutoff but did not exceed 13 Å were included in the calculations. The Particle Mesh Ewald (PME) summation method90 was employed to correct for the cutoffs of all long-range electrostatic interactions. The octahedral solvent boxes of the systems were replicated in all dimensions using periodic boundary conditions. The periodic cells of both systems were periodically adjusted as the size (volume) of the systems changed during the equilibration phases of the MD simulations. Each unit of the cell was allowed to adjust its size (volume) in response to changes that may have arisen during NPT sampling. The Amber9982 force-field was used by NAMD to assign all atomic parameters and implement all force-field equations.
Energy Minimization
The energy landscapes of both systems were searched for local energy minima to remove high energy atomic “clashes” that may have arisen between the added waters, the added ions, and the poses. Energy minimizations were executed in a step-wise manner in explicit-solvent with a 12 Å non-bonded cutoff. Each minimization step was terminated when the change in total energy of the docking pose and the change in the derivative of the gradient was negligible. SD minimizations were executed with the SANDER module of AMBER8, and CG minimizations were executed with NAMD 2.61B.91
Heating
Atomic velocities were reassigned at higher kinetic energies until the average kinetic energies of the systems were slowly, over the span of 31 picoseconds (ps) and with temperature reassignment every 1 ps, brought to 310 K. After the average kinetic energies of the systems reached 310K, the average kinetic energies were held at 310K for 19 ps by temperature reassignment every 1 ps, as necessary.
Equilibration
After heating, velocities were reassigned in both systems every 1 ps to keep the average kinetic energy at 310 K until the change in total energy as a function of time became negligible.
Production
After equilibration, both systems were allowed to evolve without temperature reassignment. The size of the periodic cell remained constant during NPT sampling. A 3 ns MD trajectory was sampled for both systems.
Pose A
The solvated docking-pose A contained a total of 46,396 atoms; which included 194 Stat3 residues (3,111 atoms), 68 compound 26 atoms, 14,405 explicit waters (43,215 atoms), and 2 Na+ ions. The system was subjected to the following minimization procedure: 1) 7,000 SD followed by 46,000 CG steps on the solvent; 2) 31 ps (15,500 time steps) of solvent heating followed by 19 ps (9,500 time steps) of temperature reassignment to 310K; 3) 493 ps (246,500 time steps) of solvent equilibration with temperature reassignment to 310K; 4) 2,000 SD followed by 6,000 CG steps on the docking-pose; 5) 1,000 CG energy minimization on the system; 6) 31 ps (15,500 time steps) of system heating followed by 19 ps (9,500 time steps) of temperature reassignment to 310K; 7) 200 ps (100,000 time steps) of system equilibration with temperature reassignment to 310K; 8) 3 ns system sampling without temperature reassignment.
Pose B
The solvated docking-pose B contained a total of 44,944 atoms; which included 194 Stat3 residues (3,111 atoms), 68 compound 26 atoms, 13,921 explicit waters (41,763 atoms), and 2 Na+ atoms. The system was subjected to the following minimization procedure: 1) 7,000 SD steps on the solvent; 2) 6,000 CG energy minimization on the solvent hydrogens; 3) 12,000 CG steps on the solvent; 4) 31 ps (15,500 time steps) of solvent heating followed by 19 ps (9,500 time steps) of temperature reassignment to 310K; 5) 810 ps (405,000 time steps) of solvent equilibration with temperature reassignment to 310K; 6) 1,000 SD followed by 6,000 CG energy minimizations on the docking-pose; 7) 31 ps (15,500 time steps) of heating with temperature reassignment to 310K (37oC); 8) 100 ps (50,000 time steps) of equilibration with temperature reassignment to 310K; 9) 200 ps (100,000 time steps) of system equilibration temperature reassignment to 310K; 10) 3 ns system sampling without temperature reassignment.
Molecular Dynamics Trajectory RMSD Clustering
For both systems, the solvent was removed after the conclusion of the 3 ns MD simulation, and 3,000 structures from the final 3 ns of the trajectory were RMSD clustered using the g_cluster module of the Gröningen Machine for Chemical Simulations 3.3 (GROMACS 3.3) program.92 The systems were RMSD clustered using the gromos algorithm described by Daura et al.93 Before the RMSD clustering, the Stat3 backbone atoms were least-squares fitted and a 1 Å cutoff was used. The representative structure from each cluster was the structure with the smallest average distance to all other structures in the cluster.
Poisson-Boltzmann and Generalized Born Electrostatics
For both systems, AMBER 880 was used to separately calculate the solvation energy of the isolated implicitly solvated Stat3, the isolated implicitly solvated 26, and the isolated implicitly solvated docking-pose. The polar (electrostatic/Coulombic) component of the solvation energy was calculated using finite-difference methods to solve the linearized Poisson-Boltzmann (PB) equation. The Generalized Born (GB) method was also used to approximate the linearized PB equation and calculate electrostatic and non-polar solvation energies. The non-polar component of the solvation energy was calculated by measuring the solvent-accessible surface area (SASA) with a 1.4 Å water probe, and subsequently multiplying the SASA value to the empirical constant 0.0072 kcal/mol/ Å2. The total solvation energy was determined by summing the non-polar and polar components. The total solvation contribution to the free-energy of binding (FEB) of 26 to Stat3 was determined by subtracting the total solvation energy of the isolated implicitly solvated Stat3 and the total solvation energy of the isolated implicitly solvated Stat3, from the total solvation energy of the isolated implicitly solvated 26. For the electrostatics calculations, the solute dielectric was set to 2, the solvent dielectric constant was set to 80, the ionic strength set to 0, and the grid spacing set to 0.25 Å. For the gas phase (vacuum) molecular mechanics energies (Coulombic/electrostatic, van der Waals, internal), the external dielectric was set to 1. MM-PBSA and MM-GBSA calculations were performed on 150 structures from every 100th ps of the final 1.5 ns of a 3 ns trajectory.
Supplementary Material
Acknowledgements
We are grateful to the National Cancer Institute (CA096652) and the M. D. Anderson Cancer Center University Cancer Fund for support of this work. We also acknowledge the NCI Cancer Center Support Grant CA016672 for the support of our NMR facility and both the M. D. Anderson Cancer Center Proteomics Facility and Raymond Jacobson of the Structural Biology Program for mass spectrometry. JMB and DWL acknowledge support from the NASA University Research Center through Texas Southern University. Funding as an Odyssey Fellow (Z.R.) was supported by the Odyssey Program and the Cockrell Foundation Award for Scientific Achievement at UTMDACC.
Glossary
Abbreviations
- ABN
azabicyclo[4.3.0]nonane-9-carboxylate
- DIC
diisopropylcarbodiimide
- Gaba
γ-amino butyric acid
- GB
generalized Borne
- Haic
5-amino-1,2,4,5,6,7-hexahydro-4-oxo-azepino[3,2,1-hi]indole-2-carboxylic acid
- ΔHaic
“dehydro-Haic” 5-amino-1,2,4,5-tetrahydro-4-oxo-azepino[3,2,1-hi]indole-2-carboxylic acid
- Met(O)
methionine sulfoxide
- Met(O2)
methionine sulfone
- pCinn
4-phosphoryloxy cinnamic acid
- pInd
5-phosphoryloxyindole-2-carboxylate
- SAR
structure activity relationship
- pTic
4-phosphoryloxy-1,2,3,4-tetrahydro-3-isoquinolinecarboxylic acid.
Footnotes
Supporting Information Available. General synthesis procedures, general HPLC procedures, a table of mass spectral characterization of compounds 5-34, HPLC chromatograms of compounds 5-34, and NMR spectra of compounds 45 and 47.
REFERENCES
- 1.Bromberg J, Darnell JE., Jr. The role of STATs in transcriptional control and their impact on cellular function. Oncogene. 2000;19:2468–2473. doi: 10.1038/sj.onc.1203476. [DOI] [PubMed] [Google Scholar]
- 2.Levy DE, Darnell JE., Jr. Stats: transcriptional control and biological impact. Nature Rev. Mol. Cell. Biol. 2002;3:651–662. doi: 10.1038/nrm909. [DOI] [PubMed] [Google Scholar]
- 3.Stark GR, Kerr IM, Williams BR, Silverman RH, Schreiber RD. How cells respond to interferons. Ann. Rev. Biochem. 1998;67:227–264. doi: 10.1146/annurev.biochem.67.1.227. [DOI] [PubMed] [Google Scholar]
- 4.Bowman T, Garcia R, Turkson J, Jove R. STATs in oncogenesis. Oncogene. 2000;19:2474–2488. doi: 10.1038/sj.onc.1203527. [DOI] [PubMed] [Google Scholar]
- 5.Buettner Ralf, Mora Linda B., Jove Richard. Activated STAT signaling in human tumors provides novel molecular targets for therapeutic intervention. Clin. Cancer Res. 2002;8:945–954. [PubMed] [Google Scholar]
- 6.Bromberg J. Stat proteins and oncogenesis. J. Clin. Invest. 2002;109:1139–1142. doi: 10.1172/JCI15617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Darnell JE., Jr. Transcription factors as targets for cancer therapy. Nature Rev. Cancer. 2002;2:740–749. doi: 10.1038/nrc906. [DOI] [PubMed] [Google Scholar]
- 8.Yu H, Jove R. The STATS of cancer — new molecular targets come of age. Nature Reviews Cancer. 2004;4:97–105. doi: 10.1038/nrc1275. [DOI] [PubMed] [Google Scholar]
- 9.Leong PL, Andrews GA, Johnson DE, Dyer KF, Xi S, Mai JC, Robbins PD, Gadiparthi S, Burke NA, Watkins SF, Grandis JR. Targeted inhibition of Stat3 with a decoy oligonucleotide abrogates head and neck cancer cell growth. P. Natl. Acad. Sci. USA. 2003;100:4138–4143. doi: 10.1073/pnas.0534764100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Chan KS, Sano S, Kiguchi K, Anders J, Komazawa N, Takeda J, DiGiovanni J. Disruption of Stat3 reveals a critical role in both the initiation and the promotion stages of epithelial carcinogenesis. J. Clin. Invest. 2004;114:720–728. doi: 10.1172/JCI21032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Xi S, Gooding WE, Grandis JR. In vivo antitumor efficacy of STAT3 blockade using a transcription factor decoy approach: implications for cancer therapy. Oncogene. 2005;24:970–979. doi: 10.1038/sj.onc.1208316. [DOI] [PubMed] [Google Scholar]
- 12.Zhang X, Zhang J, Wei H, Tian Z. STAT3-decoy oligodeoxynucleotide inhibits the growth of human lung cancer via down-regulating its target genes. Oncol. Rep. 2007;17:1377–1382. [PubMed] [Google Scholar]
- 13.Zhang X, Zhang J, Wang L, Wei H, Tian Z. Therapeutic effects of STAT3 decoy oligodeoxynucleotide on human lung cancer in xenograft mice. BMC cancer. 2007;7:149, 13. doi: 10.1186/1471-2407-7-149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Liu X, Li J.i, Zhang J. STAT3-decoy ODN inhibits cytokine autocrine expression by murine tumor cells. Cell. Mol. Immunol. 2007;4:309–313. [PubMed] [Google Scholar]
- 15.Ren Z, Cabell LA, Schaefer TS, McMurray JS. Identification of a High Affinity Phosphopeptide Inhibitor of Stat3. Bioorg. Med. Chem. Lett. 2003;13:633–636. doi: 10.1016/s0960-894x(02)01050-8. [DOI] [PubMed] [Google Scholar]
- 16.Coleman DR, IV, Ren R, Mandal PK, Cameron AG, Dyer GA, Muranjan S, Chen X, McMurray JS. Investigation of the binding determinants of phosphopeptides targeted to the SH2 domain of Stat3. Development of a high affinity peptide inhibitor. J. Med. Chem. 2005;48:6661–6670. doi: 10.1021/jm050513m. [DOI] [PubMed] [Google Scholar]
- 17.Sawyer TK. Src homology-2 domains: structure, mechanisms, and drug discovery. Biopolymers. 1998;47:243–261. doi: 10.1002/(SICI)1097-0282(1998)47:3<243::AID-BIP4>3.0.CO;2-P. [DOI] [PubMed] [Google Scholar]
- 18.Cody WL, Lin Z, Panek RL, Rose DW, Rubin JR. Progress in the development of inhibitors of SH2 domains. Curr. Pharm. Des. 2000;6:59–98. doi: 10.2174/1381612003401532. [DOI] [PubMed] [Google Scholar]
- 19.Muller G. Peptidomimetic SH2 domain antagonists for targeting signal transduction. Top. Curr. Chem. 2001;211:17–59. [Google Scholar]
- 20.Shakespeare WC. SH2 domain inhibition: a problem solved? Curr. Opin. Chem. Biol. 2001;5:409–415. doi: 10.1016/s1367-5931(00)00222-2. [DOI] [PubMed] [Google Scholar]
- 21.Metcalf CA, III, Sawyer T. Src homology-2 domains and structure-based, small-molecule library approaches to drug discovery. In: Makriyannis A, Biegel D, editors. Drug Discovery Strategies and Methods. Marcel Dekker, Inc.; New York, NY: 2004. pp. 23–59. [Google Scholar]
- 22.Shakespeare W, Yang M, Bohacek R, Cerasoli F, Stebbins K, Sundaramoorthi R, Azimioara M, Vu C, Pradeepan S, Metcalf C, III, Haraldson C, Merry T, Dalgarno D, Narula S, Hatada M, Lu X, van Schravendijk MR, Adams S, Violette S, Smith J, Guan W, Bartlett C, Herson J, Iuliucci J, Weigele M, Sawyer T. Structure-based design of an osteoclast-selective, nonpeptide Src homology 2 inhibitor with in vivo antiresorptive activity. Proc. Natl. Acad. Sci, U.S.A. 2000;97:9373–9378. doi: 10.1073/pnas.97.17.9373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Violette SM, Shalespeare WC, Bartlett C, Guan W, Smith JA, Rickles RJ, Bohacek RS, Holt DA, Baron R, Sawyer T. A Src SH2 selective binding compound inhibits osteoclast-mediated resorption. Chem. Biol. 2000;7:225–235. doi: 10.1016/s1074-5521(00)00090-9. [DOI] [PubMed] [Google Scholar]
- 24.Betageri Rajashekhar, Beaulieu Pierre L., Llinas-Brunet Montse, Ferland Jean-Marie, Cardozo Mario, Moss Neil, Patel Usha, Proudfoot John R. Pyridones as Src family SH2 domain inhibitors. PCT Int. Appl. 1999 WO 9931066. [Google Scholar]
- 25.Gay B, Suarez S, Caravatti G, Furet P, Meyer T, Schoepfer J. Selective GRB2 SH2 inhibitors as anti-Ras therapy. Int. J. Cancer. 1999;83:235–241. doi: 10.1002/(sici)1097-0215(19991008)83:2<235::aid-ijc15>3.0.co;2-b. [DOI] [PubMed] [Google Scholar]
- 26.Furet P, Garcia-Echeverria C, Gay B, Schoepfer J, Zeller M, Rahuel J. Structure-based design, synthesis and X-ray crystallography of a high affinity antagonist of the Grb2-SH2 domain containing an asparagine mimetic. J. Med. Chem. 1999;42:2359–2363. doi: 10.1021/jm991013u. [DOI] [PubMed] [Google Scholar]
- 27.Atabey N, Gao Y, Yao ZJ, Breckenridge D, Soon L, Soriano JV, Burke TR, Bottaro DP. Potent blockade of hepatocyte growth factor-stimulated cell motility, matrix invasion and branching morphogenesis by antagonists of Grb2 Src homology 2 domain interactions. J. Biol. Chem. 2001;276:14308–14314. doi: 10.1074/jbc.M010202200. [DOI] [PubMed] [Google Scholar]
- 28.Shi Z-D, Lee K, Wei C-Q, Roberts LR, Worthy KM, Fisher RJ, Burke TR., Jr Synthesis of a 5-Methylindolyl-Containing Macrocycle That Displays Ultrapotent Grb2 SH2 Domain-Binding Affinity. J. Med. Chem. 2004;47:788–791. doi: 10.1021/jm030440b. [DOI] [PubMed] [Google Scholar]
- 29.Becker S, Groner B, Muller CW. Three-dimensional structure of the Stat3β homodimer bound to DNA. Nature. 1998;394:145–151. doi: 10.1038/28101. [DOI] [PubMed] [Google Scholar]
- 30.Waksman G, Schoelson SE, Pant N, Cowburn D, Kuriyan J. Binding of a high affinity phosphotyrosyl peptide to the Sec SH2 domain: crystal structures of the complexed and peptide free forms. Cell. 1993;72:779–790. doi: 10.1016/0092-8674(93)90405-f. [DOI] [PubMed] [Google Scholar]
- 31.Xu RX, Word JM, Davis DG, Rink MJ, Willard DH, Jr, Gampe RT., Jr. Solution structure of the human pp60c-src SH2 domain complexed with a phosphorylated tyrosine pentapeptide. Biochemistry. 1995;34:2107–2121. doi: 10.1021/bi00007a003. (1995) [DOI] [PubMed] [Google Scholar]
- 32.Plummer MS, Holland DR, Shahripour A, Lunney EA, Fergus JH, Marks JS, McConnell P, Mueller WT, Sawyer TK. Design, synthesis, and cocrystal structure of a nonpeptide Src SH2 domain ligand. J Med Chem. 1997;40:3719–3725. doi: 10.1021/jm970402q. [DOI] [PubMed] [Google Scholar]
- 33.Bohacek RS, Dalgarno DC, Hatada M, Jacobsen VA, Lynch BA, Macek KJ, Merry T, Metcalf CA, III, Narula SS, Sawyer TK, Shakespeare WC, Violette SM, Weigele M. X-ray Structure of Citrate Bound to Src SH2 Leads to a High-Affinity, Bone-Targeted Src SH2 Inhibitor. J. Med. Chem. 2001;44:660–663. doi: 10.1021/jm0002681. [DOI] [PubMed] [Google Scholar]
- 34.Rahuel J, Gay B, Erdmann D, Strauss A, Garcia-Echeverria C, Furet P, Caravatti G, Fretz H, Schoepfer J, Gruetter MG. Structural basis for specificty of Grb2-SH2 revealed by a novel ligand binding mode. Nature (Struct. Biol.) 1996;3:586–589. doi: 10.1038/nsb0796-586. [DOI] [PubMed] [Google Scholar]
- 35.Ettmayer P, France D, Gounarides J, Jarosinski M, Martin MS, Rondeau JM, Sabio M, Topiol S, Weidmann B, Zurini M, Bair KW. Structural and conformational requirements for high-affinity binding to the SH2 domain of Grb2. J. Med. Chem. 1999;42:971–980. doi: 10.1021/jm9811007. [DOI] [PubMed] [Google Scholar]
- 36.Tong L, Warren TC, King J, Betageri R, Rose J, Jakes S. Crystal structures of the human p56lck SH2 domain in complex with two short phosphotyrosyl peptides at 1.0 A and 1.8 A resolution. J. Mol. Biol. 1996;256:601–610. doi: 10.1006/jmbi.1996.0112. [DOI] [PubMed] [Google Scholar]
- 37.Breeze AL, Kara BV, Barratt DG, Anderson M, Smith JC, Luke RW, Best JR, Cartlidge SA. Structure of a specific peptide complex of the carboxy-terminal SH2 domain from the p85 alpha subunit of phosphatidylinositol 3-kinase. EMBO J. 1996;15:3579–89. [PMC free article] [PubMed] [Google Scholar]
- 38.Pauptit RA, Dennis CA, Derbyshire DJ, Breeze AL, Weston SA, Rowsell S, Murshudov GN. NMR trial models: experiences with the colicin immunity protein Im7 and the p85alpha C-terminal SH2-peptide complex. Acta Crystallogr D. 2001;57(Pt10):1397–1404. doi: 10.1107/s0907444901012434. [DOI] [PubMed] [Google Scholar]
- 39.Autodock, Autogrid, Autotors. Molecular Graphics Laboratory; Scripps Research Institute; La Jolla, CA: 2007. [Google Scholar]
- 40.Pearson DA, Blanchette M, Baker ML, Guindon CA. Trialkylsilanes as scavengers for the trifluoroacetic acid deblocking of protecting groups in peptide synthesis. Tetrahedron Lett. 1989;30:2739–2742. [Google Scholar]
- 41.Perich JW, Reynolds EC. The facile one-pot synthesis of Nα -(9-fluorenylmethoxycarbonyl)-O-(O’,O”-dialkylphosphoro)-L-tyrosines using dialkyl N,N-diethylphosphoramidites. Synlett. 1991;8:577–578. [Google Scholar]
- 42.Perich JW, Johns RB. An improved one-pot synthesis of Nα-(tert-butoxycarbonyl)-O-(O’,O”-dialkylphosphoro)-L-tyrosines using dialkyl N,N-diethylphosphoramidites. Synthesis. 1989;9:701–703. [Google Scholar]
- 43.McMurray JS, Coleman DR, Wang W, Campbell ML. The Synthesis of Phosphopeptides. Biopolymers (Peptide Science) 2001;60:3–31.28. doi: 10.1002/1097-0282(2001)60:1<3::AID-BIP1001>3.0.CO;2-L. Reviewed in. [DOI] [PubMed] [Google Scholar]
- 44.De Lombaert S, Blanchard L, Stamford LB, Sperbeck DM, Grim MD, Jensen TM, Rodriguez HR. Practical Syntheses of a Novel Tricyclic Dipeptide Mimetic Based on a [6H]-Azepino Indoline Nucleus: Application to Angiotensin-Converting Enzyme Inhibition. Tetrahedron Lett. 1994;35:7513–7516. [Google Scholar]
- 45.De Lombaert S. Preparation of antihypertensive tricyclic azepine derivatives useful as inhibitors of enkephalinase and angiotensin converting enzyme (ACE). U.S. 1997. (Cont.-in-part of U.S. Ser. No. 85,223, abandoned).
- 46.Mandal PK, Kaluarachchi KK, Ogrin D, Bott SG, McMurray JS. An efficient synthesis of the constrained peptidomimetic 2-oxo-3-(N-9-fluorenyloxycarbonylamino)-1-azabicyclo[4.3.0]nonane-9-carbox ylic acid from pyroglutamic acid. J. Org. Chem. 2005;70:10128–10131. doi: 10.1021/jo0515935. [DOI] [PubMed] [Google Scholar]
- 47.Shahripour A, Plummer MS, Lunney E, Para KS, Stankovic CJ, Rubin JR, Humblet C, Fergus JH, Marks JS, Herrera R, Hubbell SE, Saltiel AR, Sawyer TK. Novel phosphotyrosine mimetics in the design of peptide ligands for pp60src SH2 domain. Bioorg. Med. Chem. Lett. 1996;6:1209–1214. [Google Scholar]
- 48.Vu CB, Corpuz EG, Pradeepan SG, Violette S, Bartlett C, Sawyer TK. Nonpeptidic SH2 inhibitors of the tyrosine kinase ZAP-70. Bioorg. Med. Chem. Lett. 1999;9:3009–3014. doi: 10.1016/s0960-894x(99)00524-7. [DOI] [PubMed] [Google Scholar]
- 49.McKinney J, Raimundo BC, Cushing TD, Yoshimura H, Ohuchi Y, Hiratate A, Fukushima H. Preparation of peptides as inhibitors of STAT function. U.S. 2002. p. 31. CODEN: USXXAM US 6426331 B1 20020730.
- 50.Mckinney J, Raimundo BC, Cushing TD, Yoshimura H, Ohuchi Y, Hiratate A, Fukushima H, Xu F, Peto C. STAT4 and STAT6 binding dipeptide derivatives. PCT Int. Appl. 2001. p. 85. CODEN: PIXXD2 WO 2001083517 A1 20011108.
- 51.Coleman DR, IV, Kaluarachchi K, Ren Z, Chen X, McMurray JS. Solid phase synthesis of phosphopeptides incorporating 2,2-dimethyloxazolidine pseudoproline analogues: evidence for trans Leu-Pro peptide bonds in Stat3 inhibitors. Int. J. Pept. Res. Ther. 2008;14:1–9. [Google Scholar]
- 52.Hanessian S, McNaughton-Smith G. A versatile synthesis of a β -turn peptidomimetic scaffold: an approach towards a designed model antagonist of the tachykinin NK-2 receptor. Bioorg. Med. Chem. Lett. 1996;6:1567–1572. [Google Scholar]
- 53.Li W, Moeller KD. Conformationally Restricted TRH Analogs: The Compatibility of a 6,5-Bicyclic Lactam-Based Mimetic with Binding to TRH-R. J. Am. Chem. Soc. 1996;118:10106–10112. [Google Scholar]
- 54.Salimbeni A, Paleari F, Canevotti R, Crisculoi M, Lippi A, Angiolini M, Belvisi L, Scolastico C, Colombo L. Design and synthesis of conformationally constrained arginal thrombin inhibitors. Bioorg. Med. Chem. Lett. 1997;7:2205–2210. [Google Scholar]
- 55.Belvisi L, Bernardi A, Checchia A, Manzoni L, Potenza D, Scolastico C, Castorina M, Cupelli A, Giannini G, Carminati P, Pisano C. Potent Integrin Antagonists from a Small Library of RGD-Including Cyclic Pseudopeptides. Org. Lett. 2001;3:1001–1004. [PubMed] [Google Scholar]
- 56.Roy S, Lombart H-G, Lubell WD, Hancock REW, Farmer SW. Exploring relationships between mimic configuration, peptide conformation and biological activity in indolizidin-2-one amino acid analogs of gramicidin S. J. Pept. Res. 2002;60:198–214. doi: 10.1034/j.1399-3011.2002.21026.x. [DOI] [PubMed] [Google Scholar]
- 57.Halab L, Becker JAJ, Darula Z, Tourwe D, Kieffer BL, Simonin F, Lubell WD. Probing Opioid Receptor Interactions with Azacycloalkane Amino Acids. Synthesis of a Potent and Selective ORL1 Antagonist. J. Med. Chem. 2002;45:5353–5357. doi: 10.1021/jm020078l. [DOI] [PubMed] [Google Scholar]
- 58.Belvisi L, Caporale A, Colombo M, Manzoni L, Potenza D, Scolastico C, Castorina M, Cati M, Giannini G, Pisano C. Cyclic RGD peptides containing azabicycloalkane reverse-turn mimics. Helv. Chim. Acta. 2002;85:4353–4368. [Google Scholar]
- 59.Boeglin D, Hamdan FF, Melendez RE, Cluzeau J, Laperriere A, Heroux M, Bouvier M, Lubell WD. Calcitonin gene-related peptide analogues with aza and indolizidinone amino acid residues reveal conformational requirements for antagonist activity at the human calcitonin gene-related peptide 1 receptor. J. Med. Chem. 2007;50:1401–1408. doi: 10.1021/jm061343w. [DOI] [PubMed] [Google Scholar]
- 60.Ho JZ, Gibson TS, Semple JE. Novel, Potent Non-Covalent Thrombin Inhibitors Incorporating P3-Lactam Scaffolds. Bioorg. Med. Chem. Lett. 2002;12:743–748. doi: 10.1016/s0960-894x(02)00010-0. [DOI] [PubMed] [Google Scholar]
- 61.Bohacek R, De Lombaert S, McMartin C, Priestle J, Gruetter M. Three-Dimensional Models of ACE and NEP Inhibitors and Their Use in the Design of Potent Dual ACE/NEP Inhibitors. J. Am. Chem. Soc. 1996;118:8231–8249. [Google Scholar]
- 62.Amblard M.l, Daffix I, Berge G, Calmes M, Dodey P, Pruneau D, Paquet J-L, Luccarini J-M, Belichard P, Martinez J. Synthesis and Characterization of Bradykinin B2 Receptor Agonists Containing Constrained Dipeptide Mimics. J. Med. Chem. 1999;42:4193–4201. doi: 10.1021/jm9901531. [DOI] [PubMed] [Google Scholar]
- 63.Bolin DR, Swain AL, Sarabu R, Berthel SJ, Gillespie P, Huby NJS, Makofske R, Orzechowski L, Perrotta A, Toth K, Cooper JP, Jiang N, Falcioni F, Campbell R, Cox D, Gaizband D, Belunis CJ, Vidovic D, Ito K, Crowther R, Kammlott U, Zhang X, Palermo R, Weber D, Guenot J, Nagy Z, Olson GL. Peptide and peptide mimetic inhibitors of antigen presentation by HLA-DR Class II MHC molecules. Design, structure-activity relationships, and x-ray crystal structures. J. Med. Chem. 2000;43:2135–2148. doi: 10.1021/jm000034h. [DOI] [PubMed] [Google Scholar]
- 64.Becker JAJ, Wallace A, Garzon A, Ingallinella P, Bianchi E, Cortese R, Simonin F, Kieffer BL, Pessi A. Ligands for □-opioid and ORL1 receptors identified from a conformationally constrained peptide combinatorial library. J. Biol. Chem. 1999;274:27513–27522. doi: 10.1074/jbc.274.39.27513. [DOI] [PubMed] [Google Scholar]
- 65.McMurray JS. Structural basis for the binding of high affinity phosphopeptides to Stat3. Biopolymers. 2008;90:69–79. doi: 10.1002/bip.20901. [DOI] [PubMed] [Google Scholar]
- 66.Shao H, Xu X, Mastrangelo MA, Jing N, Cook RG, Legge GB, Tweardy DJ. Structural requirements for signal transducer and activator of transcription 3 binding to phosphotyrosine ligands containing the YXXQ motif. J. Biol. Chem. 2004;279:18967–18973. doi: 10.1074/jbc.M314037200. [DOI] [PubMed] [Google Scholar]
- 67.Chen X, Vinkemeier U, Zhao Y, Jeruzalmi D, Darnell JE, Kuriyan J. Crystal structure of a tyrosine phosphorylated STAT-1 dimer bound to DNA. Cell. 1998;93:827–839. doi: 10.1016/s0092-8674(00)81443-9. [DOI] [PubMed] [Google Scholar]
- 68.Mao X, Ren Z, Parker GN, Sondermann H, Pastorello MA, Wang W, McMurray JS, Demeler B, Darnell JE, Jr., Chen X. Structural bases of unphosporylated Stat1 association and receptor binding. Mol. Cell. 2005;17:761–771. doi: 10.1016/j.molcel.2005.02.021. [DOI] [PubMed] [Google Scholar]
- 69.Mandal PK, Heard PA, Ren Z, Chen X, McMurray JS. Solid phase synthesis of Stat3 inhibitors incorporating O-carbamoylserine and O-carbamoylthreonine as glutamine mimics. Bioorg. Med. Chem. Lett. 2007;17:654–656. doi: 10.1016/j.bmcl.2006.10.099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.DeLano WL. The PyMOL Molecular Graphics System. DeLano Scientific LLC; San Carlos, CA, USA: http://www.pymol.org. [Google Scholar]
- 71.Turkson J, Ryan D, Kim JS, Zhang Y, Chen Z, Haura E, Laudano A, Sebti S, Hamilton AD, Jove R. Phosphotyrosyl peptides block Stat3-mediated DNA-binding activity, gene regulation and cell transformation. J. Biol. Chem. 2001;276:45443–45455. doi: 10.1074/jbc.M107527200. [DOI] [PubMed] [Google Scholar]
- 72.Turkson J, Kim JS, Zhang S, Yuan J, Huang M, Glenn M, Haura E, Sebti S, Hamilton AD, Jove R. Novel peptidomimetic inhibitors of signal transducer and activator of transcription 3 dimerization and biological activity. Mol. Cancer Ther. 2004;3:261–269. [PubMed] [Google Scholar]
- 73.Gunning PT, Katt WP, Glenn M, Siddiquee K, Kim JS, Jove R, Sebti SM, Turkson J, Hamilton AD. Isoform selective inhibition of STAT1 or STAT3 homo-dimerization via peptidomimetic probes: structural recognition of STAT SH2 domains. Bioorg Med Chem Lett. 2007;17:1875–1878. doi: 10.1016/j.bmcl.2007.01.077. [DOI] [PubMed] [Google Scholar]
- 74.Siddiquee KA, Gunning PT, Glenn M, Katt WP, Zhang S, Schroeck C, Sebti SM, Jove R, Hamilton AD, Turkson J. An oxazole-based small-molecule Stat3 inhibitor modulates Stat3 stability and processing and induces antitumor cell effects. ACS Chem Biol. 2007;2:787–798. doi: 10.1021/cb7001973. [DOI] [PubMed] [Google Scholar]
- 75.Shao H, Cheng HY, Cook RG, Tweardy DJ. Identification and Characterization of Signal Transducer and Activator of Transcription 3 Recruitment Sites within the Epidermal Growth Factor Receptor. Cancer Res. 2003;63:3923–3930. [PubMed] [Google Scholar]
- 76.Dourlat J, Valentin B, Liu W-C, Garbay C. New syntheses of tetrazolylmethylphenylalanine and O-malonyltyrosine as pTyr mimetics for the design of STAT3 dimerization inhibitors. Bioorg. Med. Chem. Letters. 2007;17:3943–3946. doi: 10.1016/j.bmcl.2007.04.107. [DOI] [PubMed] [Google Scholar]
- 77 (a).Chen J, Nikolovska-Coleska Z, Yang C-Y, Gomez C, Gao W, Krajewski K, Jiang S, Roller P, Wang S. Design and synthesis of a new, conformationally constrained, macrocyclic small-molecule inhibitor of STAT3 via ‘ click chemistry’. Bioorg. Med. Chem. Letters. 2007;17:3939–3942. doi: 10.1016/j.bmcl.2007.04.096. [DOI] [PubMed] [Google Scholar]; (b) Bernstein FC, Koetzle TF, Williams GJ, Meyer EF, Jr., Brice MD, Rodgers JR, Kennard O, Shimanouchi T, Tasumi M. The Protein Data Bank: a computer-based archival file for macromolecular structures. J. Mol. Biol. 1977;112:535–542. doi: 10.1016/s0022-2836(77)80200-3. [DOI] [PubMed] [Google Scholar]
- 78.InsightII. Accelrys Inc.; San Diego, CA: 1998. [Google Scholar]
- 79.Bolin DR, Swain AL, Sarabu R, Berthel SJ, Gillespie P, Huby NJ, Makofske R, Orzechowski L, Perrotta A, Toth K, Cooper JP, Jiang N, Falcioni F, Campbell R, Cox D, Gaizband D, Belunis CJ, Vidovic D, Ito K, Crowther R, Kammlott U, Zhang X, Palermo R, Weber D, Guenot J, Nagy Z, Olson GL. Peptide and peptide mimetic inhibitors of antigen presenting by HLA-DR class II MHC molecules. Design, structure-activity relationships, and X-ray crystal structures. J. Med. Chem. 2000;43:2135–2148. doi: 10.1021/jm000034h. [DOI] [PubMed] [Google Scholar]
- 80.Case DA, Darden TA, Cheatham TE, III, Simmerling CL, Wang J, Duke RE, Luo R, Merz KM, Wang B, Pearlman DA, Crowley M, Brozell S, Tsui V, Golhke H, Mongan J, Hornak V, Cui G, Beroza P, Schafmeister C, Caldwell JW, Ross WS, Kollman PA. Amber 8. edition 8.0 University of California; San Francisco, CA: 2004. [Google Scholar]
- 81.Jakalian A, Jack DB, Bayly CT. Fast, efficient generation of high quality atomic charges. AM1-BCC model: II Parameterization and validation. J. Comput. Chem. 2002;23:1623–1641. doi: 10.1002/jcc.10128. [DOI] [PubMed] [Google Scholar]
- 82.Wang J, Wolf RM, Caldwell JW, Kollman PA, Case DA. Development and testing of a general amber force field. J. Comput. Chem. 2004;25:1157–1174. doi: 10.1002/jcc.20035. [DOI] [PubMed] [Google Scholar]
- 83.Wang J, Cieplak P, Kollman PA. How well does a restrained electrostatic potential (RESP) perform in calculating conformational energies of organic and biological molecules? J. Comp. Chem. 2000;21:1049–1074. [Google Scholar]
- 84.Dejoux A, Cieplak P, Hannick N, Moyna G, Dupradeau F-Y. AmberFFC, a flexible program to convert AMBER and GLYCAM force fields for use with commercial molecular modeling packages. J. Mol. Model. 2001;7:422–432. [Google Scholar]
- 85.Huey R, Morris GM, Olson AJ, Goodsell DS. A semiempirical energy force field with charge-based desolvation. J. Comput. Chem. 2007;28:1145–1152. doi: 10.1002/jcc.20634. [DOI] [PubMed] [Google Scholar]
- 86.Morris GM, Goodsell DS, Halliday RS, Huey R, Hart WE, Belew RK, Olson AJ. Automated docking using a Lamarckian genetic algorithm and an empirical binding free energy function. J. Comput. Chem. 1998;19:1639–1662. [Google Scholar]
- 87.Jorgensen WL, Chandrasekar J, Madura JD. Comparison of simple potentialfunctions for simulating liquid water. J. Chem. Phys. 1983;79:926–935. [Google Scholar]
- 88.Berendsen HJC, Postma JPM, van Gunsteren WF, DiNola A, Haak JR. Molecular dynamics with coupling to an external bath. J. Chem. Phys. 1984;81:3684–3690. [Google Scholar]
- 89.Ryckaert JP, Ciccitti G, Berendsen HJC. Numerical integration of the cartesian equations of motion of a system with constrains: molecular dynamics of n-alkanes. J. Comp. Phys. 1977;23:327–341. [Google Scholar]
- 90.Essmann U, Perera L, Berkowitz ML, Darden T, Lee H, Pedersen LG. A smooth particle mesh Ewald method. J. Chem. Phys. 1995;103:8577–8593. [Google Scholar]
- 91.Phillips JC, Braun R, Wang W, Gumbart J, Tajkhorshid E, Villa E, Chipot C, Skeel RD, Kale L, Klaus S. Scalable molecular dynamics with NAMD. J. Comput. Chem. 2005;26:1781–1802. doi: 10.1002/jcc.20289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.(a) van der Spoel D, Lindahl E, Hess B, Groenhof G, Mark AE, Berendsen HJC. GROMACS: Fast, Flexible, and Free. J. Comp. Chem. 2005;26:1701–1718. doi: 10.1002/jcc.20291. [DOI] [PubMed] [Google Scholar]; (b) van der Spoel D, Lindahl E, Hess B, van Buuren AR, Apol E, Meulenhoff PJ, Tieleman DP, Sijbers ALTM, Feenstra KA, van Drunen R, Berendsen HJC. Gromacs User Manuel version 3.3. 2005 wwww.gromacs.org.
- 93.Daura X, Gademann K, Jaun B, Seebach D, van Gunsteren WF, Mark AE. Peptide Folding: When Simulation Meets Experiment. Angew. Chem. Int. Ed. 1999;38:236–240. [Google Scholar]
- 94.Schaefer TS, Sanders LK, Nathans D. Cooperative transcriptional activity of Jun and Stat3beta, a short form of Stat3. Proc. Natl. Acad. Sci. USA. 1995;92:9097–9101. doi: 10.1073/pnas.92.20.9097. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.