

Abstract
Background and Aims
Epigenetics is defined as mechanisms that regulate gene expression without base sequence alteration. One molecular basis is considered to be DNA cytosine methylation, which reversibly modifies DNA or chromatin structures. Although its correlation with epigenetic inheritance over generations has been circumstantially shown, evidence at the gene level has been limited. The present study aims to find genes whose methylation status directly correlates with inheritance of phenotypic changes.
Methods
DNA methylation in vivo was artificially reduced by treating rice (Oryza sativa ssp. japonica) seeds with 5-azadeoxycytidine, and the progeny were cultivated in the field for > 10 years. Genomic regions with changed methylation status were screened by the methylation-sensitive amplified polymorphysm (MSAP) method, and cytosine methylation was directly scanned by the bisulfite mapping method. Pathogen infection with Xanthomonas oryzae pv. oryzae, race PR2 was performed by the scissors-dip method on mature leaf blades.
Key Results
The majority of seedlings were lethal, but some survived to maturity. One line designated as Line-2 showed a clear marker phenotype of dwarfism, which was stably inherited by the progeny over nine generations. MSAP screening identified six fragments, among which two were further characterized by DNA blot hybridization and direct methylation mapping. One clone encoding a retrotransposon gag–pol polyprotein showed a complete erasure of 5-methylcytosines in Line-2, but neither translocation nor expression of this region was detectable. The other clone encoded an Xa21-like protein, Xa21G. In wild-type plants, all cytosines were methylated within the promoter region, whereas in Line-2, corresponding methylation was completely erased throughout generations. Expression of Xa21G was not detectable in wild type but was constitutive in Line-2. When infected with X. oryzae pv. oryzae, against which Xa21 confers resistance in a gene-for-gene manner, the progeny of Line-2 were apparently resistant while the wild type was highly susceptible without Xa21G expression.
Conclusions
These results indicated that demethylation was selective in Line-2, and that promoter demethylation abolished the constitutive silencing of Xa21G due to hypermethylation, resulting in acquisition of disease resistance. Both hypomethylation and resistant trait were stably inherited. This is a clear example of epigenetic inheritance, and supports the idea of Lamarckian inheritance which suggested acquired traits to be heritable.
Key words: Acquired traits, DNA methylation, epigenetic inheritance, 5-methylcytosine, Oryza sativa, Xa21 resistance gene, Xanthomanas oryzae
INTRODUCTION
Epigenetics is defined as a change in gene expression without base sequence alteration (Riggs and Porter, 1996), and is frequently found during somatic cell differentiation in animal cells. This typically occurs in clonal expansion of a single cell, leading to a diversity of cell types (Holliday, 1993). An epigenetically acquired trait within an organism is transmissible from cell to cell, and is commonly observed during ontogeny (Reik et al., 2001). This alteration is usually erased when germ cells are formed, precluding inheritance of epigenetically acquired traits by the next generation.
However, epigenetically acquired traits have been known to be sexually transmitted in plants under certain circumstances (Jablonka and Lamb, 1989). For example, upon treatments with fertilizer, flax plants (Linum usitatissimum) exhibited a variety of phenotypes, which were stably inherited by the progeny (Durrant, 1962; Durrant and Nicholas, 1970; Cullis and Kolodynska, 1975). Heritable differences have also been observed in Nicotiana rustica after a single generation of growth under special nutritional conditions (Hill, 1965). Recent analyses on flax have suggested a correlation between copy numbers of repetitive sequences and induced differences (Cullis, 2005), but underlying molecular mechanisms have so far not been completely understood. One assumption is that an environmental stimulus can induce heritable chromatin modification as an adaptive response (Jablonka and Lamb, 1989).
Recent epigenetic research suggests that DNA methylation is one of the major mechanisms. In higher eukaryotes, DNA methylation usually points to methylation of cytosines, which is catalysed by cytosine methyltransferases yielding 5-methylcytosine (m5C) after DNA replication. Whereas m5C accounts for only approximately 4 % of the total cytosine residues in mammalian DNA, it is generally much more prevalent in plant DNA, comprising up to 30 % of the total cytosines (Finnegan et al., 1998). The location of m5C also differs between mammals and plants, being found only in CpG in the former, but also in CpNpG and CpNpN (N is A, T or C) in the latter. Both CpG and CpNpG sequences are symmetric, with methylation transmitted through meiosis by the action of maintenance methyltransferases specific for each sequence. Methylation of asymmetric CpNpN is usually considered not to be heritable, being re-established in every generation (Jones et al., 2001).
The biological function of DNA methylation has been proposed to be involved in gene silencing, often being associated with hypermethylation of promoter sequences (Paszkowski and Whitham, 2001; Bird, 2002). Studies on genome-wide methylation mapping of Arabidopsis revealed heavy methylation in heterochromatin, repetitive sequences and small RNA-coding regions (Zhang et al., 2006; Zilberman et al., 2007), consistent with the reverse relationship between DNA methylation and gene expression. This study also revealed that over one-third of expressed genes were methylated in coding regions (body-methylated), and that 95 % genes were free of methylation in their promoter regions (Zhang et al., 2006; Zilberman et al., 2007). Since methylation of both DNA and histone tails was shown to be critical for maintenance or formation of heterochromatin (Tariq and Paszkowski, 2004; Peters and Schübeler, 2005; Fuchs et al., 2006), such a biased distribution of m5Cs may result in a serious modification of chromatin architecture and concomitant gene expression (Bird, 2002; Bender, 2004).
Whether or not the change of DNA methylation in somatic cells can be transmitted to germ cells is unclear. However, there is some evidence to support this idea, for example assays with methylation inhibitors, 5-azacytidine (azaC) and 5-azadeoxycytidine (azadC), have shown heritable hypomethylation in rice (Sano et al., 1990; Kumpatla et al,. 1997), flax (Fieldes, 1994), tobacco (Vyscot et al., 1995), Brassica (King, 1995), Melandrium (Janousek et al., 1996) and triticale (Amado et al., 1997). In some cases, heritable phenotypic changes were observed. A temporary suppression of the histone H1 gene in an RNAi transgenic Arabidopsis simultaneously caused altered patterns of DNA methylation and developmental defects, both being heritable by the progeny (Wierzbicki and Jerzmanowski, 2004).
DNA methylation thus appears to fulfil conditions for epigenetic inheritance, being reversibly changeable over generations, inducing no base alteration and strongly affecting gene expression through regulation of chromatin structure. However, direct evidence of its participation in stable inheritance of epigenetically acquired traits at the gene level has so far been limited. In the present study, an attempt was made to prove the cause–effect relationship between DNA methylation and epigenetic inheritance, and it was found that demethylation activated a disease resistance gene, thereby conferring a disease-resistant trait, and that both hypomethylation and acquired phenotype were stably inherited by the progeny.
MATERIALS AND METHODS
Plant materials
Rice (Oryza sativa ssp. japonica, ‘Yamada-nishiki’) was used throughout the experiments. Approximately 1000 seeds were allowed to imbibe water at 37 °C for 16 h, and were treated with 200 mL of 0·5 mm azadC (Sigma) in 20 mm Tris–HCl, pH 7·5 at 20 °C for 2 d. After washing, seeds were planted in a culture box containing sterilized soil, and cultivated at 23 °C under a 16/8 h light/dark photoperiod in a growth cabinet. While the majority of seedlings died, 35 survived, and these were transferred to pots containing soil after 2 weeks and grown further to maturity in 1997. The resulting plants were designated as P (parent) with serial line numbers, Line-1 to Line-35. Their seeds were collected and propagated in a field in subsequent years (1998–2006), and phenotypic traits were assessed by estimating plant height and day of heading (ear emergence) after sowing (defined as the day when 50 % of the population headed) (Sano et al., 1990). The first progeny were referred to as F1, and successive offspring as F2 (1999) to F9 (2006). For detailed analysis, Line-2 was selected, which showed a visible phenotypic change, dwarfism, in both the parent and the progeny. For molecular biological analyses, seeds of appropriate generations were hydroponically cultured in a growth chamber, and 2- to 3-week-old seedlings were harvested and subjected to further experiments.
Statistical analysis
The distribution frequency was analysed with SAS software (Elliot, 2000), and 95 % confidence intervals were calculated as described (Snedecor and Cochran, 1980).
DNA blot hybridization and RT–PCR
Genomic DNA was isolated by the cetyltrimethyl ammonium bromide (CTAB) method (Murray and Thompson, 1980) with modifications. A 15 µg aliquot of genomic DNA was digested with HpaII or MspI, fractionated on a 1·0 % agarose gel and blotted onto a nylon membrane according to the manufacturer's protocol. The blot was then hybridized with 32P-labelled probes in 0·5 m Church phosphate buffer, 1 mm EDTA and 7 % SDS at 65 °C for 16 h, and successively washed with 2 × SSC and 0·1 % SDS at 65 °C, for exposure to X-ray film or to a bio-imaging analyser system (BAS-2500, Fuji Film). The probes were OsMADS1 (Joen et al., 2000), 25S-17S rDNA (Ellis et al., 1990), Tos10 (Hirochika et al., 1996) and Karma (Komatsu et al., 2003), prepared by PCR with gene-specific primers and labelled with 32P using a BcaBEST™ labelling kit (Takara, Kyoto, Japan). The specific probe for the HMF2 region was prepared by PCR at positions 75 340–76 300 of the bacterial artificial chromosome (BAC) clone OJ1499_A07. RT–PCR was performed as described previously (Steward et al., 2002) using primers for Xa21G forward, 5′-ATGGGCCTCACGTGTGATAC-3′ and reverse, 5′-CAGCCTTGGCCATCAGAAGC-3′; and PR1b forward, 5′-ATGGAGGTATCCAAGCTGGC-3′ and reverse, 5′-TTAGTAAGGCCTCTGTCCGA-3′.
Methylation-sensitive amplified polymorphism analysis
Methylation-sensitive amplified polymorphisms (MSAPs) were determined according to an established method (Reyna-Lopez et al., 1997) with modifications. A 5-μg aliquot of DNA from wild-type or dwarf seedlings was successively digested with EcoRI and HpaII or MspI, ligated to adaptors containing one of the above restriction sites, and pre-amplified with the corresponding primers (Reyna-Lopez et al., 1997) (the EcoRI adaptor was 3′-CATCTGACGCATGGTTAA-5′; the EcoRI primer with an additional A at the 3′ end was 5′-GACTGCGTACCAATTCA-3′; the HpaII/MspI adapter was 3′-AGTACTCAGGACGAGC-5′; and the HpaII/MspI primer was 5′-ATCATGAGTCCTGCTCGG-3′). The resulting PCR products were diluted 4-fold with water and used for the second selective amplification with the same primers but containing three or four additional selection nucleotides at the 3′ end (Reyna-Lopez et al., 1997) [the EcoRI primers with three additional nucleotides (underlined) were 5-GACTGCGTACCAATTCAGA-3′ and 5′-GACTGCGTACCAATTCAAG-3; the HpaII/MspI primer with four additional nucleotides (underlined) was 5′-ATCATGAGTCCTGCTCGGTCAA-3′]. Products were electrophoretically fractionated on 3 % agarose S (Nippon Gene, Toyama, Japan) or 3 % NuSieve 3 : 1 agarose (Camrex Bio Science, Rockland, ME, USA), and fragments that were differentially amplified between wild-type and dwarf lines were isolated and re-amplified with the same set of primers. The resulting fragments were used to probe differential methylation at genomic loci, and then cloned into the pGEM-T Easy vector™ (Promega, Madison, WI, USA) to determine the nucleotide sequence.
Bisulfite mapping
Total DNA was extracted from seedling leaves by the CTAB method (Murray and Thompson, 1980) and subjected to direct methylation mapping. The DNA was digested with restriction enzymes (EcoRI and KpnI) and recovered by ethanol precipitation after phenol–chloroform extraction, and the bisulfite modification reaction was performed as described (Steward and Sano, 2004) with slight modifications. The primers for PCR were designed for predicted sequences after modification, in which C and m5C were assumed to be converted into Y (C or T) in the forward and into R (A or G) in the reverse direction. For HMF2 (gag–pol protein), the modified DNA was amplified at regions I and II with sequence-specific forward primers (for region I, 5′- GAGYGAGATGATGGGTAYTGAAG-3′; for region II, 5′-GAYGGGTAGYTGAYGAGGAAGAAGGT-3′) and the same reverse primer for both regions (5′-AATACAAAAAACAAAAACAAAATAACTTAA-3′). The pairs of primers specific for regions I and II amplified a 347 and a 612 bp fragment, respectively. Eight and 12 clones for the wild type, and ten clones for each generation of F3–F7 were subjected to sequence determination for regions I and II, respectively. For HMF5 (Xa21G), the modified DNA was amplified at regions I, II and III (Fig. 2A) by PCR with sequence-specific primers (region I forward, 5′-GTGGAAYGAGAATYTTGAAAGG-3′; region I reverse, 5′-CATAARRAATTATCCCTRTC-3′; region II forward, 5′-AAAAYATTAGTGATATGGTA-3′; region II reverse, 5′-CTTRTCTRRAATRCCCCCTTC-3′; region III forward, 5′-GGAYGTTTGGGTTTAAGA-3′; region III reverse, 5′-CTCCARRATCACTATCCC-3′). More than 20 clones for the wild type or for Line-2 (F9) were subjected to sequence determination. Amplified PCR products were ligated to the pGEM-T Easy vector™ (Promega) and cloned into Escherichia coli DH5α (Stratagene, La Jolla, CA, USA). Sequences were determined with an ABI PRISM BigDye™ Terminator DNA sequencing kit and a 3100 Genetic Analyzer automated sequencer (Applied Biosystems). The conversion efficiency of cytosine into uracil was complete, as all the cytosines in Line-2 were converted into thymine after PCR (see Fig. 3A for HMF2, and Fig. 5A for HMF5, in which two regions were amplified from the same DNA sample treated with bisulfite).
Fig. 2.
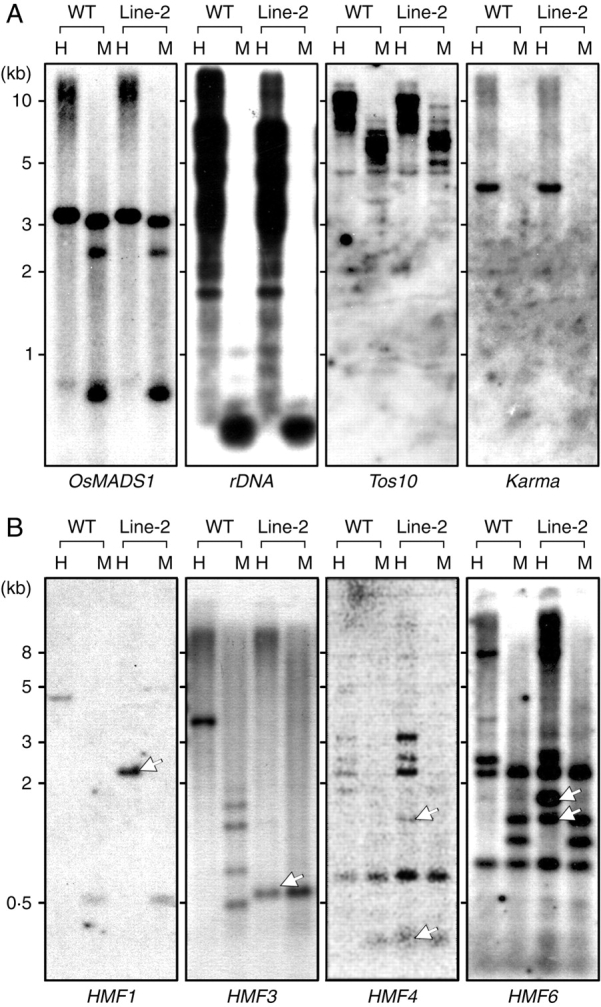
Methylation analysis by DNA blot hybridization. (A) Methylation status in known genes. Genomic DNA was isolated from wild type (WT) and the F3 progeny of Line-2 plants, and a 15 µg aliquot was digested with either HpaII (H) or MspI (M). After electrophoretic fractionation on a 0·8 % agarose gel, DNA was blotted onto a nylon membrane, and subjected to hybridization with probes of known genes as indicated; OsMADS1, 25S-17S rDNA, Tos10 or Karma. (B) Methylation status in MSAP fragments. Genomic DNA from the wild type (WT) and the F7 progeny of Line-2 plants was subjected to hybridization with DNA probes as identified; HMF1, HMF3, HMF4 or HMF6. Fragments with a changed pattern are indicated by arrows (lower panels).
Fig. 3.
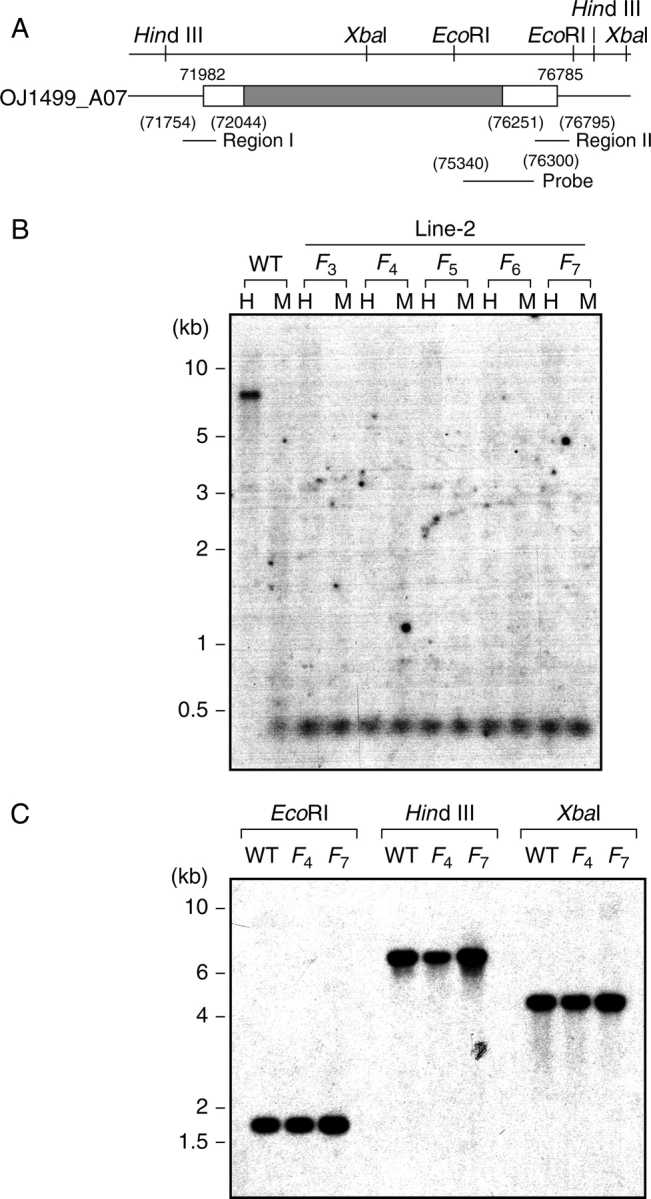
Methylation analysis of the HMF2 locus. (A) Genomic organization of the retrotransposon region containing the HMF2 sequence. The number indicates the position on the BAC clone OJ1499_A07, and the restriction site position for the indicated enzymes is given on the upper panel. The positions of regions I and II, and of the hybridization probe are indicated by horizontal bars. The translated and untranslated regions are shown by filled and open blocks, respectively. (B) Inheritance of demethylation. Aliquots of 15 µg of genomic DNA isolated from wild-type (WT) plants and the indicated progeny of Line-2 (F3–F7) were digested with either HpaII (H) or MspI (M), and analysed by DNA blot hybridization using a unique 960 bp fragment (positions 75 340–763 00) as the probe, as shown in (A). Note that the identified fragment of 0·3 kb was the largest among CCGG-cleaved fragments which hybridized to the probe. (C) Copy number estimation and transposition. Aliquots of 15 µg of genomic DNA isolated from wild-type (WT) plants and progeny (F4 and F7) were digested with EcoRI, HindIII or XbaI, and analysed by DNA blot hybridization using a unique 960 bp fragment (positions 75 340–763 00) as the probe as described above. A slight difference in intensity among HindIII-cut fragments could be due to a slight difference in amounts of total DNA loaded.
Fig. 5.

Methylation analysis of HMF5 (Xa21G). (A) Genomic organization of the Xa21 locus. A 4 kb genomic locus is illustrated with the exons (box) and introns (line) indicated. Exons (E) are numbered in Arabic letters starting from the transcription initiation site (+1 which corresponds to nucleotide position 25 208 591 of chromosome 2). CCGG sites are indicated by vertical bars over the sequence, and mapping and probed regions are indicated by horizontal bars. The regions for direct methylation mapping are located at nucleotide positions –369 to + 144 (region I), + 484 to + 808 (region II) and + 2043 to + 2413 (region III). (B) Phylogenetic relationship between Xa21G and other Xa21-related proteins. The proteins compared are D (U72726), B (Xa21) (U37133·1), C (U72723), A1 (U72725), E (U72724), A2 (U72727), F (U72728) and G (NM-001053967). (C) Inheritance of demethylation. Aliquots of 15 µg of genomic DNA isolated from the wild type (WT) and the indicated progeny (F3–F9) of Line-2 plants were digested with either HpaII (H) or MspI (M), and analysed by DNA blot hybridization using a unique 1 kb fragment (positions 329–1341) as the probe.
Pathogen infection
Healthy leaves from 3-month-old plants were inoculated with Xanthomonas oryzae pv. oryzae, race PR2 by the scissors-dip method (Kauffman et al., 1973). The leaf blade was cut with a pair of scissors, which had previously been dipped into a bacterial solution, at a position approximately two-thirds of the way from the tip, and incubated at 23 °C for up to 12 d. After infection, a 20 cm piece from the cut surface was harvested, homogenized and suspended in 10 mL of water, aliquots of which were used for estimation of bacterial numbers after appropriate dilution.
RESULTS
Heritable phenotypic changes
Among approx. 1000 germinated seeds initially treated with azadC, the majority died at the three-leaf stage (about 3 weeks after treatment). The surviving seedlings numbered only 35, which were then cultured to maturity. The stature of 33 of them was apparently normal, being indistinguishable from that of the wild-type plants, but two showed apparent dwarfism, which was thought to be convenient to use as a marker phenotype for further analysis. One line (Line-2) was subsequently selected and successively propagated every year, and its traits were recorded (Fig. 1A). Although the extent varied with the year due to culture conditions, the Line-2 sample and its progeny consistently showed at least a 28 % reduction in stem length (Fig. 1B). The heading time was also shortened, being 10–14 d earlier in Line-2 than in controls (Fig. 1B). Since plant growth ceases after ear emergence (heading) in rice, the correlation between the two was examined by plotting the stem length against the day of heading. Line-2 clearly demonstrated independence from the wild-type group (Fig. 1C), suggesting the observed alteration to be fixed. The results indicated that a pulse treatment of germinated seeds with azadC had induced phenotypic changes evident at maturity, and that such changes were stably inherited by the progeny. As this phenotype did not segregate even in the F1 generation, and as seeds from all progeny germinated equally and grew to maturity, it was conceivable that the change was homozygous without detrimental effects on growth.
Fig. 1.

Phenotypic properties. (A) Features of mature rice plants (Oryza sativa ssp. japonica, ‘Yamadanishiki’). The wild-type (WT, right) and F7 progeny of Line-2, (Line-2, left) were cultivated under standard field conditions, and photographed at maturity. (B) Distribution of the stem length and heading days. Fifty to 130 plants each of the wild type and Line-2 were measured every year. As the representative, values for 54 F8 Line-2 plants (Line-2) and 36 wild-type plants (WT), both harvested in 2005, are depicted for heading days (ear emergence, upper panel) and stem length (lower panel) based on the stem–leaf plot obtained from the tests for normality with 95 % confidence. The mean for the ear emergence was 101·8 ± 0·70 d for the wild type and 93·1 ± 0·98 d for Line-2. The mean for the stem length of the wild type and the Line-2 plants was 95·2 ± 4·14 and 71·4 ± 3·6 cm, respectively. (C) Relationship between the stem length and the ear emergence (heading days). The mean value for each item (ear emergence and stem length) in each year (1998–2005) was statistically calculated as described above and plotted. The Line-2 (red) samples were F2 (1998) to F8 (2005) (the total number was 429), and wild-type plants (green) from the corresponding years (the total number was 1017) were also measured. The horizontal and vertical axes indicate stem length and ear emergence, respectively. Note that each spot does not necessarily represent an individual plant due to many overlaps.
Methylation of known gene loci
Since azadC is a powerful inhibitor of DNA methylation in vivo, it is conceivable that the m5C content in Line-2 was reduced in comparison with that in the wild-type counterpart. In order to examine this possibility, the DNA methylation status of several gene loci was analysed by DNA blot hybridization. DNA samples from each generation were differentially digested with HpaII and MspI, targeting C*CGG and *CCGG, respectively (the asterisk indicates m5C), and probed with appropriate DNA fragments (Fig. 2A). The examined regions were OsMADS1 as the coding sequence, which was reported to be closely related to rice dwarfism (Joen et al., 2000), rDNA (Ellis et al., 1990) as the repeat sequence, and Tos10 (Hirochika et al., 1996) and karma (Komatsu et al., 2003) as transposable elements. Resulting restriction patterns in these loci showed no distinct changes between wild type and all generations of Line-2, although the MspI pattern in the Tos10 locus differed slightly (F3 is shown as the representative in Fig. 2A). However, these analyses principally indicated that the examined regions were not significantly demethylated. Transcripts of OsMADS1 and karma were not detectable in seedlings of either the wild type or Line-2 as checked by RT–PCR (data not shown).
MSAP screening
In order to determine further whether or not demethylation actually occurred in Line-2, MSAP screening was carried out; using this method, loci with altered methylation levels can be clearly identified. To this end, the method was modified using specific primers containing an additional four bases at the 3′ end, resulting in a large reduction in numbers of amplified fragments, which could easily be fractionated on ordinary agarose gels (data not shown). DNA samples from Line-2 (F7) and wild-type plants were amplified by this method after cleavage with MspI or HpaII, and compared for their migration patterns. The results initially identified > 20 fragments which were present in the controls but absent in Line-2. After confirming differential methylation by DNA blot hybridization and determining the nucleotide sequences, six clones were finally selected and designated as HMF (hypomethylated fragment) with serial numbers (Table 1). Although the isolated fragments were short, database searching allowed identification of their locations in the rice genome. The methylation status of genomic loci of the identified regions was examined at CCGG sites by DNA blot hybridization using DNA samples from Line-2 (F7) and wild-type plants. The results showed that all regions were demethylated in Line-2, giving shorter DNA fragments than in the control upon digestion with HpaII (HMF1, 3, 4 and 6 in Fig. 2B, HMF2 in Fig. 3A and HMF5 in Fig. 5A).
Table 1.
Clones identified by MSAP screening
| Clone* | Length (bp) | Chromosome | Position | Best match | Accession no. |
|---|---|---|---|---|---|
| HMF1 | 333 | 5 | 28 088 962-28 089 294 | Non-coding region | AP008211 |
| HMF2 | 247 | 8 | 1 472 037–1 472 276 | Putative gag–pol polyprotein | AK071597 |
| 1 467 283–1 467 525 | |||||
| HMF3 | 243 | 7 | 21 479 052 –21 479 154 | Non-coding region | AP008213 |
| 21 478 859 –21 478 908 | |||||
| HMF4 | 246 | 2 | 10 392 556–10 392 774 | Non-coding region | AP008208 |
| HMF5 | 172 | 2 | 2 509 043–25 209 127 | Putative protein kinase Xa21, receptor type precursor | AK065018 |
| HMF6 | 113 | 3 | 18 747 145–18 747 231 | Expressed protein | AK110714 |
*HMF1, located between two coding regions for unknown proteins (AAT77390 and AAT77391); HMF2, two copies on chromosome 8; HMF3, located upstream of a hypothetical protein similar to NBS-LRR protein (XM_478535); HMF4, located between two coding regions for putative Xet3 proteins (P0669G10·1 and P0669G10·3); HMF5, a pseudogene (J1792_D02·2) for a gag–pol polyprotein was found 3·8 kb downstream; HMF6, the putative protein contains a proline-rich domain.
Demethylation at retrotransposon locus
A rice genome search revealed that a region spanning approx. 250 bases containing the HMF2 sequence was duplicated on either side of the coding region of the gag–pol polyprotein (Fig. 3A). Because of this duplication, a unique 960 bp probe was prepared inside the two HMF2 regions to avoid multiple patterns on DNA blot hybridization analysis. Upon digestion with HpaII, the control wild type showed most HpaII fragments to be 7 kb (Fig. 3B). Since > 72 CCGG sites are available in the HMF2 region, the pattern indicates only particular CCGG sites to be susceptible to HpaII, suggesting almost all CCGG sequences to be methylated. In the offspring of Line-2, both HpaII and MspI produced exclusively a 0·3 kb fragment (Fig. 3B), indicating complete demethylation through generations. Since the gag–pol polyprotein constitutes part of a retrotransposon, and since the activation of retrotransposons is frequently related to the methylation status (Bender, 2004), the possibility of transposition of the HMF2 locus was examined using EcoRI, HindIII and XbaI restriction enzymes, each differentially yielding different sized fragments if transposition took place. Hybridization assays resulted in the same restriction patterns with these three enzymes (Fig. 3C), suggesting no transposition in this region. The hybridization intensity was also similar among samples, suggesting no increase in copy numbers. The result is consistent with northern hybridization findings, in which transcripts for the gag–pol polyprotein were not detectable in any samples examined (data not shown).
The methylation status was directly examined by the bisulfite mapping method. Since a 250 bp region within HMF2 was duplicated (Fig. 3A), both were independently subjected to bisulfite analysis. The 5′-proximal region of 250 bp was designated as region I. The 3′-proximal region was 510 bp, in which 250 bp from the 3′ end was the same as region I. This fragment was designated as region II (Fig. 3A). DNA samples from wild type and each generation of F3–F7 were treated with bisulfite, amplified with region-specific primers by PCR, cloned and subjected to sequence determination (Fig. 4A). When a 60 base sequence was randomly selected, and aligned, it was evident that the majority of cytosines remained intact in the wild-type control, indicating them to be originally m5C in both regions I and II (Fig. 4A, upper panel). In contrast, all cytosines were converted into thymine in all generations of Line-2, indicating them to be originally cytosines (Fig. 4A, lower panel). When the distribution and frequency of m5C were estimated from the ratio between cytosines and thymines, m5C was found to be evenly distributed in the wild-type control (Fig. 4B). In contrast, no m5C was found in Line-2 throughout the progeny, indicating complete erasure of m5C and its stable transmission (Fig. 4B). In the wild-type DNA, the frequency of m5C in methylatable CpG was 96·7 and 92·1 % in regions I and II, respectively. The frequency in CpNpG was 48·2 % in region I and 53·2 % in region II. However, methylation of CpNpN was low, at only 6·7 % in region I and 35·5 % in region II. These experiments indicated the methylation status to differ between regions I and II, the latter being more heavily methylated at all methylatable sites, and that, once erased, the demethylated status was stably maintained by the offspring. Moreover, the unmethylated status at CpNpN was found to be stably inherited over generations in Line-2.
Fig. 4.
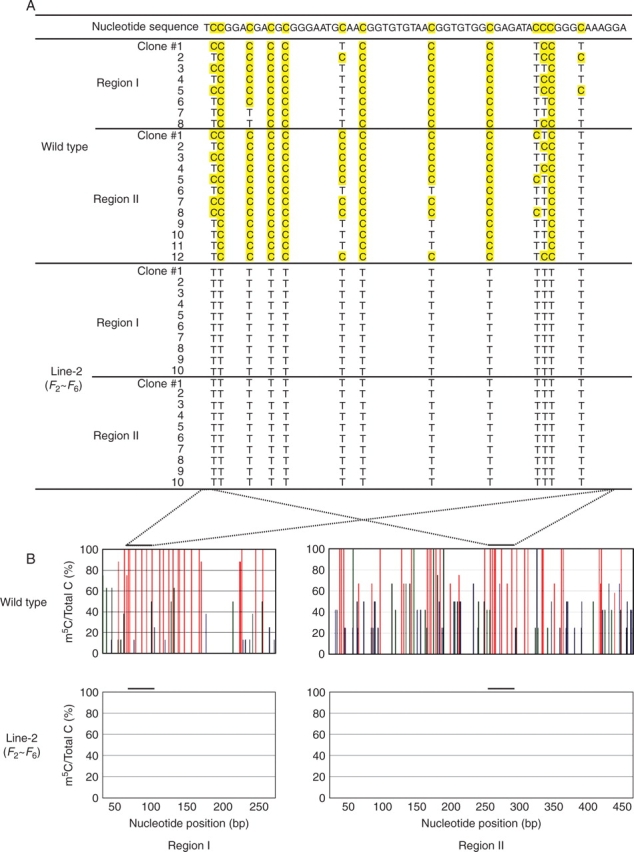
Direct methylation mapping of the HMF2 region. (A) Identification of m5C. After bisulfite treatment, the 290 and 544 bases in regions I and II, respectively, were amplified by PCR, cloned and sequenced. Cytosines (C) and m5C in the original sequence were converted into thymine (T) and C, respectively. The original nucleotide sequence of the wild type without bisulfite treatment is shown on the top line with shading of the methylatable cytosines. The converted bases are indicated by T or C (shaded) in each clone originating from bisulfite-treated wild-type or dwarf progeny. Eight and 12 clones were analysed for regions I and II, respectively, for the wild-type control. For progeny analysis, ten clones for each region were prepared from each generation (F3–F7). The clone number is given on the left. (B) Distribution and frequency of m5Cs. Histograms represent the percentage of m5Cs over the total cytosines (vertical axis) at positions containing CpG (red), CpNpG (green) and CpNpN (blue) sequences (N is A, C or T) in the top strand at the indicated nucleotide positions (horizontal axis) in the wild type (upper panel) and Line-2 progeny (F3–F7) (lower panel). The bar on the top indicates the sequence shown in (A). A negative result for the progeny means no m5C could be assigned to these regions.
Demethylation at disease resistance locus
HMF5 was found to be a part of Os02g0615800, a gene for a putative Xa21-like protein (Fig. 5A). The Xa21 is a rice disease resistance gene encoding a receptor-like kinase, and a member of a multigene family which consists of at least seven members, A1, A2, B (Xa21), C, D, E and F (Song et al., 1997). Phylogentic analysis indicated that the presently identified clone was rather distantly related to those members, and it was designated as Xa21G (Fig. 5B). The methylation status of the 5′ end region of this gene was examined by DNA blot hybridization, using MspI and HpaII. DNA from wild-type plants yielded multiple fragments with diverse size upon digestion by each enzyme, indicating partial methylation at several CCGG sites (Fig. 5C). In contrast, DNA from Line-2 was predominantly cleaved into a single 1·3 kb fragment by both enzymes, clearly indicating demethylation at CCGG sites located at positions 75 and 1243 (Fig. 5A, C). Since this pattern was common in F3–F9, it was evident that the demethylated status was faithfully maintained through the generations.
The methylation frequency of individual cytosine residues in the Xa21G locus was then determined by direct methylation mapping using DNA from wild-type and F9 plants. Three regions were examined: region I comprises > 492 bp including the 5′ end of cDNA and promoter sequence, which contained the CG island; region II comprises 324 bp located in the coding sequence; and region III comprising 370 bp covering an exon and intron (Fig. 5A). After bisulfite treatment, fragments corresponding to each region were amplified from the same DNA sample by PCR, cloned, and the sequence of independent clones was determined. The results revealed particular features. First, in the wild type, region I was heavily methylated, while regions II and III were almost completely free of methylation (Fig. 6A, B) (Table 2). Secondly, in the wild type, all cytosines in region I were completely methylated in 90 % of the examined clones, but unmethylated in the remaining 10 %, showing ‘all or nothing’ methylation in a clone (Fig. 6A and Table 2). Thirdly, in Line-2, methylation in all regions was completely erased (Fig. 6A, B, and Table 2). The results indicated that Xa21G is classified into a group of promoter-methylated and body-unmethylated genes (Zhang et al., 2006), and that Xa21G in Line-2 was totally stripped of methylation.
Fig. 6.
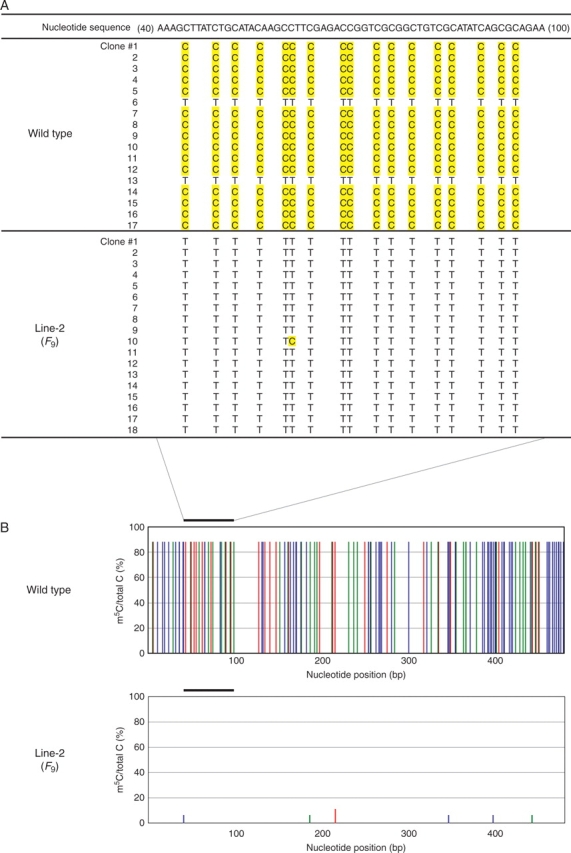
Direct methylation mapping of Xa21G. (A) Identification of m5C in the promoter region containing the CG island. After bisulfite treatment, the 492 bases in region I were amplified by PCR, cloned and sequenced. Cytosines (C) and m5C in the original sequence were converted into thymine (T) and C, respectively. The original nucleotide sequence of the wild type without bisulfite treatment is shown on the top line with shading of the methylatable cytosines. The converted bases are indicated by T or C (shaded) in each clone originating from bisulfite-treated wild type or F9 of Line-2. Seventeen and 18 clones were analysed for the wild type and Line-2, respectively. The clone number is given on the left. (B) Distribution and frequency of m5Cs. Histograms represent the percentage of m5Cs over the total cytosines (vertical axis) at positions containing CpG (red), CpNpG (green) and CpNpN (blue) sequences (N is A, C or T) in the top strand at the indicated nucleotide positions (horizontal axis) in the wild type (upper panel) and F9 of Line-2 (lower panel). The bar on the top indicates the sequence shown in (A). A negative result for Line-2 means that almost no m5C could be assigned to this region.
Table 2.
Methylation frequency in the Xa21G locus
| Region (bp) | Sample | No. of clones | m5C/total C (%) |
|---|---|---|---|
| I (513) | WT | 17 | 1830/2074 (88·2) |
| Line-2 | 18 | 7/2196 (0·3) | |
| II (324) | WT | 8 | 3/624 (0·5) |
| Line-2 | 7 | 0/546 (0) | |
| III (370) | WT | 16 | 5/1104 (0·4) |
| Line-2 | 12 | 0/828 (0) |
The numbers of m5Cs in the total methylatable Cs in the total clones examined are presented as numbers observed and as percentages (%) in parentheses. For example, the 2074 total Cs in region I from the wild type were calculated by multiplying 17 clones by 122 methylatable Cs in 513 bases of region I. WT, wild type; Line-2 is represented by the F9 progeny.
Acquisition of disease resistance traits
Expression analysis by RT–PCR showed that, under non-stressed conditions, transcripts of Xa21G were not detectable in the wild type, while they were clearly accumulated in Line-2 (Fig. 7A). Since Xa21 is a resistance gene against the pathogenic bacterium, X. oryzae pv. oryzae (Ronald, 1997), its transcript induction was examined before and after bacterial inoculation of healthy leaves. Transcripts were not detectable in the wild type control, but were heavily accumulated in Line-2 (Fig. 7B). Successful infection was confirmed by expression of a marker gene of pathogen response, PR1b, transcripts of which were equally induced in both wild-type and Line-2 leaves only after pathogen infection (Fig. 7B). In contrast, Xa21G transcripts were not induced in the wild type, but were heavily accumulated in Line-2 (Fig. 7B), indicating that Xa21G was constitutively silenced in the wild type even under pathogen attack, while it was constitutively expressed in Line-2. Subsequent infection bioassay showed that wild-type plants were highly susceptible, developing severe necrosis with lesion length up to 15 cm, which were comparable with other susceptible strains (Song et al., 1995; Wang et al., 1996). In contrast, Line-2 was stably resistant, showing the average lesion length to be less than one-third that of the control throughout generations (Fig. 7C, D). This resistance was also comparable with reported resistant lines (Song et al., 1995; Wang et al., 1996). The number of bacteria which grew in infected areas was reduced up to 1/30 in Line-2 in comparison with the control (Fig. 7E). These results suggested that Line-2 acquired the disease-resistant trait, which was absent in the wild type, and that such resistance was due to activated Xa21G.
Fig. 7.

Expression of Xa21G. (A) Transcript accumulation profile. Total RNA was isolated from healthy leaves of wild type and Line-2 progeny (F3–F9), and subjected to RT–PCR with specific primers for Xa21G. As the PCR control, the actin gene was similarly amplified. (B) Response to pathogen challenge. Total RNA was isolated from healthy leaves of wild type and Line-2 (F9), which were treated (+) or not treated (–) with Xanthomonas oryzae pv. oryzae (Xoo) for 14 d, and subjected to RT–PCR with specific primers for Xa21G and PR-1. As the control, actin cDNA and genomic DNA were amplified. (C) Resistance to X. oryzae pv. oryzae infection. Mature healthy leaf blades from the wild type (WT) or the indicated progeny of Line-2 (F3–F9) were inoculated with X. oryzae pv. oryzae, incubated for 16 d and photographed. Two representative samples from each plant are shown. The scale in cm is shown on the left. (D) Lesion length (cm) was measured for 3–5 independent samples 16 d after inoculation as depicted in (C) with the standard deviation. (E) Quantification of X. oryzae cells. Inoculated leaves from the indicated plants with X. oryzae were cut 20 cm from the inoculated surface after incubation for the indicated number of days, and propagated cells were counted. Values are expressed in colony-forming units (CFU) per leaf on a logarithmic scale with the standard deviation from measurements of 3–5 independent inoculations.
DISCUSSION
This report documents that genes constantly silenced due to hypermethylation can be transcriptionally activated by demethylation, resulting in conferring new phenotypes, and that changes in both methylation and phenotype are stably inherited.
Non-random demethylation
The initial finding that the majority of azadC-treated seedlings were lethal suggested that demethylation was a random process that affected a number of gene loci, many of which were essential for survival. Line-2 thus appeared to have fortuitously escaped such ‘lethal hits’, perhaps due to incomplete demethylation. To examine the demethylation status in Line-2, the methylation patterns were initially examined at known gene loci by DNA blot hybridization. However, no clear difference was detected between wild-type and Line-2 plants, suggesting demethylation to have been induced locally. Subsequent MSAP screening yielded six differentially methylated fragments, among which two were characterized in this study. HMF2 is located in the coding sequence of a gag–pol polyprotein, which constitutes the LTR (long terminal repeat) retrotransposon. Retrotransposons account for > 15 % of the rice genome, yet their activity is almost completely suppressed. One suppression mechanism is proposed to be DNA methylation (Hirochika et al., 2000), and DNA blot hybridization assays indicated complete demethylation at CCGG sites, with this pattern being stably inherited up to the F7 generation. The direct methylation mapping confirmed this, showing a total erasure of m5C not only from CpG, but also from CpNpG and CpNpN, and its stable transmission through generations. Such a demethylation implies a reactivation of the retrotransposon (Tompa et al., 2002), accompanying gene translocation and expression. Experimentally, however, no trace of translocation was detected in any generation, suggesting that the demethylation was in fact not sufficient for retrotransposon reactivation.
HMF5 is located in the Xa21G locus. The Xa21 genes constitute the gene-for-gene resistance system against X. oryzae pv. oryzae, which induces severe bacterial blight disease in rice (Ronald, 1997). At least seven gene members have been found in the rice genome on chromosome 11 (see Fig. 2B), among which five were inactive (Wang et al., 1998), one (Xa21D) was partially active and only Xa21 exhibited resistance activity against all seven virulent races of the bacterium (Wang et al., 1996, 1998). However, Xa21 itself originated from wild rice, Oryza longistaminata, and was introduced into cultivated rice, O. sativa ssp. indica (‘IR24’) by crossing to confer strong resistance (Khush et al., 1991). Consequently, the other member of cultivated rice, O. sativa ssp. japonica, to which the present material belongs, does not possess this particular gene, resulting in susceptibility to the disease (see Fig. 4A). Nevertheless, japonica rice possesses an Xa21-like gene (Xa21G) on chromosome 2, although its expression appeared to be repressed under all circumstances. It should be noted, however, that the Xa21G locus in 10 % of the DNA population or cell population was not methylated, presumably resulting in its expression. This is consistent with the presence of its expressed sequence tag (AK065018) but, since RT–PCR did not detect the transcripts, the expression level could be low, which is conceivably not sufficient to confer full resistance.
The present study revealed that the observed inactivation was due to methylation of all cyosines in the promoter region containing dense CpG islands. In mammalian cells, CpG islands were proposed to be epigenetically regulated by methylation (Carninci et al., 2006), consistent with the present finding. However, methylation of all cytosines may affect not only CpG islands, but also DNA structure itself, stably forming a specific conformation (Zacharias et al., 1988; 1990), which seriously interferes with protein interaction (Rachal et al., 1989; Klimasauskas et al., 1994). This may cause almost irreversible silencing of genes.
Biological significance
Demethylation at the promoter region reactivated the gene function, and conferred the disease-resistant trait. Xa21 is believed to serve as a specific receptor/recognizer of pathogen signals, which are further transduced into multiple cellular components to activate the defence reaction (Song et al., 1995). The present results suggest that, despite being equipped with a gene-for-gene resistance system, japonica rice is unable to utilize it due to the absence of the first component, Xa21. This is the reverse direction against evolution, in which natural selection plays important roles. Why was such an important gene silenced? One clue is the presence of a sequence (646 bp) encoding a transposon-like, gag–pol polyprotein fragment at 4·4 kb downstream of the Xa21G locus. Transposable elements are usually selectively and heavily methylated, thereby being prevented from actively jumping over the genome (Hirochika et al., 2000). It is conceivable that during inactivation of invading sequences by methylation, genes located in the vicinity were also methylated by an over-defensive response. Under natural conditions, such plants might have been diminished due to susceptibility to disease, but cultivation by humans possibly rescued them from extinction.
Both the hypomethylated status of Xa21G and the disease-resistant trait were stably inherited by the progeny with a cause–effect relationship. However, the question arises of whether or not activation of Xa21G alone was responsible for the observed resistance. A database search of O. sativa ssp. japonica (cv. Nipponbare) identified two Xa21G-related genes on chromosome 2, located 20 kb upsteam of the Xa21G locus (chromosome position 25 208 591), one at position 25 184 975 and the other at position 25 173 621. However, RT–PCR showed that neither gene was expressed in wild type and Line-2 plants even under pathogen challenge (unpubl. obs.). The database search also revealed several rather distantly related sequences mainly on chromosome 11, perhaps corresponding to those in indica rice (Song et al., 1997). Currently their expression in Line-2 is not known, but the possibility of their contribution to the observed resistance appears to be low, as the majority of Xa21-related genes on chromosome 11 were biologically inactive, even if overexpressed in transgenic rice (Wang et al., 1998). Judging from available data, it is speculated that the predominant resistance gene in japonica rice is Xa21G.
Maintenance of methylation
A curious feature is that methylation was not random in the Xa21G locus, being concentrated only in the promoter sequence and absent in the gene-body sequence. How can such a biased methylation occur? The simplest explanation is that the methylation pattern in the mother strand is faithfully recognized and copied to the daughter strand during DNA replication. To date, maintenance of methylation patterns in plants has been considered to be mediated through type 1 DNA methyltransferase (Met1) and chromomethylase (CMT), recognizing symmetric CpG and CpNpG in the mother strand, respectively (Wada, 2005). However, the present finding pointed to methylation of all cytosine residues, suggesting other mechanism(s). One possibility is involvement of domains rearranged methylase (DRM), which is capable of methylating all cytosines regardless of the sequence context (Wada et al., 2003), and methylated DNA-binding domain (MBD) proteins, which recognize m5C in DNA (Zamach and Grafi, 2007). It is tempting to speculate on the presence of a methylation machinery, in which MBD scans m5C, and associated DRM methylates corresponding cytosines.
Another notable feature is that methylation/demethylation patterns are faithfully transmitted to the offspring. In mammalian cells, the methylation pattern is completely erased in the germline, and is reconstituted during subsequent development (Bird, 2002). In plants, however, it is thought that the DNA methylation pattern is not erased during gametogenesis, and therefore that it is not re-established in every generation (Kinoshita et al., 2004, 2007). This and the present results strongly suggest that methylation patterns in somatic cells are maintained in germ cells, and, if this is the case, a system such as the methylation machinery described above could be one possible mechanism.
Lamarckian inheritance
The present observation was obtained by artificial erasure of methylation by 5-azadC treatment. Since the majority of treated seedlings were lethal, a vast demethylation could have been detrimental for survival, and only those which were affected at non-essential genes might have survived. Dwarfism and disease resistance are such examples, suggesting that demethylation has occurred at limited genomic regions in these lines. Whether or not such a selective demethylation at a particular gene locus takes place under natural conditions is of particular interest with respect to epigenetic regulation. To date, a change in DNA methylation patterns has occasionally been reported during the vegetative stage of several plant species in response to environmental conditions, including mechanical stress (Galaud et al., 1993), short days (Lizal and Relichova, 2001), heavy metals (Aina et al., 2004; Choi and Sano, 2007), cold (Steward et al., 2002) and pathogen infection (Wada et al., 2004; Pavet et al., 2006). In the case of the toadflax (Linaria vulgaris), a mutant in which the fundamental symmetry of the flower is changed was identified 250 years ago by Linnaeus. The gene responsible for flower development, Lcyc, was found to be heavily methylated and silenced in this mutant, but demethylated in revertants with normal flower phenotype (Cubas et al., 1999). It was thus suggested that methylation/demethylation does occur under natural conditions, and that heritable epigenetic mutations play a significant role in evolution. The present finding substantiates this idea, showing that gene expression is flexibly tuned by methylation, allowing plants to gain or lose particular traits which are heritable as far as methylation patterns of corresponding genes are maintained. This is in support of the concept of Lamarckian inheritance, suggesting that acquired traits are heritable.
ACKNOWLEDGEMENTS
We thank Dr Thomas Jacobs (University of Illinois at Urbana-Champaign) for help in statistical analysis, and Ms Yuka Yamamoto (Nara Institute of Science and Technology) for preparation of the manuscript. This work was partly supported by grants from the Japan Society for the Promotion of Science.
LITERATURE CITED
- Aina R, Sgorbati S, Santagostino A, Labra M, Ghiani A, Citterio S. Specific hypomethylation of DNA is induced by heavy metals in white clover and industrial hemp. Physiologia Plantarum. 2004;121:472–480. [Google Scholar]
- Amado L, Abranches R, Neves N, Viegas W. Development-dependent inheritance of 5-azacytidine-induced epimutations in triticale: analysis of rDNA expression patterns. Chromosome Research. 1997;5:445–450. doi: 10.1023/a:1018460828720. [DOI] [PubMed] [Google Scholar]
- Bender J. DNA methylation and epigenetics. Annual Review of Plant Biology. 2004;55:41–68. doi: 10.1146/annurev.arplant.55.031903.141641. [DOI] [PubMed] [Google Scholar]
- Bird A. DNA methylation patterns and epigenetic memory. Genes and Development. 2002;16:6–21. doi: 10.1101/gad.947102. [DOI] [PubMed] [Google Scholar]
- Carninci P, Sandelin A, Lenhard B, Katayama S, Shimokawa K, Ponjavic J, et al. Genome-wide analysis of mammalian promoter architecture and evolution. Nature Genetics. 2006;38:626–635. doi: 10.1038/ng1789. [DOI] [PubMed] [Google Scholar]
- Choi C-S, Sano H. Abiotic-stress induces demethylation and transcriptional activation of a gene encoding a glycerophosphodiesterase-like protein in tobacco plants. Molecular Genetics and Genomics. 2007;277:589–600. doi: 10.1007/s00438-007-0209-1. [DOI] [PubMed] [Google Scholar]
- Cubas P, Vincent C, Coen E. An epigenetic mutation responsible for natural variation in floral symmetry. Nature. 1999;401:157–161. doi: 10.1038/43657. [DOI] [PubMed] [Google Scholar]
- Cullis CA. Mechanisms and control of rapid genomic changes in flax. Annals of Botany. 2005;95:201–206. doi: 10.1093/aob/mci013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cullis CA, Kolodynska K. Variation in the isozymes of flax (Linum usitatissimum) genotrophs. Biochemical Genetics. 1975;13:686–697. doi: 10.1007/BF00484926. [DOI] [PubMed] [Google Scholar]
- Durrant A. The environmental induction of heritable change in. Linum. Heredity. 1962;17:27–61. [Google Scholar]
- Durrant A, Nicholas DB. An unstable gene in flax. Heredity. 1970;25:513–527. [Google Scholar]
- Elliot RJ. Learning SAS in the computer lab. Pacific Grove, CA: Dexbury Press; 2000. [Google Scholar]
- Ellis TH, Delseny M, Lee D, Burcham KW. Methylated and under-methylated rDNA repeats are interspersed at random in two higher plant species. Plant Molecular Biology. 1990;14:73–80. doi: 10.1007/BF00015656. [DOI] [PubMed] [Google Scholar]
- Fieldes MA. Heritable effects of 5-azacytidine treatments on the growth and development of flax (Linum usitatissimum) genotrophs and genotypes. Genome. 1994;37:1–11. doi: 10.1139/g94-001. [DOI] [PubMed] [Google Scholar]
- Finnegan EJ, Genger RK, Peacock WJ, Dennis ES. DNA methylation in plants. Annual Review of Plant Physiology and Plant Molecular Biology. 1998;49:223–247. doi: 10.1146/annurev.arplant.49.1.223. [DOI] [PubMed] [Google Scholar]
- Fuchs J, Demidov D, Houben A, Schubert I. Chromosomal histone modification patterns – from conservation to diversity. Trends in Plant Science. 2006;11:199–208. doi: 10.1016/j.tplants.2006.02.008. [DOI] [PubMed] [Google Scholar]
- Galaud JP, Gaspar T, Boyer N. Inhibition of internode growth due to mechanical stress in Bryonia dioica: relationship between changes in DNA methylation and ethylene metabolism. Physiologia Plantarum. 1993;87:25–30. [Google Scholar]
- Hill J. Environmental induction of heritable changes in. Nicotiana rustica. Nature. 1965;207:732–734. [Google Scholar]
- Hirochika H, Sugimoto K, Otsuki Y, Tsugawa H, Kanda M. Retrotransposons of rice involved in mutations induced by tissue culture. Proceedings of the National Academy of Sciences of the USA. 1996;93:7783–7788. doi: 10.1073/pnas.93.15.7783. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hirochika H, Okamoto H, Kakutani T. Silencing of retrotransposons in Arabidopsis and reactivation by the ddm1 mutation. Plant Cell. 2000;12:357–368. doi: 10.1105/tpc.12.3.357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holliday R. DNA methylation: molecular biology and biological significance. Basel: Birkhauser Verlag; 1993. Epigenetic inheritance based on DNA methylation. In: Jost JP, Saluz HP, eds; pp. 452–468. [Google Scholar]
- Jablonka E, Lamb MJ. The inheritance of acquired epigenetic variations. Journal of Theoretical Biology. 1989;139:69–83. doi: 10.1016/s0022-5193(89)80058-x. [DOI] [PubMed] [Google Scholar]
- Janousek B, Siroky J, Vyskot B. Epigenetic control of sexual phenotype in a dioecious plant. Melandrium album. Molecular Genetics and Genomics. 1996;250:483–490. doi: 10.1007/BF02174037. [DOI] [PubMed] [Google Scholar]
- Joen J-S, Lee S, Jung K-H, Yang W-S, Yi G-H, Oh B-G, An G. Production of transgenic plants showing reduced heading date and plant height by ectopic expression of rice MADS-box genes. Molecular Breeding. 2000;6:581–592. [Google Scholar]
- Jones L, Ratcliff F, Baulcombe DC. RNA-directed transcriptional gene silencing in plants can be inherited independently of the RNA trigger and requires Met1 for maintenance. Current Biology. 2001;11:747–757. doi: 10.1016/s0960-9822(01)00226-3. [DOI] [PubMed] [Google Scholar]
- Kauffman HE, Reddy APK, Hsieh SPV, Merca SD. An improved technique for evaluating resistance to rice varieties of. Xanthomonas oryzae. Plant Disease Reports. 1973;57:537–541. [Google Scholar]
- Khush GS, Bacalangco E, Ogata T. A new gene for resistance to bacterial blight from. O. longistaminata. Rice Genetics Newsletter. 1991;7:121–122. [Google Scholar]
- King GJ. Morphological development in Brassica olerancea is modulated by in vivo treatment with 5-azacytidine. Journal of Horticultural Science and Biotechnology. 1995;70:333–342. [Google Scholar]
- Kinoshita T, Miura A, Choi Y, Kinoshita Y, Cao X, Jacobsen SE, et al. One-way control of FWA imprinting in Arabidopsis endosperm by DNA methylation. Science. 2004;303:521–523. doi: 10.1126/science.1089835. [DOI] [PubMed] [Google Scholar]
- Kinoshita Y, Saze H, Kinoshita T, Miura A, Soppe WJ, Koornneef M, et al. Control of FWA gene silencing in Arabidopsis thaliana by SINE-related direct repeats. Plant Journal. 2007;49:38–45. doi: 10.1111/j.1365-313X.2006.02936.x. [DOI] [PubMed] [Google Scholar]
- Klimasauskas S, Kumar S, Roberts RJ, Cheng X. HhaI methyltransferase flips its target base out of the DNA helix. Cell. 1994;76:457–369. doi: 10.1016/0092-8674(94)90342-5. [DOI] [PubMed] [Google Scholar]
- Komatsu M, Shimamoto K, Kyozuka J. Two-step regulation and continuous retrotransposition of the rice LINE-type retrotransposon. Karma. Plant Cell. 2003;15:1934–1944. doi: 10.1105/tpc.011809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kumpatla SP, Teng W, Buchholz AG, Hall TC. Epigentic transcriptional silencing and 5-azacytidine-mediated reactivation of a complex transgene in rice. Plant Physiology. 1997;115:361–373. doi: 10.1104/pp.115.2.361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lizal P, Relichova J. The effect of day length, vernalization and DNA demethylation on the flowering time in. Arabidopsis thaliana. Physiologia Plantarum. 2001;113:121–127. [Google Scholar]
- Murray MG, Thompson WF. Rapid isolation of high molecular weight plant DNA. Nucleic Acids Research. 1980;8:4321–4325. doi: 10.1093/nar/8.19.4321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paszkowski J, Whitham SA. Gene silencing and DNA methylation processes. Current Opinion in Plant Biology. 2001;4:123–129. doi: 10.1016/s1369-5266(00)00147-3. [DOI] [PubMed] [Google Scholar]
- Pavet V, Quintero C, Cecchini NM, Rosa AL, Alvarez ME. Arabidopsis displays centromeric DNA hypomethylation and cytological alterations of heterochromatin upon attack by. Pseudomonas syringae. Molecular Plant-Microbe Interaction. 2006;19:577–587. doi: 10.1094/MPMI-19-0577. [DOI] [PubMed] [Google Scholar]
- Peters AH, Schübeler D. Methylation of histones: playing memory with DNA. Current Opinion in Plant Biology. 2005;17:230–238. doi: 10.1016/j.ceb.2005.02.006. [DOI] [PubMed] [Google Scholar]
- Rachal MJ, Yoo H, Becker FF, Lapeyre JN. In vitro DNA cytosine methylation of cis-regulatory elements modulates c-Ha-ras promoter activity in vivo. Nucleic Acids Research. 1989;17:5135–5147. doi: 10.1093/nar/17.13.5135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reik W, Dean W, Walter J. Epigenetic reprogramming in mammalian development. Science. 2001;293:1089–1093. doi: 10.1126/science.1063443. [DOI] [PubMed] [Google Scholar]
- Reyna-Lopez GE, Simpson J, Ruiz-Herrera J. Differences in DNA methylation patterns are detectable during the dimorphic transition of fungi by amplification of restriction polymorphism. Molecular Genetics and Genomics. 1997;253:703–710. doi: 10.1007/s004380050374. [DOI] [PubMed] [Google Scholar]
- Riggs AD, Porter TN. Overview of epigenetic mechanism. In: Russo VEA, Martienssen RA, Riggs AD, editors. Epigenetic mechanisms of gene regulation. Cold Spring Harbor, NY: Cold Spring Harbor Laboratory Press; 1996. pp. 29–45. [Google Scholar]
- Ronald PC. The molecular basis of disease resistance in rice. Plant Molecular Biology. 1997;35:179–186. [PubMed] [Google Scholar]
- Sano H, Kamada I, Youssefian S, Katsumi M, Wabiko H. A single treatment of rice seedlings with 5-azacytidine induces heritable dwarfism and undermethylation of genomic DNA. Molecular Genetics and Genomics. 1990;220:441–447. [Google Scholar]
- Snedecor GW, Cochran WG. Statistical methods. 7th edn. Ames, IA: Iowa State University Press; 1980. [Google Scholar]
- Song W-Y, Wang GL, Chen LL, Kim HS, Pi LY, Holsten T, et al. A receptor kinase-like protein encoded by the rice disease resistance gene, Xa21. Science. 1995;270:1804–1806. doi: 10.1126/science.270.5243.1804. [DOI] [PubMed] [Google Scholar]
- Song W-Y, Pi L-Y, Wang G-L, Gardner J, Holsten T, Ronald PC. Evolution of the rice Xa21 disease resistance gene family. Plant Cell. 1997;9:1279–1283. doi: 10.1105/tpc.9.8.1279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Steward N, Sano H. Measuring changes in chromatin using micrococcal nuclease. In: Tollefsbol T, editor. Epigenetics protocols. Totowa: Humana Press; 2004. pp. 65–75. [DOI] [PubMed] [Google Scholar]
- Steward N, Ito M, Yamakuchi Y, Koizumi N, Sano H. Periodic DNA methylation in maize nucleosomes and demethylation by environmental stress. Journal of Biological Chemistry. 2002;277:37741–37746. doi: 10.1074/jbc.M204050200. [DOI] [PubMed] [Google Scholar]
- Tariq M, Paszkowski J. DNA and histone methylation in plants. Trends in Plant Science. 2004;6:244–251. doi: 10.1016/j.tig.2004.04.005. [DOI] [PubMed] [Google Scholar]
- Tompa R, McCallum CM, Delrow J, Henikoff JG, van Steensel B, Henikoff S. Genome-wide profiling of DNA methylation reveals transposon targets of CHROMOMETHYLASE3. Current Biology. 2002;12:65–68. doi: 10.1016/s0960-9822(01)00622-4. [DOI] [PubMed] [Google Scholar]
- Vyskot B, Kaukolova B, Kovarik A, Sachambula L, Reynolds D, Bezdek M. Meiotic transmission of a hypomethylated repetitive DNA family in tobacco. Theoretical and Applied Genetics. 1995;91:659–664. doi: 10.1007/BF00223294. [DOI] [PubMed] [Google Scholar]
- Wada Y. Physiological functions of plant DNA methyltransferases. Plant Biotechnology. 2005;22:71–80. [Google Scholar]
- Wada Y, Ohya H, Yamaguchi Y, Koizumi N, Sano H. Preferential de novo methylation of cytosine residues in non-CpG sequences by a domains rearranged DNA methyltransferase from tobacco plants. Journal of Biological Chemistry. 2003;278:42386–42393. doi: 10.1074/jbc.M303892200. [DOI] [PubMed] [Google Scholar]
- Wada Y, Miyamoto K, Kusano T, Sano H. Association between up-regulation of stress-responsive genes and hypomethylation of genomic DNA in tobacco plants. Molecular Genomics and Genetics. 2004;271:658–666. doi: 10.1007/s00438-004-1018-4. [DOI] [PubMed] [Google Scholar]
- Wang G-L, Song W-Y, Ruan D-L, Sideris S, Ronald PC. The cloned gene, Xa21, confers resistance to multiple Xanthomonas oryzae pv. oryzae isolates in transgenic plants. Molecular Plant-Microbe Interaction. 1996;9:850–855. doi: 10.1094/mpmi-9-0850. [DOI] [PubMed] [Google Scholar]
- Wang GL, Ruan DL, Song WY, Sideris S, Chen L, Pi LY, et al. Xa21D encodes a receptor-like molecule with a leucine-rich repeat domain that determines race-specific recognition and is subject to adaptive evolution. Plant Cell. 1998;10:765–779. doi: 10.1105/tpc.10.5.765. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wierzbicki AT, Jerzmanowski A. Suppression of histone H1 genes in Arabidopsis results in heritable developmental defects and stochastic changes in DNA methylation. Genetics. 2004;169:997–1008. doi: 10.1534/genetics.104.031997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zacharias W, Caserta M, O'Conner TR, Larson JE, Wells RD. Cytosine methylation as an effector of right-handed to left-handed DNA structural transitions. Nucleic Acids Research. 1988;74:221–224. doi: 10.1016/0378-1119(88)90291-0. [DOI] [PubMed] [Google Scholar]
- Zacharias W, Jaworski A, Wells RD. Cytosine methylation enhances Z-DNA formation. in vivo. Journal of Bacteriology. 1990;172:3278–3283. doi: 10.1128/jb.172.6.3278-3283.1990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zamach A, Grafi G. Methyl-CpG-binding domain proteins in plants: interpreters of DNA methylation. Trends in Plant Science. 2007;12:80–85. doi: 10.1016/j.tplants.2006.12.004. [DOI] [PubMed] [Google Scholar]
- Zhang X, Yazaki J, Sundaresan A, Cokus S, Chan SW-L, Chen H, et al. Genome-wide high-resolution mapping and functional analysis of DNA methylation in arabidopsis. Cell. 2006;126:1–13. doi: 10.1016/j.cell.2006.08.003. [DOI] [PubMed] [Google Scholar]
- Zilberman D, Gehring M, Tran RK, Ballinger T, Henikoff S. Genome-wide analysis of Arabidopsis thaliana DNA methylation uncovers an interdependence between methylation and transcription. Nature Genetics. 2007;39:61–69. doi: 10.1038/ng1929. [DOI] [PubMed] [Google Scholar]