

Abstract
Auditory neuropathy is a hearing disorder characterized by normal function of outer hair cells, evidenced by intact cochlear microphonic (CM) potentials and otoacoustic emissions (OAEs), with absent or severely dys-synchronized auditory brainstem responses (ABRs). To determine if selective lesions of inner hair cells (IHCs) and auditory nerve fibers (ANFs) can account for these primary clinical features of auditory neuropathy, we measured physiological responses from chinchillas with large lesions of ANFs (about 85%) and IHCs (45% loss in the apical half of the cochlea; 73% in the basal half). Distortion product OAEs and CM potentials were significantly enhanced, whereas summating potentials and compound action potentials (CAPs) were significantly reduced. CAP threshold was elevated by 7.5 dB, but response synchrony was well preserved down to threshold levels of stimulation. Similarly, ABR threshold was elevated by 5.6 dB, but all waves were present and well synchronized down to threshold levels in all animals. Thus, large lesions of IHCs and ANFs reduced response amplitudes but did not abolish or severely dys-synchronize CAPs or ABRs. Pathologies other than or in addition to ANF and IHC loss are likely to account for the evoked potential dys-synchrony that is a clinical hallmark of auditory neuropathy in humans.
Keywords: Auditory neuropathy, inner hair cells, auditory nerve, carboplatin, auditory brainstem response, cochlea, auditory evoked potentials
Introduction
Auditory neuropathy (AN) is a hearing disorder characterized by severely abnormal or absent auditory brainstem responses (ABRs) in the presence of normal outer hair cell (OHC) function as shown from intact cochlear microphonic (CM) potentials or otoacoustic emissions (OAEs) (Starr et al., 1996). Auditory thresholds may be normal, or hearing loss ranging from mild to severe may be present. However, synchronous firing of neurons at stimulus onset, a necessary condition for generating an ABR, is always impaired, and this is believed to account for the difficulties individuals with AN invariably have in understanding speech (Kraus et al., 2000).
AN appears to be much more common than was initially thought. Among infants and children with permanent sensorineural hearing loss, prevalence of AN has been reported to range from 5% to 24%, with 10–14% being the most frequently reported prevalence (Berg et al., 2005; Foerst et al., 2006; Ngo et al., 2006). Management of AN is challenging for audiologists and speech therapists. The vast majority of infants and children with AN do not develop speech and language normally, even if they are fitted properly with hearing aids (Berlin et al., 1995, 2003; Sininger, 2002; Sininger et al., 2001; Starr et al., 2000). Cochlear implantation has been successful for some but not all children with AN, and it is far from being an accepted method for management (Mason et al., 2003; Peterson et al., 2003; Shallop et al., 2001). Understanding the pathology underlying AN may help in managing and treating the disorder.
Based on the clinical manifestations of AN, it is likely that the condition has multiple causes. Among postulated pathologies are lesions of inner hair cells (IHCs) or auditory nerve fibers (ANFs) (Harrison, 1998; Salvi et al., 1999; Starr, 2001; Starr et al., 1996) or demyelination of the auditory nerve (El-Badry et al., 2007a,b). Whether IHC or ANF lesions alone can produce the clinical manifestations that are hallmarks of AN is the focus of the current study.
Chinchillas injected systemically with carboplatin, a widely used platinum-based chemotherapy agent, provide a model system for determining if selective IHC and ANF lesions can account for AN. Numerous studies have shown that carboplatin rapidly destroys both IHCs and ANFs in the chinchilla cochlea, while leaving OHCs intact and functioning (Ding et al., 1999; Takeno et al., 1994; Trautwein et al., 1996; Wake et al., 1994, 1996; Wang et al., 2003). We used carboplatin-treated chinchillas to determine if selective IHC and ANF lesions could produce physiological abnormalities paralleling the clinical features of AN in humans. Distortion product OAEs (DPOAEs), summating potentials (SPs), compound action potentials (CAPs), and ABRs were recorded in eight chinchillas before and 2 wk after carboplatin injection (i.e., before and after IHC and ANF lesions). These physiological measures were chosen because they reflect the function of the cochlea, auditory nerve and auditory brainstem, and they represent most of the diagnostic criteria of AN. We were particularly interested in determining if IHC and ANF loss would result in absent or dys-synchronous ABRs in the presence of intact CMs and OAEs, since ABR abnormalities are invariably present in humans with AN.
Materials and Methods
Subjects and electrode implantation surgery
Eight adult chinchillas (Chinchilla laniger) obtained from a vendor licensed by the U.S. Department of Agriculture (Jarr Chinchilla Inc., Hubbard, OH) underwent surgery for implantation of recording electrodes. Animals were anesthetized with an intramuscular injection of ketamine (50 mg/kg) and acepromazine (0.3 mg/kg), and placed on a Deltaphase® Isothermal Pad (Braintree Scientific, Inc., Braintree, MA), with the head secured in a stereotaxic apparatus. A postauricular incision was made to expose the right bulla and a small hole was made in the dorsal portion for insertion of a silver-ball electrode. The electrode was placed against the round window niche and fixed to the bulla with dental cement. The dorsal cranium was then exposed and a small hole was drilled in the rostral cranium for implantation of a ground electrode, which was fixed to the skull using dental cement. Buprenorphine (0.05 mg/kg) was administered subcutaneously for 3 days after surgery, and animals were allowed to recover for at least 14 days before electrophysiological testing was initiated.
Electrophysiology, DPOAE recording, and carboplatin injection
Electrical activity and DPOAEs were recorded in response to stimuli (4 kHz tone bursts for CM, SP, CAP and DPOAE; clicks for ABR) presented to the right ears of the animals through ER-2 insert earphones (Etymotic Research, Inc., Elk Grove Village, IL). CMs, SPs and CAPs were recorded from the round window electrode. ABRs were recorded from subdermal needle electrodes inserted at the scalp midline (noninverting), posterior to the stimulated right ear (inverting), and at the midline of the back (common).
CM, SP, CAP, and ABR recording utilized commercial hardware and software (Tucker Davis Technologies, Inc., Alachua, FL). Stimuli were generated digitally using an array processor (AP2), and a 16-bit digital-to-analog (D/A) converter (DA 3–4). The signals were attenuated with a programmable attenuator (PA5), then routed through a headphone buffer (HB7) and delivered to the animal’s right ear. Repetition rates were 6.1/s for the CM, 21/s for the SP, and 2.1/s for the CAP. Stimulus duration was 50 ms for the CM, 15 ms for the SP, and 5 ms for the CAP. Rise/fall time was shaped with a cos2 window of two cycles of the 4 kHz stimulus. The stimuli were presented with an alternating polarity, starting at a level of 80 dB SPL and decreasing in 5 dB steps. ABRs were recorded in response to alternating polarity clicks of 100-μs duration, presented at a repetition rate of 20.1/s, at levels starting at 90 dB SPL and decrementing in 5 dB steps.
All testing was conducted in a single-walled sound booth. For CM, SP, CAP, and DPOAE tests, awake animals were placed in a custom-designed animal restraint (Snyder and Salvi, 1994). For ABR recordings, animals were anesthetized with an intramuscular injection of ketamine (30 mg/kg) and acepromazine (0.1 mg/kg) and placed on a heating pad in the sound booth. Responses were band-pass filtered (100–15,000 Hz for the CM; 5–300 Hz for the SP; 100–3000 Hz for the CAP and ABR), amplified (10,000 X for the CAP, CM, and SP and 50,000 X for the ABR) using a bioamplifier (DB4), and digitized using an analog-to-digital (A/D) converter (AD1). The responses were recorded over an epoch of 20 ms for the SP and CAP, and 60 ms for the CM, and 100 sweeps were averaged at each stimulus level. For the ABR, responses were recorded over a 12.5 ms epoch and 1000 sweeps were averaged at each stimulus level.
Input-output functions for the DPOAEs were obtained using two primary tones (f1 and f2, where f2/f1 = 1.2 and f1 frequency was 4 kHz) generated using two D/A converters (16 bits, 100 kHz) on two separate signal-processing boards in a computer. The output of each D/A converter was low-pass filtered (roll-off 90 dB between 20 and 24 kHz) and routed to a computer controlled attenuator and buffer amplifier, and then to the insert earphones, which were coupled to a low-noise ER10B microphone (Etymotic Research, Inc., Elk Grove Village, IL) through a narrow tube. Stimulus level was incremented in 5 dB steps from 0 to 80 dB SPL. The output of the microphone was delivered to an A/D converter (16 bit) on a signal-processing board and sampled for 500 ms at a sampling rate of 31 kHz (15,500 samples at each stimulus level).
After baseline measures were obtained, each animal received a single intraperitoneal injection of carboplatin (LKT Laboratories, Inc., St. Paul, MN; 75 mg/kg). Animals were retested 2 wk after injection, then euthanized for histology.
Histology
Chinchillas were anesthetized with an intramuscular injection of ketamine (30 mg/kg) and acepromazine (0.1 mg/kg) and perfused intracardially with phosphate buffered saline (PBS, pH 7.4) followed by 2.5% glutaraldehyde in PBS. The animals were then decapitated and the right bulla was quickly removed. The round window and oval window were opened and the cochlea was immersed in fixative (2.5% glutaraldehyde in PBS) for at least 24 h at 4 °C.
Histological procedures have been described in detail previously (Ding et al., 2001a,b). Briefly, the basilar membrane was carefully dissected from the right cochlea for surface preparations and stained with hematoxylin. Missing hair cells across the entire basilar membrane were counted and cochleograms, showing the percent of IHC and OHC loss as a function of percent distance from the apex, were constructed for all right ears. Position in the cochlea was translated to frequency using Greenwood’s generalized cochlear frequency-place map (Greenwood, 1990). The right cochleas (minus the organ of Corti) were then decalcified (Decal, Baxter Scientific Products, Deerfield, IL, USA) and embedded in Epon 812 for counts of myelinated nerve fibers in the habenula perforatae. After polymerization, sections were cut parallel to the modiolar axis at a thickness of 1.5 μm, mounted on glass slides and stained with 0.5% toluidine blue. Myelinated ANFs were counted in three habenula perforatae located in the second turn of each cochlea. The average number of myelinated ANFs in cochleas of the eight carboplatin-treated chinchillas was compared to ANF counts from cochleas of normal chinchillas used in a previous study (Ding et al., 2001a).
All procedures regarding the use and care of animals in this study were reviewed and approved by the Institutional Animal Care and Use Committee at the University at Buffalo.
Results
Figure 1 shows IHC and OHC losses caused by carboplatin, averaged across all eight chinchillas. OHC loss was negligible, averaging 1.6 ± 1.0% (mean ± standard error of the mean) across the entire cochlea. IHC loss generally increased from the apical end of the basilar membrane to the basal region up to approximately 75% distance from the apex. Peak losses of 85–90% occurred in the cochlear region located approximately 60–75% distance from the apex, which encodes frequencies between 3 and 6 kHz (Greenwood, 1990). The average IHC loss was 45.0 ± 7.8% in the apical half of the cochlea and 72.7 ± 7.4% in the basal half.
Figure 1.
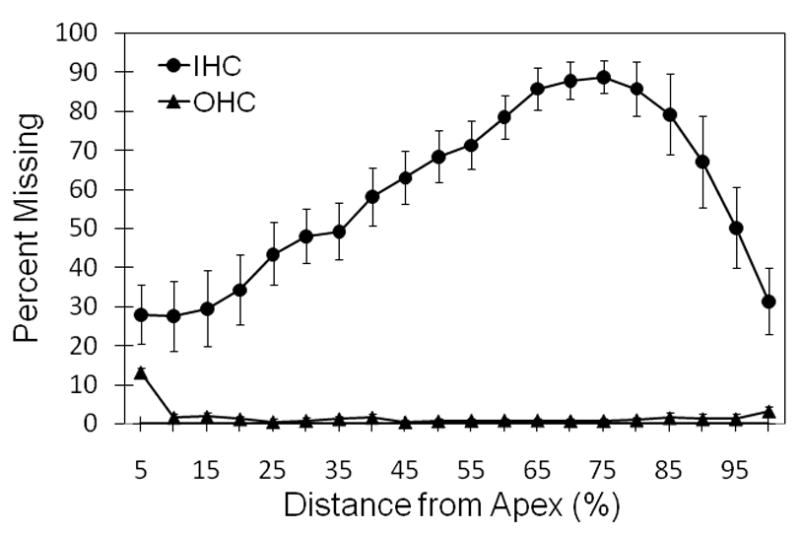
Mean cochleogram of the eight carboplatin-treated chinchillas showing loss of IHCs (solid circles) and OHCs (solid triangles) as a function of place along the basilar membrane (referenced to the apex).
Figure 2 shows myelinated ANFs within the habenula perforatae in a normal chinchilla (left panel) and in a chinchilla treated with carboplatin (right panel). The average number of myelinated ANFs in the carboplatin-treated chinchillas (n = 8) was 10.9 ± 3.5 per habenula perforata. Compared to an average of 74.2 ± 6.6 ANFs per channel in normal chinchillas (Ding et al., 2001a), this corresponds to a loss of approximately 85%.
Figure 2.

ANFs within the habenula perforatae in a normal chinchilla (left panel) and in a chinchilla 2 wk after carboplatin injection (right panel).
The input-output functions of the physiological measures before and 2 wk after carboplatin injection are shown in Fig. 3. After IHC and ANF lesions were induced, CM potentials (Fig. 3A) and DPOAEs (Fig. 3B) were enhanced, whereas SPs (Fig. 3C) and CAPs (Fig. 3D) were depressed relative to pre-injection values. Amplitudes in response to 70, 75 and 80 dB SPL stimuli were averaged to facilitate the following pre- versus post-lesion comparisons. The average CM amplitude increased by 41.4 ± 25.3% and average DPOAE level increased by 5.4 ± 2.9 dB SPL relative to the pre-injection values. The average SP amplitude decreased by 57.3 ± 17.7% and the CAP decreased by 75.9 ± 10.7%.
Figure 3.
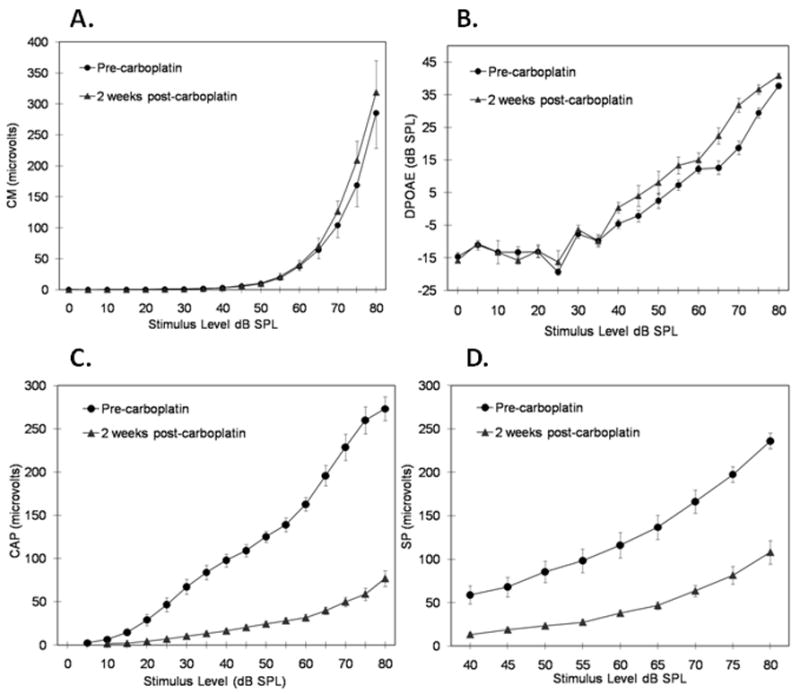
Input-output functions of the physiological measures at 4 kHz before (solid circles) and 2 wk after (solid triangles) carboplatin injection. (A) cochlear microphonics; (B) distortion product OAEs; (C) summating potential; (D) compound action potential. All points are averages for all eight chinchillas, with the following exceptions: Pre-carboplatin: CM: 0 dB SPL, n = 2; 5 dB SPL, n = 4; CAP: 5 dB SPL, n = 2; 10 dB SPL, n = 7. Post-carboplatin: CM: 0 dB SPL, n = 4; 5 dB SPL, n = 5; CAP: 10 dB SPL, n = 3; 15 dB SPL, n = 3; 20 dB SPL, n = 7.
The effects of IHC and ANF lesions on DPOAE levels and CM, CAP, and SP amplitudes were analyzed using two-way ANOVAs, with Time (pre-lesion, post-lesion) and Stimulus Level as repeated measures. Analyses were restricted to stimulus levels for which data from all eight animals were available. The stimulus levels included in each analysis were 10–80 dB SPL for CM, 25–80 dB SPL for CAP, 45–80 dB SPL for SP, and 0–80 dB SPL for DPOAE. For each ANOVA, the interaction between Time and Level was statistically significant and effect sizes (η2) were large to very large: F(14,98) = 2.94, p =.001, η2 =.30 for CM potentials; F(11,77) = 69.22, p <.001, η2=.91 for CAPs, F(7,49) = 45.92, p <.001, η2 =.87 for SPs, and F(16,112) = 4.96, p <.001, η2 =.42 for DPOAEs. Despite the significant interaction for CM potentials, pairwise comparisons at 70, 75 and 80 dB SPL did not reach statistical significance (p values.06); however, a trend for enhancement of the CM after carboplatin is clearly apparent (see Fig. 3A). Pairwise comparisons for CAPs and SPs showed significant reductions post-carboplatin at all tested levels (p values ≤.002). Pairwise comparisons for DPOAEs showed significant enhancements at all levels above 35 dB SPL (p values ≤.015), with the anomalous exception of 60 dB SPL (p =.07).
Mean CAP threshold, averaged across all eight animals, was 9.4 ± 3.2 dB SPL prior to carboplatin, and 16.9 ± 5.9 dB SPL after IHC and ANF lesions were induced; this elevation of 7.5 dB was statistically significant, t(7) = −3.55, p =.009. Despite the threshold elevation, CAPs were well synchronized in all animals. Mean ABR threshold was 33.1 ± 2.6 dB SPL prior to carboplatin and 38.8 ± 5.8 dB SPL after IHC and ANF lesions were induced; this difference of 5.7 dB was statistically significant, t(7) = −3.21, p =.015. Despite the threshold elevation, all eight animals had synchronized ABRs; furthermore, as shown in Fig. 4 for four animals, all waves of the ABR were preserved and the response replicated well at all levels down to threshold.
Figure 4.
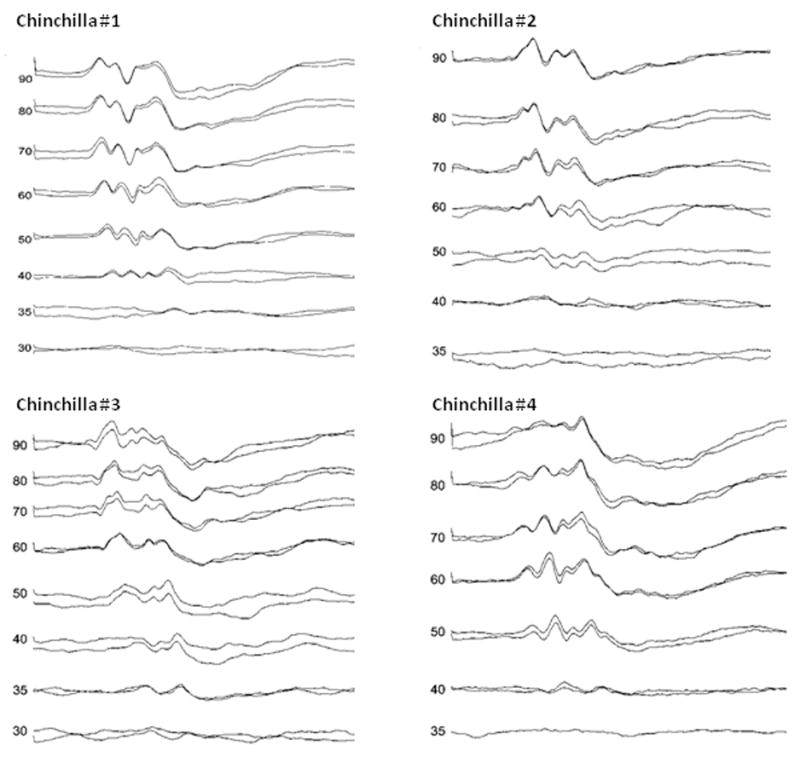
ABRs of four different chinchillas 2 wk after carboplatin injection. Numbers to the left of each ABR are stimulus level in dB SPL. Two ABRs were collected at each stimulus level above threshold, accounting for double traces. Note the excellent replication of responses at each level.
Figure 5 presents data from the chinchilla with the greatest IHC loss in the basal half of the cochlea. The cochleogram for this animal (Fig. 5A) shows total loss of IHCs in the 4 kHz region of the cochlea. Despite the IHC loss, ABRs measured before carboplatin (Fig. 5B) and after carboplatin (Fig. 5C) had similar morphology, amplitude, latency and repeatability.
Figure 5.
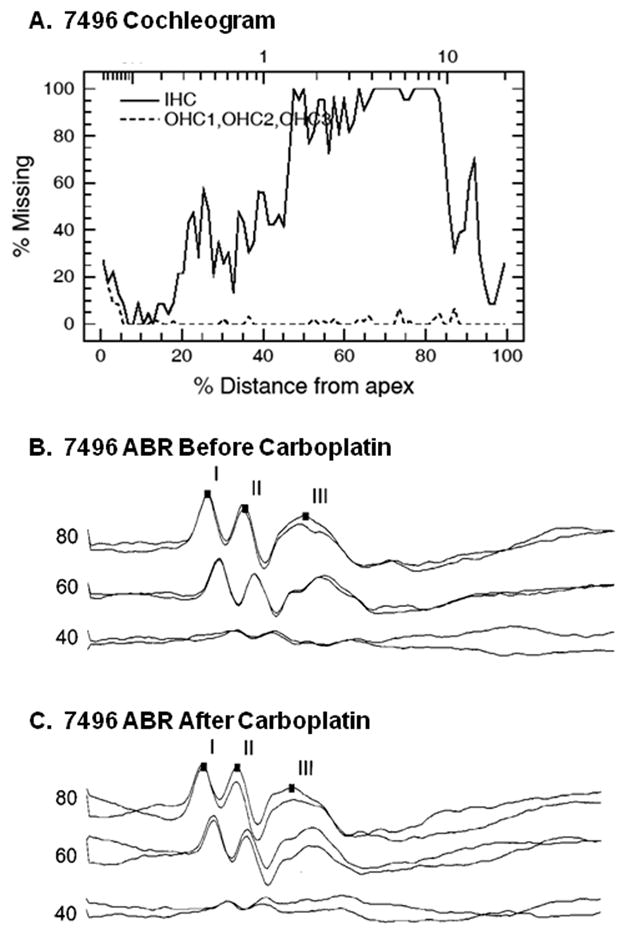
Cochleogram and ABRs from one chinchilla with massive loss of IHCs in the basal half of the cochlea. (A) Cochleogram showing IHC and OHC losses as a function of position along the basilar membrane. (B) ABRs measured before carboplatin injection. (C) ABRs measured 2 wk after carboplatin destroyed hair cells. Numbers to the left of each ABR are stimulus levels in dB SPL. Two ABRs were collected at each stimulus level, accounting for double traces.
Discussion
Clinically, AN manifests by preserved OHC activity in the presence of severe abnormality of the ABR beyond that expected from the hearing loss. In the majority of cases of AN, the ABR is absent. Consistent with this, the CAP is either absent or severely dys-synchronized in all cases of AN (Santarelli and Arslan 2002; Santarelli et al 2008; Starr et al., 1996, 2000). Among speculated pathologies underlying AN are lesions of the IHCs and ANFs (Harrison, 1998; Salvi et al., 1999; Starr, 2001). In the current study, we evaluated acute IHC and ANF lesions as potential pathologies underlying AN. Both CAPs and ABRs were found to be intact and well synchronized despite 85% loss of ANFs and nearly 60% loss of IHCs across the entire cochlea (73% in the basal half). As shown in Fig. 4, all waves of the ABR, including the most peripheral Wave I, were present and waveforms replicated well at all levels down to threshold. These results are in marked contrast to the hallmark criterion for the diagnosis of AN, which is gross abnormality or absence of the ABR. One interpretation of our data is that IHC and ANF losses are not sufficient pathologies to account for the hallmark clinical manifestation of AN. Alternatively, if IHC or ANF lesions are sufficient to abolish the neural synchrony necessary for generating CAPs and ABRs, then it appears that losses must exceed the large losses observed in the current study or, possibly, that chronicity of damage plays a role.
Effects of IHC and ANF Loss on CAPs and ABRs
Recording the CAP from the round window depends on synchronous firing of ANFs, intact synapses between IHCs and afferent ANFs, and intact IHCs. With marked loss of the IHCs and ANFs, substantial reduction of the CAP amplitude was an expected oucome, based on previous studies (El-Badry et al., 2007a; Qiu et al., 2000; Wang et al., 1997). Reduction of the CAP amplitude simply represents the loss of the sensory and neural elements contributing to the generation of the CAP. Despite the marked reduction in the CAP amplitude, threshold was elevated only by 7.5 dB on average. This result indicates that the CAP threshold is relatively insensitive to marked neural and IHC loss in the presence of intact OHCs. Similar results were reported by others (El-Badry et al., 2007a; Qiu et al., 2000; Salvi et al., 2000) and can be explained by the finding that the remaining ANFs have normal thresholds, tuning, and characteristic frequencies (Wang et al., 1997). The current results indicate that just 15% of the ANFs were enough to maintain near normal CAP threshold in the presence of intact OHCs.
The ABR is a far field evoked potential consisting of several peaks representing activation of specific generators along the auditory pathway from the cochlea to regions of the midbrain. As with the CAP, the ABR depends on synchronous firing of the auditory neurons at each generator site, beginning at the level of the auditory nerve. Despite the massive loss of ANFs and IHCs induced by carboplatin in the current study, all animals had fully intact ABRs, including the most peripheral waves (Figs. 4 and 5). Moreover, synchrony of the response was preserved down to threshold levels of stimulation in all animals and threshold was elevated by less than 6 dB, on average (0–15 dB for individual animals). Our findings of intact CAPs and ABRs with small threshold elevations despite the massive ANF and IHC losses indicate that the synchrony of the remaining neurons was not affected by the loss of the majority of the sensory and neural elements in the presence of intact OHCs. This finding strongly suggests that the dys-synchrony in neural responses in AN arises from pathologies other than IHC or ANF loss.
Effects of IHC and ANF Loss on CM potentials, SPs and DPOAEs
Massive loss of IHCs and ANFs in the presence of intact OHCs was associated with significant enhancement of DPOAEs and CM potentials. Interestingly, some, but not all, AN subjects show larger amplitude DPOAEs and CMs compared to normal controls (Starr et al., 2000, 2001b). Enhancement of DPOAEs and CMs in AN can be explained by hyperpolarization of the OHCs associated with disruption of normal activity in the medial olivocochlear (MOC) efferent system (Berlin et al., 1993; Starr, 2001). OHC hyperpolarization could result from loss of tonic MOC efferent feedback to the OHCs due to IHC or ANF loss and reduced input from the afferent arm of the feedback loop (Jock et al., 1996; McFadden, 2007; Takeno et al., 1994), or to some other mechanism such as loss of MOC efferent fibers themselves. In light of our findings, IHC or ANF loss should be considered as possible pathologies in the clinical subset of AN patients who have enhanced DPOAEs or CMs.
Recent studies have shown IHCs to be the major source for the SP, with no contribution from the ANFs (Durrant et al., 1998; El-Badry et al., 2007a; Zheng et al., 1997). Results of these studies also suggest that decline in the SP may serve as a specific marker of IHC damage in ears with intact OHCs. Our current results are consistent with previous suggestions, as shown by the reduction of SP amplitude by more than half after IHC loss in the carboplatin-treated chinchillas. If IHC pathology is the mechanism underlying AN, subjects with AN would be expected to have absent or reduced SP. However, numerous studies (e.g., Santarelli and Arslan, 2002; Santarelli et al., 2008; Sheykholeslami et al., 2001; Starr et al., 2001b) found that the SP is normal or even larger than normal in human subjects with AN. In light of our findings, it must be considered unlikely that AN patients with normal or enhanced SP have significant IHC loss.
Implications for mechanisms underlying AN
A consensus among investigators is that AN involves dys-synchrony of the neural discharge within the auditory system, resulting in severe impairment in temporal coding (Kraus et al., 2000; Starr et al., 1996; Zeng et al., 1999). Disruption of the neural synchrony and temporal deficits account for the clinical presentation of AN, specifically the absent ABR, absent acoustic reflex, and the severe impairment of speech discrimination, as well as impaired performance on tests that require precise encoding of temporal cues such as gap detection, sound localization, and masking level difference (Kraus et al., 2000; Zeng et al., 1999, 2005). Therefore, any pathological mechanism that impairs temporal encoding and neural synchrony might be responsible for AN. Although the functional aspects of the experimental pathology tested in the present study may not totally represent the mechanisms underlying a dys-synchrony that develops in a more chronic fashion in many humans with AN, the current results show that substantial IHC and ANF losses do not produce dys-synchrony of CAPs or ABRs, and are therefore unlikely to be sufficient pathologies underlying AN. However, two other candidate mechanisms for evoked potential dys-synchrony that cannot be ruled out by the current study are dysfunctional synapses between the IHCs and auditory dendrites (Starr et al., 1991) and demyelination of the auditory nerve (El-Badry et al., 2007a,b; Starr et al., 1996, 2001a).
Acknowledgments
The authors gratefully acknowledge the invaluable assistance of Dalian Ding, Center for Hearing and Deafness, State University of New York at Buffalo, for performing all histology. This research was supported by NIH/NIDCD grant P01 DC03600 (SLM).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Berg AL, Spitzer JB, Towers HM, Bartosiewicz C, Diamond BE. Newborn hearing screening in the NICU: profile of failed auditory brainstem response/passed otoacoustic emission. Pediatrics. 2005;116:933–8. doi: 10.1542/peds.2004-2806. [DOI] [PubMed] [Google Scholar]
- Berlin CI, Hood LJ, Hurley A, Wen H. Hearing aids: only for hearing- impaired patients with abnormal otoacoustic emissions. In: Berlin CI, editor. Hair cells and Hearing Aids. Singular Pub Group; San Diego CA: 1995. pp. 99–111. [Google Scholar]
- Berlin CI, Hood L, Cecola L, Lackson D, Szabo P. Does type I afferent neuron dysfunction reveal itself through lack of effenrent supression? Hear Res. 1993:40–50. doi: 10.1016/0378-5955(93)90199-b. [DOI] [PubMed] [Google Scholar]
- Berlin CI, Hood L, Morlet T, Rose K, Brashears S. Auditory neuropathy/dys-synchrony: diagnosis and management. Ment Retard Dev Disabil Res Rev. 2003;9:225–31. doi: 10.1002/mrdd.10084. [DOI] [PubMed] [Google Scholar]
- Butinar D, Zidar J, Leonardis L, Popovic M, Kalaydjieva L, Angelicheva D, Sininger Y, Keats B, Starr A. Hereditary auditory, vestibular, motor, and sensory neuropathy in a Slovenian Roma (Gypsy) kindred. Ann Neurol. 1999;46:36–44. doi: 10.1002/1531-8249(199907)46:1<36::aid-ana7>3.0.co;2-j. [DOI] [PubMed] [Google Scholar]
- Ding DL, Wang J, Salvi R. Selective loss of inner hair cells and type-I ganglion neurons in carboplatin-treated chinchillas. Mechanisms of damage and protection. Ann N Y Acad Sci. 1999;884:152–70. doi: 10.1111/j.1749-6632.1999.tb08640.x. [DOI] [PubMed] [Google Scholar]
- Ding DL, McFadden SL, Salvi RJ. Calpain immunoreactivity and morphological damage in chinchilla inner ears after carboplatin. J Assoc Res Otolaryngol. 2001a;3:68–79. doi: 10.1007/s101620020004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ding DL, McFadden SL, Salvi RJ. Cochlear hair cell densities and inner ear staining techniques. In: Willott JF, editor. Handbook of Mouse Auditory Research: From Behavior to Molecular Biology. CRC Press LLC; Boca Raton, FL: 2001b. pp. 189–204. [Google Scholar]
- Durrant JD, Wang J, Ding DL, Salvi RJ. Are inner or outer hair cells the source of summating potentials recorded from the round window? J Acoust Soc Am. 1998;104:370–7. doi: 10.1121/1.423293. [DOI] [PubMed] [Google Scholar]
- El-Badry MM, McFadden S. Electrophysiological correlates of progressive sensorineural pathology in carboplatin-treated chinchillas. Brain Res. 2007a;1134:112–30. doi: 10.1016/j.brainres.2006.11.078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- El-Badry MM, Ding D, McFadden SL, Eddins AC. Physiological effects of auditory nerve myelinopathy in chinchillas. Eur J Neurosci. 2007b;25:1437–46. doi: 10.1111/j.1460-9568.2007.05401.x. [DOI] [PubMed] [Google Scholar]
- Foerst A, Beutner D, Lang-Roth R, Huttenbrink KB, von Wedel H, Walger M. Prevalence of auditory neuropathy/synaptopathy in a population of children with profound hearing loss. Int J Pediatr Otorhinolaryngol. 2006;70:1415–22. doi: 10.1016/j.ijporl.2006.02.010. [DOI] [PubMed] [Google Scholar]
- Greenwood DD. A cochlear frequency-position function for several species--29 years later. J Acoust Soc Am. 1990;87:2592–605. doi: 10.1121/1.399052. [DOI] [PubMed] [Google Scholar]
- Harrison RV. An animal model of auditory neuropathy. Ear Hear. 1998;19:355–61. doi: 10.1097/00003446-199810000-00002. [DOI] [PubMed] [Google Scholar]
- Jock BM, Hamernik RP, Aldrich LG, Ahroon WA, Petriello KL, Johnson AR. Evoked-potential thresholds and cubic distortion product otoacoustic emissions in the chinchilla following carboplatin treatment and noise exposure. Hear Res. 1996;96:179–90. doi: 10.1016/0378-5955(96)00058-5. [DOI] [PubMed] [Google Scholar]
- Kraus N, Bradlow AR, Cheatham MA, Cunningham J, King CD, Koch DB, Nicol TG, McGee TJ, Stein LK, Wright BA. Consequences of neural asynchrony: a case of auditory neuropathy. J Assoc Res Otolaryngol. 2000;1:33–45. doi: 10.1007/s101620010004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mason JC, De Michele A, Stevens C, Ruth RA, Hashisaki GT. Cochlear implantation in patients with auditory neuropathy of varied etiologies. Laryngoscope. 2003;113:45–9. doi: 10.1097/00005537-200301000-00009. [DOI] [PubMed] [Google Scholar]
- McFadden SL. Biochemical bases of hearing. In: Campbell KCM, editor. Pharmacology and Ototoxicity for Audiologist. Thomson Delmar; Australia: 2007. pp. 86–123. [Google Scholar]
- Ngo RY, Tan HK, Balakrishnan A, Lim SB, Lazaroo DT. Auditory neuropathy/auditory dys-synchrony detected by universal newborn hearing screening. Int J Pediatr Otorhinolaryngol. 2006;70:1299–1306. doi: 10.1016/j.ijporl.2005.12.004. [DOI] [PubMed] [Google Scholar]
- Peterson A, Shallop J, Driscoll C, Breneman A, Babb J, Stoeckel R, Fabry L. Outcomes of cochlear implantation in children with auditory neuropathy. J Am Acad Audiol. 2003;14:188–201. [PubMed] [Google Scholar]
- Qiu C, Salvi R, Ding D, Burkard R. Inner hair cell loss leads to enhanced response amplitudes in auditory cortex of unanesthetized chinchillas: evidence for increased system gain. Hear Res. 2000;139:153–71. doi: 10.1016/s0378-5955(99)00171-9. [DOI] [PubMed] [Google Scholar]
- Salvi RJ, Ding D, Wang J, Jiang HY. A review of the effects of selective inner hair cell lesions on distortion product otoacoustic emissions, cochlear function and auditory evoked potentials. Noise Health. 2000;2:9–26. [PubMed] [Google Scholar]
- Salvi RJ, Wang J, Ding D, Stecker N, Arnold S. Auditory deprivation of the central auditory system resulting from selective inner hair cell loss: animal model of auditory neuropathy. Scand Audiol Suppl. 1999;51:1–12. [PubMed] [Google Scholar]
- Santarelli R, Arslan E. Electrocochleography in auditory neuropathy. Hear Res. 2002;170:32–47. doi: 10.1016/s0378-5955(02)00450-1. [DOI] [PubMed] [Google Scholar]
- Santarelli R, Starr A, Michalewski HJ, Arslan E. Neural and receptor cochlear potentials obtained by trnastympanic electrocochleography in auditory neuropathy. Clinical Neurophysiol. 2008;119:1028–41. doi: 10.1016/j.clinph.2008.01.018. [DOI] [PubMed] [Google Scholar]
- Shallop JK, Peterson A, Facer GW, Fabry LB, Driscoll CL. Cochlear implants in five cases of auditory neuropathy: postoperative findings and progress. Laryngoscope. 2001;111:555–62. doi: 10.1097/00005537-200104000-00001. [DOI] [PubMed] [Google Scholar]
- Sheykholeslami K, Kaga K, Kaga M. An isolated and sporadic auditory neuropathy (auditory nerve disease): report of five patients. J Laryngol Otol. 2001;115:530–34. doi: 10.1258/0022215011908423. [DOI] [PubMed] [Google Scholar]
- Sininger Y. Identification of auditory neuropathy in infants and children. Semin Hear. 2002;23:193–200. [Google Scholar]
- Sininger Y, Oba S. Patients with auditory neuropathy: Who are they and what can they hear? In: Sininger Y, Starr A, editors. Auditory neuropathy: A new perspective on hearing disorders. Singular/Thomson Learning; San Diego: 2001. pp. 15–35. [Google Scholar]
- Snyder D, Salvi R. A novel chinchilla restraint device. Lab Anim. 1994;23:42–4. [Google Scholar]
- Starr A. The neurology of auditory neuropathy. In: Sininger Y, Starr A, editors. Auditory neuropathy: A new perspective on hearing disorders. Singular/Thomson Learning; San Diego: 2001. pp. 37–49. [Google Scholar]
- Starr A, Sininger YS, Pratt H. The varieties of auditory neuropathy. J Basic Clin Physiol Pharmacol. 2000;11:215–30. doi: 10.1515/jbcpp.2000.11.3.215. [DOI] [PubMed] [Google Scholar]
- Starr A, Sininger Y, Nguyen T, Michalewski HJ, Oba S, Abdala C. Cochlear receptor (microphonic and summating potentials, otoacoustic emissions) and auditory pathway (auditory brain stem potentials) activity in auditory neuropathy. Ear Hear. 2001b;22:91–9. doi: 10.1097/00003446-200104000-00002. [DOI] [PubMed] [Google Scholar]
- Takeno S, Harrison RV, Ibrahim D, Wake M, Mount RJ. Cochlear function after selective inner hair cell degeneration induced by carboplatin. Hear Res. 1994;75:93–102. doi: 10.1016/0378-5955(94)90060-4. [DOI] [PubMed] [Google Scholar]
- Trautwein P, Hofstetter P, Wang J, Salvi R, Nostrant A. Selective inner hair cell loss does not alter distortion product otoacoustic emissions. Hear Res. 1996;96:71–82. doi: 10.1016/0378-5955(96)00040-8. [DOI] [PubMed] [Google Scholar]
- Wake M, Takeno S, Ibrahim D, Harrison R. Selective inner hair cell ototoxicity induced by carboplatin. Laryngoscope. 1994;104:488–93. doi: 10.1288/00005537-199404000-00016. [DOI] [PubMed] [Google Scholar]
- Wake M, Anderson J, Takeno S, Mount RJ, Harrison RV. Otoacoustic emission amplification after inner hair cell damage. Acta Otolaryngol. 1996;116:374–81. doi: 10.3109/00016489609137860. [DOI] [PubMed] [Google Scholar]
- Wang J, Ding D, Salvi RJ. Carboplatin-induced early cochlear lesion in chinchillas. Hear Res. 2003;181:65–72. doi: 10.1016/s0378-5955(03)00176-x. [DOI] [PubMed] [Google Scholar]
- Wang J, Powers NL, Hofstetter P, Trautwein P, Ding D, Salvi R. Effects of selective inner hair cell loss on auditory nerve fiber threshold, tuning and spontaneous and driven discharge rate. Hear Res. 1997;107:67–82. doi: 10.1016/s0378-5955(97)00020-8. [DOI] [PubMed] [Google Scholar]
- Zeng FG, Kong YY, Michalewski HJ, Starr A. Perceptual consequences of disrupted auditory nerve activity. J Neurophysiol. 2005;93:3050–63. doi: 10.1152/jn.00985.2004. [DOI] [PubMed] [Google Scholar]
- Zeng FG, Oba S, Garde S, Sininger Y, Starr A. Temporal and speech processing deficits in auditory neuropathy. Neuroreport. 1999;10:3429–35. doi: 10.1097/00001756-199911080-00031. [DOI] [PubMed] [Google Scholar]
- Zheng XiY, Ding DL, McFadden SL, Henderson D. Evidence that inner hair cells are the major source of cochlear summating potential. Hear Res. 1997:76–88. doi: 10.1016/s0378-5955(97)00127-5. [DOI] [PubMed] [Google Scholar]