

Abstract
The melanocortin 4 receptor (MC4R) is a G-protein-coupled receptor (GPCR) and a key molecule in the regulation of energy homeostasis. At least 159 substitutions in the coding region of human MC4R (hMC4R) have been described experimentally; over 80 of those occur naturally, and many have been implicated in obesity. However, assessment of the presumably functionally essential residues remains incomplete. Here we have performed a complete in silico mutagenesis analysis to assess the functional essentiality of all possible nonnative point mutants in the entire hMC4R protein (332 residues). We applied SNAP, which is a method for quantifying functional consequences of single amino acid (AA) substitutions, to calculate the effects of all possible substitutions at each position in the hMC4R AA sequence. We compiled a mutability score that reflects the degree to which a particular residue is likely to be functionally important. We performed the same experiment for a paralogue human melanocortin receptor (hMC1R) and a mouse orthologue (mMC4R) in order to compare computational evaluations of highly related sequences. Three results are most salient: 1) our predictions largely agree with the available experimental annotations; 2) this analysis identified several AAs that are likely to be functionally critical, but have not yet been studied experimentally; and 3) the differential analysis of the receptors implicates a number of residues as specifically important to MC4Rs vs. other GPCRs, such as hMC1R.—Bromberg, Y., Overton, J., Vaisse, C., Leibel, R. L., Rost, B. In silico mutagenesis: a case study of the melanocortin 4 receptor.
Keywords: MC4R, MC1R, SNAP, active functional site, obesity, diabetes
The melanocortin 4 receptor (MC4R), one of 5 melanocortin receptors (MCRs) in the G-protein coupled receptor (GPCR) superfamily, is expressed primarily in the hypothalamus, particularly in the paraventricular nucleus and the lateral hypothalamus, and at high levels in the brain stem, notably the nucleus of the solitary tract (1). Endogenous melanocortin ligands (e.g., α-MSH) activate the MCRs by stimulating the formation of cAMP and the subsequent activation of protein kinase A. Additional cAMP-dependent and -independent signaling pathways involving MAPK (2), Ca2+ mobilization (3), and protein kinase C (4) have also been reported. In vitro, the MC4R has been shown to be both constitutively active (5) and physically recycled (6); the receptor is internalized from the plasma membrane into an intracellular compartment and subsequently transported back to the cell surface, where the molecule is reused for signaling.
The MC4R, and two other members of the melanocortin family (the MC1R and MC3R), are unique in that they are the only members of the GPCR family known to have endogenous antagonists (“inverse agonists”; see refs. 7, 8): Agouti signaling protein (ASIP) and Agouti-related protein (AgRP). ASIP, a peptide normally expressed in skin cells, antagonizes the melanocortin 1 receptor (MC1R) in the hair follicle, causing pigment-type switching from eumelanin (black) to pheomelanin (yellow) (9). When overexpressed ectopically, ASIP blocks MC4R and MC1R signaling in the brain and the hair follicle (10), respectively. AgRP, released by neurons in the arcuate nucleus of the hypothalamus, is a natural antagonist of the MC3R and MC4R (11). The molecular mechanisms by which ASIP and AgRP act as inverse agonists remain unclear (8).
MC4R is involved in weight regulation: mice homozygous for a null allele of the Mc4r are obese because of hyperphagia and decreased energy expenditure (12). In humans, MC4R mutations constitute the most commonly identified cause of monogenic obesity (∼2.5–3% of all cases of severe human obesity, BMI>40; see refs. 13, 14). Over 80 nonsynonymous mutations in the MC4R coding sequence have been reported, yet analyses of their consequences for protein structure and function remain limited. Dominantly inherited nonsense and missense mutations that completely disable MC4R represent the most common apparent molecular mechanism for obesity; however, recessive modes of inheritance are also encountered (15).
Experimental studies have elucidated some of the structure-function aspects of MC4R and found that all of its conserved residues are involved in agonist binding and signaling (16). In most cases, however, the exact residues related to any specific function remain unknown. Single amino acid (AA) mutations associated with obesity most often result in intracellular retention of the mutant receptor, followed by receptors with impaired ligand-binding capacity and receptors with defective signal transduction (17). In addition, obese humans segregating for MC4R alleles with nonsynonymous AA variants that appear to function normally in vitro have been reported (17).
A comprehensive mutagenesis survey that substitutes all MC4R residues by each of the 19 nonnative AAs and tests the functional consequences is not experimentally feasible. In silico modeling may provide insights into the relationships among sequence, structure, and function. The most reliable technique for such modeling and structure prediction is comparative modeling (18). This technique finds a protein with a structure (template) that is similar to the protein to be modeled (target) and then copies the coordinates of the backbone of the template onto the target (19, 20); unaligned residues typically remain without a model. The two proteins with known structure that are most sequence-similar to MC4R are rhodopsin (RHO; 24% sequence ID in 143 aligned residues; ref. 21) and the β2-andrenegic receptor (B2AR; 24% sequence ID in 307 aligned residues; ref. 22). Structural models for MC4R (and other GPCRs) have been primarily based on the structure of RHO. However, because of the substantial sequence differences between the two receptors, RHO-based MC4R models are not sufficiently accurate to provide reliable per-residue data regarding function. Models based on B2AR might be more useful, because this receptor is more similar in sequence to MC4R. However, there are no comprehensive and convincing B2AR-based structural models for MC4R available.
To reduce the level of detail, rather than mapping residues onto 3-dimensional (3-D) coordinates, we limited our analyses to a 1-dimensional perspective including information regarding the location of putative transmembrane helices (Fig. 1). Even at this reduced resolution, however, minor differences remain in the details of protein structure: for example, the locations of the membrane helices are not exact but are resolved to a few residues. This minor uncertainty also affects the accuracy of our predictions of the regions in the extracellular and cytoplasmic regions.
Figure 1.

Important hMC4R residues. One-dimensional structure of the hMC4R receptor (adapted from Govaerts et al.; ref. 42). At present, no reliable 3-D structure at higher level of detail is available. Circles represent constituent AAs (1-letter alphabet). Shading reflects nominal importance of residues according to SNAPMAT score (light gray circles represent scores above default threshold D>0; dark gray, D>10; black, D>20). Green outlines reflect potentially active residues in the sequence (as annotated by Swiss-Prot) and red shows experimentally determined active residues (Supplemental Table 9). Experimentally determined activity is reported as assigned by authors of corresponding publications; i.e., only those residues are highlighted in red that are reported by authors of referenced papers to be directly involved in protein function (ligand binding, signaling, etc.). Arrows indicate 12 novel high-confidence predictions of functionally important sites not reported in the literature (see Discussion).
For each human MC4R (hMC4R) AA residue, we used SNAP (Supplemental Fig. 1 and ref. 23) to predict the functional consequences of exchanging the native AA with each of the 19 other naturally occurring AAs. SNAP is a neural network-based method that uses evolutionary conservation and structure/function relationships to predict the functional consequences of AA substitutions. SNAP was optimized on the Protein Mutant Database (PMD) (24). Cross-validation tests indicate that SNAP identifies >80% of the nonneutral variants with 77% accuracy and >76% of the neutral mutations with 80% accuracy at its default threshold (hMC4R was not used in the development of SNAP). SNAP performed only slightly worse for the subset of Swiss-Prot (25) annotated transmembrane proteins in the test set, identifying 74% of the nonneutral variants at 73% accuracy and 72% of the neutral mutations with 74% accuracy. The advantage of SNAP is that it can be applied to serial analyses of the effects of substitution at a specific position of all 19 nonnative AAs. This paper reports the first attempt to perform such an analysis of a membrane protein.
Our comprehensive in silico mutagenesis predicted functionally essential residues in hMC4R, mouse MC4R (mMC4R), and human MC1R (hMC1R) without relying on experimental information, and without knowledge of the specific function in which a given residue is involved. To assess the sensitivity and specificity of our computational approach we compared experimentally determined functional consequences of mutations to our predictions. We also examined the specific prediction differences and similarities between the receptors providing some insight into their molecular physiology.
MATERIALS AND METHODS
Data set
Melanocortin receptor sequences were taken from Swiss-Prot: MC4R_HUMAN and MC4R_MOUSE are the Swiss-Prot identifiers for the human (hMC4R) and mouse (mMC4R) melanocortin-4 receptors (332 residues), respectively. MSHR_HUMAN is the entry for the human melanocortin-1 receptor (hMC1R; 317 residues). We studied 159 substitutions in hMC4R for which experimental data were available (Supplemental Table 1). In addition, to compare predictions of functionally important residues that correspond in sequence between hMC1R and hMC4R and human and mouse MC4Rs, we aligned each pair of receptors using ClustalW (26) (Fig. 2).
Figure 2.

Alignment between hMC4R and hMC1R. We aligned hMC4R and hMC1R by ClustalW (Thompson et al.; ref. 26) to compare functional classifications of their constituent AAs. Yellow highlighting demarcates structural regions (membrane, extracellular, cytoplasmic) shown in Fig. 1. Color of residues reflects SNAPMAT score cutoffs: red, cutoff > 20; blue, cutoff > 10; green, cutoff > 0. Higher scores indicate higher specificity but lower sensitivity (Eq. 1). Residues that differ in functional class assignment (important vs. not important) between the 2 receptors are outlined by dashes.
Computing residue mutability scores
SNAP predicts the functional consequences of single AA substitutions. As input, SNAP uses sequence-derived features and biophysical properties of both mutant and wild-type (WT) residues (Supplemental Fig. 1). The method is described in detail elsewhere (23). For the three melanocortin receptors we obtained SNAP predictions for all 19 nonnative substitutions at each sequence position. We considered three different ways of computing residue mutability: 1) using only SNAP scores of to-Ala substitutions, 2) computing the arithmetic mean of all SNAP scores for the 19 nonnative substitutions at each position, and 3) computing the arithmetic mean of the SNAP scores for substitutions of interchangeable residues at each position. Interchangeability of the residues was defined by the substitutions scoring at least 0 in the PHAT (27) (transmembrane residue) and BLOSUM62 (28) (all other) substitution matrices (Supplemental Table 4).
Threshold for predicted effects
The neural networks that comprise SNAP have two output units: one is optimized to give high values for neutral variants, and the other is optimized to be high for nonneutral variants. Computing the difference between these two, we recorded the nominal effect of each substitution as a single score D, with D ranging from −100 (strong prediction for neutral) to +100 (strong prediction for nonneutral). By design, a threshold D = 0 distinguishes neutral (D≤0) from nonneutral (D>0) variants. This is the default in the sense that the neural networks are trained to solve the problem at this threshold; D = 0 is also the threshold that users of the SNAP server (29) see by default. D can be moved higher or lower to generate more or less reliable predictions. For example, considering as nonneutral only mutations with D > 20, we increase the accuracy (see Eq. 1 below) of predicting nonneutral effects at the expense of the coverage (i.e., more nonneutrals predicted correctly, but fewer predicted overall).
We analyzed different thresholds for defining the neutrality of mutations (e.g., at SNAP score cutoff 0, 10, and 20). As expected from the results of the original SNAP publication (23), higher thresholds result in greater specificity (“accuracy”) in identifying functionally nonneutral substitutions while potentially reducing the total number of nonneutrals identified (lower sensitivity/“coverage”; Eq. 1).
Evaluating predictions
We organized previously reported experimental observations on hMC4R AA variants into 4 groups according to their reported effects on function (Supplemental Table 1): agonist binding (and signaling response to agonists), antagonist binding, constitutive activity, and cell surface expression. We computed specificity and sensitivity (where TP=true positives, nonneutral predictions on functionally disruptive mutations; FP=false positives, nonneutral predictions of functionally neutral mutations; and FN=false negatives, neutral predictions of functionally disruptive mutations):
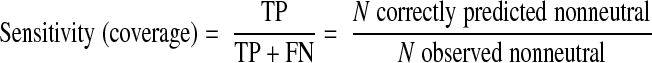 |
1 |
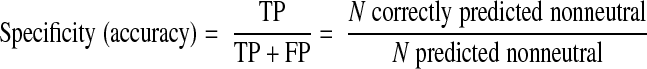 |
We also compiled functional annotations for the entire set of variants in order to compute the total number of functions nominally affected by each mutation (function count: 0 if no study reports an effect, n=1, … 4 if function was reported as affected in N of the 4 groups). If multiple reports on a particular variation differed, the mutation was classified as nonneutral with respect to that function: that is, a single report of a functionally disruptive mutation was sufficient to classify the variant as nonneutral for the affected function regardless of other existing annotations reporting neutrality of the variant. In addition, we noted whether the given function count was obtained on the basis of a single or multiple reports. We also captured performance in predicting the effects of hMC4R mutations by computing Q2, the 2-state per-residue accuracy defined by
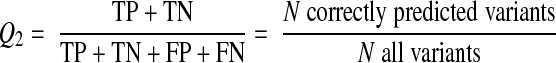 |
2 |
RESULTS
Standard SNAP performed for hMC4R as expected
hMC4R data were not used to develop SNAP; it was, therefore, particularly important that our hMC4R results be consistent with published performance estimates for other molecules (23). SNAP was trained on a collection of variants in different types of proteins (>80,000 mutations in >6000 proteins); transmembrane proteins were less common than water-soluble globular proteins. However, many of the mutations that affect the function of hMC4R in vitro map to the transmembrane regions. Integral transmembrane proteins evolve under structural constraints that differ substantially from those of globular water-soluble proteins (30, 31). The analyses described here constituted an explicit vetting of SNAP on a highly annotated transmembrane protein.
SNAP correctly identified many functionally important substitutions in hMC4R (Supplemental Table 1) at an overall 2-state accuracy of 73% (Q2, Eq. 2; specificity= 82%, sensitivity=82%, Eq. 1). This Q2 was identical to that expected of the SNAP performance for transmembrane proteins, but slightly lower than expected for overall SNAP (79%; ref. 23). This reduction in accuracy was largely due to false positives (21 of 159), that is, variants predicted to be functionally disruptive, although no experiment confirms this prediction. Of these 21, 13 (62%) had been tested experimentally only once, and 18 (86%) were not exhaustively tested for effects on all functions of hMC4R. Thus, although some of these false positives are likely mistakes, others might be correct predictions that await experimental verification.
Predictions for variants reported in multiple studies were better (Q2=80%; specificity=92%, sensitivity=86%) than predictions for residues annotated by a single experiment (Q2=67%; specificity=71%, sensitivity=76%). SNAP identified mutations altering agonist binding and expression better than mutations affecting antagonist binding and constitutive activity (Supplemental Table 2). Residues important for several aspects of function (i.e., for function count≥2, ≥3, =4) were predicted at higher accuracies (Q2=84, 88, and 100%, respectively). Performance was best at the default SNAP score cutoff of D = 0.
Comprehensive in silico mutagenesis performs well for hMC4R
We used SNAP to perform a comprehensive in silico mutagenesis by systematically mutating all residues. Our first approach of changing all residues to alanine (all-to-A; SNAPALA; Fig. 3A) predicted 65% of all hMC4R residues as likely to affect function (at D=0; Supplemental Table 3). Our second approach of all-to-all (SNAPMEAN; Fig. 3B) annotated 81% as functionally important (Supplemental Table 3). While these numbers may appear high, note that the lipid bilayer alone imposes strong constraints: about half of all hMC4R residues are embedded in the membrane as helices and are largely hydrophobic. Mutation of any of these to a nonhydrophobic residue is very likely to affect structure and disrupt function.
Figure 3.

Mutability of hMC4R residues compiled from various measures. Mutability scores for each residue in the hMC4R sequence can be assessed in a number of ways: A) SNAPALA: SNAP scores of X-to-A (alanine) transversion; B) SNAPMEAN: arithmetic mean SNAP score of 19 possible AA substitutions at each position; C) SNAPMAT: mean SNAP score over mutations that are generally neutral in proteins as annotated by the PHAT and BLOSUM62 matrices (Supplemental Table 4). For actual prediction scores, see Supplemental Table 10.
Conservation of a sequence position’s biophysical properties (i.e., hydrophobicity, size, or charge) in alignment is a very good indicator of its value in the maintenance of proper protein structure or function. However, the lack of diversity at a given position beyond the retention of basic residue characteristics (i.e., only a single residue is found in all aligned sequences) is likely an indicator of another level of importance such as direct participation in function (e.g., binding residue). We sought to extend these structure-function distinctions by identifying all mutations predicted to affect function even if the transitions were likely structurally benign. We implemented this strategy in our third approach, namely, the SNAPMAT score (Fig. 3C; Materials and Methods; Supplemental Table 4) that is most sensitive to mutations disruptive to function but not structure. The SNAPMAT score identified 58% of residues as important for function (Fig. 1 and Supplemental Table 3).
Although SNAPMAT selected fewer residues than SNAPALA and SNAPMEAN, it picked up all but one of the experimentally documented important residues. As the overall number of residues predicted to be functional still appeared intuitively high, we applied a more stringent threshold (D=10 or 20; Fig. 1). This approach predicted at least 118 residues to be functionally important (∼36% at D=20). When we focused only on these 118 (Fig 1, black circles) we recapitulated many of the known functionally important residues (15/18), while implicating a number of potentially crucial variants that have not been studied experimentally.
The fractions of residues localized to the three different cellular locations (membrane, extracellular, intracellular) differed considerably from the corresponding fractions of residues predicted to be functionally important. There were disproportionately more functionally important residues in the transmembrane region than in the other two regions (Supplemental Tables 5 and 6; P=0.051). About 57% of predicted functionally important hMC4R residues have been annotated as transmembrane, while only ∼50% of all hMC4R residues are in membrane helices. This overrepresentation of important residues in the membrane regions was confirmed at higher thresholds (SNAPMAT D>20; P=0.015).
Importance of residues identical between hMC1R and hMC4R predicted differentially
We compared the SNAP predictions of residue essentiality between hMC4R and hMC1R. These two proteins share ∼45% sequence identity over a 335-residue region (depending on alignment; Fig. 2). First, we established that SNAP’s performance for the mutations in the aligned region was similar to that for the entire hMC4R (Q2=76% for aligned regions; 73% for all mutations).
At this level of sequence similarity, two proteins typically have similar 3-D structures (32), while their functions may differ considerably (33). Any attempt to predict the 3-D structures by comparative modeling would have to use the same template for both receptors, because the two possible templates (B2AR and RHO) are approximately equidistant from both. By default, all identical residues between hMC1R and hMC4R would be modeled the same. This issue would remain even if we had a 3D structure for one of the proteins and built the model of the other from that template. In contrast, our SNAP-based approach predicted different classes of importance to function for 23% of (non-gap-aligned) residues. Forty percent of these were predicted to be important to their respective protein functions, with SNAPMAT > 10. These residues are potentially the most critical in defining the structural basis for functional differences between hMC4R and hMC1R.
Overall, SNAPMAT predicted slightly more functionally important residues in hMC4R than in hMC1R: 35 residues were predicted as important (at D=0) for hMC1R but not for hMC4R (2 gap-aligned; mean SNAPMAT=12); 42 residues were predicted to be important for hMC4R only (3 gap-aligned; Table 1 and Fig. 2; mean SNAPMAT=9).
TABLE 1.
Comparison of predictions of residue essentiality for protein function
| Condition | Aligned residues | Predicted alike | Predicted different
|
||||
|---|---|---|---|---|---|---|---|
| hMC4R
|
hMC1R
|
||||||
| Important | Neutral | Important | Neutral | Important | Neutral | ||
| Identical | 152 | 114 | 24 | 6 | 8 | 8 | 6 |
| Different | 162 | 38 | 66 | 33 | 25 | 25 | 33 |
| Gapa | 21 | 3 | 15 | 2 | 1 | ||
| Total | 335 | 152 | 90 | 42 | 48 | 35 | 40 |
|
|
|
|
|
hMC4R
|
mMC4R
|
||
| Identical | 311 | 168 | 118 | 19 | 6 | 6 | 19 |
| Different | 21 | 2 | 12 | 5 | 2 | 2 | 5 |
| Total | 332 | 170 | 130 | 24 | 8 | 8 | 24 |
Table shows correspondence of predictions of function essentiality of residues in 2 proteins. A particular pair of aligned residues was predicted either to have the same functional effect (predicted alike) or to have a different effect on function (predicted different). In both cases, we show the number of like/different predictions that classify receptor residues as functionally important or not (neutral). Functional annotation was based on the SNAPMAT score (D=0). Here, 314 residues aligned between hMC4R and hMC1R. For 21 positions, a residue in one protein aligned to a gap in the other (18 gaps in hMC1R and 3 in hMC4R). For instance, 114 aligned residues are identical between the 2 receptors and are predicted to be functionally important. For another 42 residues, importance is specific to hMC4R; of these, 6 are AAs identical to hMC1R, 33 differ, and 3 are gap-aligned (visual representation of these data in Fig. 2). Similarly, 332 residues aligned between MC4R in humans and mice. Here, 168 aligned residues are identical between the 2 receptors and are predicted to be functionally important. For another 24 residues, importance is specific to human; of these, 19 are AAs identical to mouse and 5 differ. aGaps mark positions for which one of the 2 sequences has an insertion, i.e., has no correspondence in the other protein.
Importance of residues identical between mouse and human MC4Rs predicted similarly but not identically
Finally, we compared the SNAP predictions between MC4R orthologues in mice and humans (Table 1). hMC4R and mMC4R are 94% identical in sequence over an ungapped alignment of 332 residues. As expected, >90% of the residues in the two proteins were classified into the same importance class. Of the remaining ∼10% (32 residues), there were more (24 in humans vs. 8 in mice) residues classified as important to function of the human sequence. However, for the majority of these the reliability of prediction was low (i.e., only 6 of the 32 were predicted important to their respective protein functions, with SNAPMAT>10). For one of these (classified as important for humans), the annotation of neutrality for the corresponding mouse residue was unreliable (SNAPMAT>−10). These results suggest identical function for the two proteins, albeit with some (5 residue) differences (e.g., slightly different levels of signaling or ligand affinity).
DISCUSSION
SNAP predictions provide information beyond known experimental annotations for hMC4R
Overall, for hMC4R, SNAP differentiated well between the functionally disruptive variants and neutral sites (∼86% of substitutions considered nonneutral by two or more experimental studies were correctly predicted). Predictions of nonneutral variations were better when multiple rather than single experiments reported an effect (multiple Q2=80%; single Q2=67%; Supplemental Table 7). Several explanations are plausible. First, stronger effects should be easier to observe and to predict. The strength of the predictions is proportional to that of the related experimental effect (23, 34), so variants disrupting function on several levels were more accurately predicted (function count; see Results). Second, single experiments may miss relevant phenotypes (no effect on αMSH binding does not imply no effect on AgRP binding). Third, mutations for which numerous experimental annotations agree are more likely to be in the “unquestionable” group of functional disruptions.
Although unambiguous experimental status of computationally implicated variants is critical to vetting analytic strategies, ambiguous (or absent) results for variants with strong computational “signal” provide candidates for future experimental studies. For instance, the T112M variant has been reported to slightly increase agonist binding while decreasing hMC4R cell surface expression (35). In contrast, another study found that cell surface expression and binding affinity of this variant resembled WT (36). Association studies have not explicitly linked this variant to obesity in humans, so T112M is considered to be a functionally benign polymorphism (37). SNAP predicted this variant to be functionally disruptive with high confidence (SNAP=37). Computational analysis in such instances can point to variants warranting additional or de novo experimental vetting.
Another example of the utility of computational analysis in deconvoluting ambiguous results is the analysis of V103I (suggested by association studies to be protective against obesity; see ref. 38). In vitro, the variant has agonist-binding affinities that are similar to the WT, but, according to one study, lower than WT affinity for the antagonist, AGRP (39). Our analysis suggests that the variant is likely neutral, an inference supported by a number of other in vitro studies (Supplemental Table 1). In contrast to in vitro experiments that assess particular aspects of function, SNAP computational analyses are designed to capture multiple aspects in a single assessment. Our prediction of functional neutrality for V103I might justify reducing its priority for further in vitro studies. A similar approach could be taken to stratifying other variants for more extensive studies, for example, variants that are associated with obesity but do not seem to affect receptor function in vitro (e.g., P48S and D37V, see Supplemental Table 1; SNAP predictions: neutral and nonneutral, respectively). In these scenarios, we believe that SNAP can generate a rank order of mutations for further study.
Additional last-minute data confirm estimates
Within the past month, a new set of hMC4R variants has been experimentally tested (40, 41). SNAP correctly predicted the effects of 14 of the 18 novel variants (Q2=78%; similar to performance on experimental data confirmed by several groups). SNAP predicted no variant incorrectly as neutral. The 4 incorrect predictions were nonneutral classifications for variants experimentally determined not to have any effect on agonist binding. Three of these 4 were found in obese carriers but not in the controls (41). Since only one molecular function of these receptor variants was evaluated, some of our 4 “incorrect” predictions might turn out to be right. One of the correctly assigned variants (P299S; assigned to the severe functional disruption class; ref. 41) affected a position that we predicted to be extremely important for receptor function (see below). Since we did not have access to these data while compiling the estimates presented in this work, the new data strongly support the validity of our analysis.
Computational annotation of important hMC4R regions agrees with experimental evidence
The N terminus (residues 1–41) has been implicated experimentally as crucial for the constitutive activity of the receptor (5, 42); many of our predictions of important sites for which there are no experimental data fall into this region (15 residues of 41 total in the N terminus). Extracellular loops 2 and 3 (Fig. 1) have been implicated in AGRP binding (43); our assessment found them to be very sensitive to substitution (Fig. 1). Also captured was the importance of the cytoplasmic loops 1 and 2 (implicated in G-protein coupling; ref. 44) and loop 3 (involved in maintaining constitutive activity; ref. 45). In each of the transmembrane helices, involved in α-MSH binding (16), AGRP binding (46), and constitutive activity (47), at least 38% of the residues were computationally classified as functionally important. Similarly, the C-terminal region, required for proper receptor cell-surface localization (48) and MAPK signaling (2), contained several predicted important residues. Not surprisingly, this high-level sequence review covers the entire length of the protein without any extended region that is not implicated. Note that mutations related to obesity are also found in all regions of hMC4R (Supplemental Table 1).
One aspect of our analysis is puzzling: It is commonly assumed that most residues deeply buried in transmembrane regions are unlikely to be very sensitive to hydrophobicity-maintaining substitutions. With SNAPMAT we explicitly designed a method that underweighs the effects of such structure-disrupting mutations. Nevertheless, this method predicted a higher proportion of functionally important residues in the membrane than outside (Supplemental Table 5). This finding was confirmed by the fact that many of the residues experimentally characterized as crucial for function appear to be buried in the membrane (Fig. 1).
Establishing differences between two similar melanocortin receptors: hMC1R vs. hMC4R
To what extent can the specificity of predictions regarding structure-function relationships be improved by comparisons of related proteins? We addressed this question by comparing hMC4R and hMC1R. SNAP relies heavily on evolutionary differences among protein families, so inputs are inherently similar for similar proteins. Indeed, we predicted the same functional effects for 242 of the 314 (77%) residues aligned between hMC4R and hMC1R (at D=0). Residues in the hMC4R-hMC1R alignment can be split into 5 classes (Table 1 and Fig. 2): The first class is the 21 gap-aligned residues mentioned in the Results. The other 4 are the combinations of 2 × 2 possibilities: the aligned AAs can be identical or not, and the predictions of functional importance can be identical or not.
Identical residue, identical prediction
Here 138 of 152 aligned residues (91%) that are identical in the two sequences were predicted to be in the same functional class (either both neutral or both nonneutral). Of these, functionally important AAs are likely to perform the same function in both proteins. For example, the DRY motif (hMC4R residues 146–148, hMC1R:141–143) suppresses receptor activity in the absence of ligands (49) and is conserved in most GPCRs. In melanocortin receptors, mutations in this motif render the proteins unstable, reduce α-MSH binding, and produce an increase in constitutive activity (50). The residues of the DRY motif are predicted functionally important in both receptors.
Identical residue, different prediction
The remaining 14 of 152 aligned identical residues were not predicted to have identical functional consequences. We focus our discussion on those with the highest difference in the predicted functional effect (>10). Two such residues were predicted to be important for hMC4R but not for hMC1R (L59 and L265; aligned to hMC1R:L53 and hMC1R:L261), and 6 were predicted as important for hMC1R but not for hMC4R (R8, I120, L202, A222, L243, T244; aligned to hMC4R:R7, L125, L207, A227, L247, and T248). Experimental data confirmed our prediction regarding the hMC4R:L265A substitution, which reduces antagonist binding (Supplemental Table 1); the corresponding substitution in hMC1R:L261A does not (51). Conversely, for one of the residues, hMC4R:R7, our prediction was incorrect: this residue is important for hMC4R constitutive activity (5). Other experimental evidence was less conclusive: e.g., AAs hMC1R:L120/hMC4R:L125 are not directly involved in protein function, but are close to hMC1R:D121/hMC4R:D126 that are involved in ligand-receptor interactions (52, 53).
Different AA, identical prediction
We predicted 106 aligned nonidentical residues to be in the same functional class (38 were functionally important). To analyze these 38, we looked at predictions of functional consequences of point-mutating hMC4R residues into hMC1R and vice versa. This procedure suggested that some of these residues may have different functions in the two proteins: e.g., mutating 16 of these in hMC4R, and 22 in hMC1R, by exchanging them for corresponding residues in the other receptor, was predicted to affect function. These fell into 3 categories (Supplemental Table 8): function disruption of 1) hMC1R on INSERTION of the hMC4R residue, 2) of hMC4R on INSERTION of the hMC1R residue, and 3) both receptors on SWAPPING the AAs between the two. Several of these predictions are supported experimentally, e.g., hMC1R:C35 is involved in ensuring proper folding and membrane insertion (54); swapping this AA to the corresponding hMC4R tyrosine is likely to disrupt protein function. Moreover, the C35Y mutation in hMC1R has been associated with red hair color (55), suggesting gain of function. In hMC4R, even a seemingly (biophysically) nondisruptive substitution of V95I leads to a loss of function (Supplemental Table 1); however, in hMC1R the G89V mutation in the corresponding position is considered a functionally neutral polymorphism (56). These findings suggest that aligned residues of the same functional class that differ between proteins and are not interchangeable are involved in distinct functions of the two receptors.
Different AA, different prediction
Nonidentical residues aligned at corresponding positions and predicted to differ in their functional importance are promising candidates for experimental follow-up studies of the receptor-specific functions. Fifty-six residues fell into this category; 33 were classified as important exclusively for hMC4R and 25 as exclusively important for hMC1R. Nine of the hMC4R exclusives (27% of 33) were in the N terminus, while only 3 of the MC1R exclusives were N terminal. This result is consistent with functional studies, suggesting that the N-terminal region of hMC4R is functionally important (5, 42). In fact, almost all high-scoring exclusives of hMC4R were confined to the N-terminal region. In contrast, the high-scoring exclusives for hMC1R were spread across the entire protein sequence (Fig. 1).
Establishing cross-species similarity in protein function: human vs. mouse MC4R
Although the orthologous proteins in mice and humans are generally assumed to carry the same function, SNAP predictions of importance differed significantly for 5 of the residues in the two sequences: 2 were assigned to be important for mMC4R (m:H7, L229 aligned to h:R7, L229) and 3 for hMC4R (h:S36, T112, C319 aligned to m:P36, T112, F319). For one of these (h:R7), we have previously noted that the SNAP prediction of neutrality is incorrect (the residue is important to protein constitutive activity). On the other hand, h:T112 has been shown in mutagenesis experiments (Supplemental Table 1) to be important to various aspects of function in hMC4R, consistent with the SNAP prediction. Similarly, the h:S36Y mutant (present in an obese individual, but not in the control group) has been shown to have no effect on agonist binding but slightly impaired signaling (57). Overall, although there are anecdotal reports of some differences in MC4R function among organisms in certain assays, we were unable to find any experimental data either confirming or refuting this claim. Thus, we find that a true evaluation of the accuracy of the minor functional distinction is not possible.
Twelve new predictions to guide future experiments
Our hMC4R in silico mutagenesis yielded a sizeable set of residues predicted to be functionally important. To identify the most reliable predictions, we used a very stringent cutoff of SNAPMAT ≥ 50 for hMC4R; at this level we identified 22 residues (Table 2; for comparison purposes, we also include the most reliable neutrality predictions: 5 residues with SNAPMAT≤−50). Interestingly, while there was modest correlation between SNAP predictions and evolutionary and biophysical features of the affected positions, the relationship was not clearcut. For instance, while many of the nonneutral assignments highlighted evolutionarily conserved residues, there were 6 (27%) predictions affecting nonconserved positions (including 3 confirmed by experimental data). There were only 2 extracellular positions in our set, and both were predicted neutral. However, the rest of the highlighted residues (both important and neutral) were fairly evenly distributed in the membrane and the cytoplasmic regions of the sequence. All residues predicted to be neutral were uncharged, but so were the majority of functionally important residues. Positions assigned neutral status were less likely to be buried, and 3 of the neutral positions (60%) were predicted to be more flexible (Bn>0; Table 2) in comparison to only 2 of the important ones (9%). All of these correlations are consistent with the biological reasoning behind annotating positions based on functional importance, However, SNAP’s nonlinear logic seemed to be more efficient in detecting the distinctions between functionally neutral and important residues.
TABLE 2.
Characteristics of high-scoring residues of hMC4R
| Res | Status | Effect | SNAPMAT | Loc | Hyd | Chg | Size | Cons | SSt | SA | Bn | |
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| L31 | Predicted | Neu | −58 | Ex | F | U | L | Position | E | B | −0.6 | |
| N62 | Mut: S | Imp | 66 | Tr | L | U | M | Res: 98% | L | B | −0.4 | |
| N72 | Predicted | Imp | 50 | Cy | L | U | M | Position | L | I | 0.3 | |
| P78 | Predicted | Imp | 57 | Cy | U | U | S | Res: 63% | H | B | −0.3 | |
| D90 | Confirmed | Imp | 59 | Tr | L | N | M | Res: 85% | H | B | −0.7 | |
| G98 | Predicted | Neu | −54 | Tr | U | U | S | Position | L | B | 0.3 | |
| T110 | Predicted | Neu | −56 | Ex | U | U | M | None | L | I | 1.7 | |
| D146 | Confirmed | Imp | 58 | Cy | L | N | M | Res: 79% | L | B | −0.6 | |
| R147 | Confirmed | Imp | 64 | Cy | L | P | L | Res: 91% | H | B | −0.5 | |
| Y157 | Mut: S | Imp | 57 | Cy | U | U | L | Res: 61% | L | B | −0.5 | |
| H158 | Predicted | Imp | 54 | Cy | U | P | L | None | E | B | −0.4 | |
| V163 | Predicted | Neu | −56 | Cy | F | U | L | None | H | I | −0.1 | |
| R165 | Mut: A/Q/W | Imp | 63 | Cy | L | P | L | None | H | B | −0.3 | |
| W174 | Predicted | Imp | 55 | Tr | F | U | L | Res: 45% | H | B | −0.9 | |
| F201 | Conflict: A | Imp | 57 | Tr | F | U | L | Res: 56% | H | B | −0.7 | |
| Y212 | Predicted | Imp | 52 | Tr | U | U | L | Position | H | B | −0.6 | |
| H214 | Conflict: A | Imp | 53 | Tr | U | P | L | None | L | B | 0.1 | |
| M215 | Predicted | Imp | 52 | Tr | F | U | L | Position | L | B | −0.5 | |
| A219 | Mut: V | Imp | 54 | Cy | U | U | S | Position | H | B | −0.4 | |
| H222 | Predicted | Imp | 59 | Cy | U | P | L | None | H | B | −0.7 | |
| C257 | Predicted | Imp | 62 | Tr | F | U | L | Res: 62% | H | B | −0.6 | |
| W258 | Mut: A | Imp | 61 | Tr | F | U | L | None | H | B | −0.7 | |
| C271 | Mut: A/Y/R | Imp | 53 | Tr | F | U | L | None | E | B | −0.7 | |
| N294 | Predicted | Imp | 52 | Tr | L | U | M | Position | L | B | −0.4 | |
| P299 | Mut: H | Imp | 52 | Tr | U | U | S | Res: 96% | L | B | −0.4 | |
| Y302 | Predicted | Imp | 60 | Tr | U | U | L | Res: 94% | E | B | −1.1 | |
| G324 | Predicted | Neu | −55 | Cy | U | U | S | None | L | E | 2.2 |
Table shows various features of residues predicted with high reliability as either neutral or important to hMC4R function (SNAPMAT≤−50 or SNAPMAT≥50, respectively). Res column indicates the WT residue and its position in sequence. Status indicates experimental status of the residue: predicted, no experimental data; conflict: X, experimental functional annotation of effect of substitution of WT residue by X is in conflict with prediction (Supplemental Table 1); Mut: X, experimental functional annotation of effect of substitution of WT residue by X confirms prediction (Supplemental Table 1); confirmed, experimental data (including but not limited to mutagenesis) confirms prediction (Supplemental Table 9). Effect prediction is derived from the SNAPMAT score (Neu, neutral/negative; Imp, important to function/positive). Loc indicates cellular location of the residue (Ex, extracellular; Tr, transmembrane; Cy, cytoplasmic), taken from the Swiss-Prot (25) MC4R_HUMAN entry. Rest of the values are extracted as described for SNAP method (Supplemental Fig. 1 and ref. 23). Hyd inciates residue hydrophobicity (F, hydrophobic; U, unassigned; L, hydrophilic), Chg indicates charge (N, negative; U, uncharged; P, positive), and Size indicates surface area (S, small; M, medium; L, large). Cons indicates conservation (whether the residue is conserved in alignment). Values are Res: X% (WT residue conserved in X%≥45% of sequences; position (residue not conserved, but at most 3 highest scoring residues make up ≥55% of the distribution), none (position is not conserved). Predicted values are SSt, 3-state secondary structure (H, helix; L, loop; E, extended strand), SA, 3-state solvent accessibility [B, buried (0–9% solvent accessible), I, intermediate (9–36%), E, exposed (>36%)], and Bn, normalized B value reflecting residue flexibility [where values are −3 (rigid)≤Bn≤3 (flexible)].
With respect to the accuracy of annotation, 3 of the 22 residues classified as important have been experimentally implicated in receptor function: D90 (involved in AGRP inverse agonism) (47) and D146 and R147 located within the DRY motif (50). For 7 others (of the 22), experimental mutagenesis studies report functional effects (N62S, Y157S, R165A/Q/W, A219V, W258A, C271A/Y/R, and P299H; Supplemental Table 1). Excluding previously documented variants with functional consequence left 12 novel predictions in our set of 22 AA residues. For two of these (F201A and H214A; Supplemental Table 1), there is no measurable experimental effect reported. This result may imply a prediction error on our part. However it is also possible that other variants may not be neutral at this site. Significant levels of conservation in the aligned sequences (Table 2, 56% contain a Phe) confirm this suspicion for F201. The variants may also affect an aspect of function not yet examined in the in vitro studies. If so, the negative experimental result and the SNAP prediction could both be correct, supporting more explicit use of predictions in the design of future experiments. These 2 residues and the other 10 very strong predictions (N72, P78, H158, W174, Y212, M215, H222, C257, N294, and Y302), for which no evidence exists in the literature, should have high priority for experimental vetting to increase understanding of structure/sequence-function relationships for this physiologically important receptor.
CONCLUSIONS
SNAP is a computational technique designed to evaluate the functional consequences of single AA residue substitutions. We used SNAP to compute scores over all possible substitutions at each AA residue in MC4R and introduced an optimal mutability scoring function, which identified residues that are important for the function of this receptor. Our results were generally consistent with available in vitro experimental annotations. In addition, we compared sequence-function similarities and differences between hMC4R, mMC4R, and hMC1R to gain insight into the molecular bases of receptor specific functions. We also identified 12 predicted functionally important AAs in hMC4R that have not been studied experimentally. Experimental examination of these residues might contribute to our insight into the function of this physiologically important receptor.
Increasingly, use of genome-wide association scans and deep-sequencing approaches will generate large numbers of novel and otherwise unvetted candidate genes and alleles for consideration as contributors to human physiological and disease phenotypes. Computational approaches such as the one used here will likely be helpful in prioritizing such sequence variants for further analysis. Ultimately, the development of techniques to analyze noncoding DNA sequences in comparable ways will be critical to such efforts.
Supplementary Material
Acknowledgments
We thank Cedric Govaerts (University of California, San Francisco) for providing us with an annotated data set of hMC4R variants and Guy Yachdav (Columbia University) for help with maintenance of the SNAP web server. We also thank all those who deposit their experimental results into public databases. The work of Y.B. and B.R. was supported by grant R01-LM07329-01 from the National Library of Medicine; that of J.D.O. and R.L.L. by National Institutes of Health (NIH) grant RO1-DK52431-15; and that of C.V. by NIH grants RO1-DK60540 and RO1-DK068152 and by the American Heart Association’s Established Investigator Award.
References
- Kishi T, Aschkenasi C J, Lee C E, Mountjoy K G, Saper C B, Elmquist J K. Expression of melanocortin 4 receptor mRNA in the central nervous system of the rat. J Comp Neurol. 2003;457:213–235. doi: 10.1002/cne.10454. [DOI] [PubMed] [Google Scholar]
- Patten C S, Daniels D, Suzuki A, Fluharty S J, Yee D K. Structural and signaling requirements of the human melanocortin 4 receptor for MAP kinase activation. Regul Pept. 2007;142:111–122. doi: 10.1016/j.regpep.2007.02.005. [DOI] [PubMed] [Google Scholar]
- Newman E A, Chai B X, Zhang W, Li J Y, Ammori J B, Mulholland M W. Activation of the melanocortin-4 receptor mobilizes intracellular free calcium in immortalized hypothalamic neurons. J Surg Res. 2006;132:201–207. doi: 10.1016/j.jss.2006.02.003. [DOI] [PubMed] [Google Scholar]
- Wachira S J, Hughes-Darden C A, Taylor C V, Ochillo R, Robinson T J. Evidence for the interaction of protein kinase C and melanocortin 3-receptor signaling pathways. Neuropeptides. 2003;37:201–210. doi: 10.1016/s0143-4179(03)00026-x. [DOI] [PubMed] [Google Scholar]
- Srinivasan S, Lubrano-Berthelier C, Govaerts C, Picard F, Santiago P, Conklin B R, Vaisse C. Constitutive activity of the melanocortin-4 receptor is maintained by its N-terminal domain and plays a role in energy homeostasis in humans. J Clin Invest. 2004;114:1158–1164. doi: 10.1172/JCI21927. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mohammad S, Baldini G, Granell S, Narducci P, Martelli A M, Baldini G. Constitutive traffic of melanocortin-4 receptor in Neuro2A cells and immortalized hypothalamic neurons. J Biol Chem. 2007;282:4963–4974. doi: 10.1074/jbc.M608283200. [DOI] [PubMed] [Google Scholar]
- Adan R A, Kas M J. Inverse agonism gains weight. Trends Pharmacol Sci. 2003;24:315–321. doi: 10.1016/S0165-6147(03)00130-5. [DOI] [PubMed] [Google Scholar]
- Chai B X, Neubig R R, Millhauser G L, Thompson D A, Jackson P J, Barsh G S, Dickinson C J, Li J Y, Lai Y M, Gantz I. Inverse agonist activity of agouti and agouti-related protein. Peptides. 2003;24:603–609. doi: 10.1016/s0196-9781(03)00104-9. [DOI] [PubMed] [Google Scholar]
- Dinulescu D M, Cone R D. Agouti and agouti-related protein: analogies and contrasts. J Biol Chem. 2000;275:6695–6698. doi: 10.1074/jbc.275.10.6695. [DOI] [PubMed] [Google Scholar]
- Lu D, Willard D, Patel I R, Kadwell S, Overton L, Kost T, Luther M, Chen W, Woychik R P, Wilkison W O, Cone R D. Agouti protein is an antagonist of the melanocyte-stimulating-hormone receptor. Nature. 1994;371:799–802. doi: 10.1038/371799a0. [DOI] [PubMed] [Google Scholar]
- Ollmann M M, Wilson B D, Yang Y K, Kerns J A, Chen Y, Gantz I, Barsh G S. Antagonism of central melanocortin receptors in vitro and in vivo by agouti-related protein. Science. 1997;278:135–138. doi: 10.1126/science.278.5335.135. [DOI] [PubMed] [Google Scholar]
- Huszar D, Lynch C A, Fairchild-Huntress V, Dunmore J H, Fang Q, Berkemeier L R, Gu W, Kesterson R A, Boston B A, Cone R D, Smith F J, Campfield L A, Burn P, Lee F. Targeted disruption of the melanocortin-4 receptor results in obesity in mice. Cell. 1997;88:131–141. doi: 10.1016/s0092-8674(00)81865-6. [DOI] [PubMed] [Google Scholar]
- Farooqi I S, Keogh J M, Yeo G S, Lank E J, Cheetham T, O'Rahilly S. Clinical spectrum of obesity and mutations in the melanocortin 4 receptor gene. N Engl J Med. 2003;348:1085–1095. doi: 10.1056/NEJMoa022050. [DOI] [PubMed] [Google Scholar]
- Lubrano-Berthelier C, Le Stunff C, Bougneres P, Vaisse C. A homozygous null mutation delineates the role of the melanocortin-4 receptor in humans. J Clin Endocrinol Metab. 2004;89:2028–2032. doi: 10.1210/jc.2003-031993. [DOI] [PubMed] [Google Scholar]
- Farooqi I S, Yeo G S, Keogh J M, Aminian S, Jebb S A, Butler G, Cheetham T, O'Rahilly S. Dominant and recessive inheritance of morbid obesity associated with melanocortin 4 receptor deficiency. J Clin Invest. 2000;106:271–279. doi: 10.1172/JCI9397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen M, Cai M, Aprahamian C J, Georgeson K E, Hruby V, Harmon C M, Yang Y. Contribution of the conserved amino acids of the melanocortin-4 receptor in [corrected] [Nle4,D-Phe7]-alpha-melanocyte-stimulating [corrected] hormone binding and signaling. J Biol Chem. 2007;282:21712–21719. doi: 10.1074/jbc.M702285200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tao Y X. Molecular mechanisms of the neural melanocortin receptor dysfunction in severe early onset obesity. Mol Cell Endocrinol. 2005;239:1–14. doi: 10.1016/j.mce.2005.04.012. [DOI] [PubMed] [Google Scholar]
- Moult J, Fidelis K, Kryshtafovych A, Rost B, Hubbard T, Tramontano A. Critical assessment of methods of protein structure prediction-round VII. Proteins. 2007;69:3–9. doi: 10.1002/prot.21767. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kopp J, Schwede T. Automated protein structure homology modeling: a progress report. Pharmacogenomics. 2004;5:405–416. doi: 10.1517/14622416.5.4.405. [DOI] [PubMed] [Google Scholar]
- Marti-Renom M A, Stuart A C, Fiser A, Sanchez R, Melo F, Sali A. Comparative protein structure modeling of genes and genomes. Annu Rev Biophys Biomol Struct. 2000;29:291–325. doi: 10.1146/annurev.biophys.29.1.291. [DOI] [PubMed] [Google Scholar]
- Henderson R, Baldwin J M, Ceska T A, Zemlin F, Beckmann E, Downing K H. Model for the structure of bacteriorhodopsin based on high-resolution electron cryo-microscopy. J Mol Biol. 1990;213:899–929. doi: 10.1016/S0022-2836(05)80271-2. [DOI] [PubMed] [Google Scholar]
- Rasmussen S G, Choi H J, Rosenbaum D M, Kobilka T S, Thian F S, Edwards P C, Burghammer M, Ratnala V R, Sanishvili R, Fischetti R F, Schertler G F, Weis W I, Kobilka B K. Crystal structure of the human beta2 adrenergic G-protein-coupled receptor. Nature. 2007;450:383–387. doi: 10.1038/nature06325. [DOI] [PubMed] [Google Scholar]
- Bromberg Y, Rost B. SNAP: predict effect of non-synonymous polymorphisms on function. Nucleic Acids Res. 2007;35:3823–3835. doi: 10.1093/nar/gkm238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kawabata T, Ota M, Nishikawa K. The Protein Mutant Database. Nucleic Acids Res. 1999;27:355–357. doi: 10.1093/nar/27.1.355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boeckmann B, Bairoch A, Apweiler R, Blatter M C, Estreicher A, Gasteiger E, Martin M J, Michoud K, O'Donovan C, Phan I, Pilbout S, Schneider M. The SWISS-PROT protein knowledgebase and its supplement TrEMBL in 2003. Nucleic Acids Res. 2003;31:365–370. doi: 10.1093/nar/gkg095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thompson J D, Higgins D G, Gibson T J. CLUSTAL W: improving the sensitivity of progressive multiple sequence alignment through sequence weighting, position-specific gap penalties and weight matrix choice. Nucleic Acids Res. 1994;22:4673–4680. doi: 10.1093/nar/22.22.4673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ng P C, Henikoff J G, Henikoff S. PHAT: a transmembrane-specific substitution matrix. Predicted hydrophobic and transmembrane. Bioinformatics. 2000;16:760–766. doi: 10.1093/bioinformatics/16.9.760. [DOI] [PubMed] [Google Scholar]
- Henikoff S, Henikoff J G. Amino acid substitution matrices from protein blocks. Proc Natl Acad Sci U S A. 1992;89:10915–10919. doi: 10.1073/pnas.89.22.10915. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bromberg Y, Yachdav G, Rost B. SNAP predicts effect of mutations on protein function. Bioinformatics. 2008;24:2397–2398. doi: 10.1093/bioinformatics/btn435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Von Heijne G. Membrane protein structure prediction: hydrophobicity analysis and the positive-inside rule. J Mol Biol. 1992;225:487–494. doi: 10.1016/0022-2836(92)90934-c. [DOI] [PubMed] [Google Scholar]
- Punta M, Forrest L R, Bigelow H, Kernytsky A, Liu J, Rost B. Membrane protein prediction methods. Methods. 2007;41:460–474. doi: 10.1016/j.ymeth.2006.07.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rost B. Twilight zone of protein sequence alignments. Protein Eng. 1999;12:85–94. doi: 10.1093/protein/12.2.85. [DOI] [PubMed] [Google Scholar]
- Ofran Y, Punta M, Schneider R, Rost B. Beyond annotation transfer by homology: novel protein-function prediction methods to assist drug discovery. Drug Discov Today. 2005;10:1475–1482. doi: 10.1016/S1359-6446(05)03621-4. [DOI] [PubMed] [Google Scholar]
- Bromberg Y, Rost B. Comprehensive in silico mutagenesis highlights functionally important residues in proteins. Bioinformatics. 2008;24:207–212. doi: 10.1093/bioinformatics/btn268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nijenhuis W A, Garner K M, van Rozen R J, Adan R A. Poor cell surface expression of human melanocortin-4 receptor mutations associated with obesity. J Biol Chem. 2003;278:22939–22945. doi: 10.1074/jbc.M211326200. [DOI] [PubMed] [Google Scholar]
- Tao Y X, Segaloff D L. Functional analyses of melanocortin-4 receptor mutations identified from patients with binge eating disorder and nonobese or obese subjects. J Clin Endocrinol Metab. 2005;90:5632–5638. doi: 10.1210/jc.2005-0519. [DOI] [PubMed] [Google Scholar]
- Carroll L, Voisey J, van Daal A. Gene polymorphisms and their effects in the melanocortin system. Peptides. 2005;26:1871–1885. doi: 10.1016/j.peptides.2004.12.031. [DOI] [PubMed] [Google Scholar]
- Geller F, Reichwald K, Dempfle A, Illig T, Vollmert C, Herpertz S, Siffert W, Platzer M, Hess C, Gudermann T, Biebermann H, Wichmann H E, Schafer H, Hinney A, Hebebrand J. Melanocortin-4 receptor gene variant I103 is negatively associated with obesity. Am J Hum Genet. 2004;74:572–581. doi: 10.1086/382490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xiang Z, Litherland S A, Sorensen N B, Proneth B, Wood M S, Shaw A M, Millard W J, Haskell-Luevano C. Pharmacological characterization of 40 human melanocortin-4 receptor polymorphisms with the endogenous proopiomelanocortin-derived agonists and the agouti-related protein (AGRP) antagonist. Biochemistry. 2006;45:7277–7288. doi: 10.1021/bi0600300. [DOI] [PubMed] [Google Scholar]
- Tan K, Pogozheva I D, Yeo G S, Hadaschik D, Keogh J M, Haskell-Leuvano C, O'Rahilly S, Mosberg H I, Farooqi I S. Functional characterization and structural modeling of obesity-associated mutations in the melanocortin 4 receptor. Endocrinology. 2009;150:114–125. doi: 10.1210/en.2008-0721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Calton M A, Erson E A, Skhiri M, Zhang S, Kane J P, Malloy M J, Pullinger C R, Bromberg Y, Pennacchio L A, Dent R, McPherson R, Ahituv N, Vaisse C. Association of functionally significant melanocortin-4 but not melanocortin-3 receptor mutations with severe adult obesity in a large North American case control study. Hum Mol Genet. 2009;18:1140–1147. doi: 10.1093/hmg/ddn431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Govaerts C, Srinivasan S, Shapiro A, Zhang S, Picard F, Clement K, Lubrano-Berthelier C, Vaisse C. Obesity-associated mutations in the melanocortin 4 receptor provide novel insights into its function. Peptides. 2005;26:1909–1919. doi: 10.1016/j.peptides.2004.11.042. [DOI] [PubMed] [Google Scholar]
- Yang Y, Chen M, Lai Y, Gantz I, Yagmurlu A, Georgeson K E, Harmon C M. Molecular determination of agouti-related protein binding to human melanocortin-4 receptor. Mol Pharmacol. 2003;64:94–103. doi: 10.1124/mol.64.1.94. [DOI] [PubMed] [Google Scholar]
- Burstein E S, Spalding T A, Brann M R. The second intracellular loop of the m5 muscarinic receptor is the switch which enables G-protein coupling. J Biol Chem. 1998;273:24322–24327. doi: 10.1074/jbc.273.38.24322. [DOI] [PubMed] [Google Scholar]
- Kim D H, Shin S W, Baik J H. Role of third intracellular loop of the melanocortin 4 receptor in the regulation of constitutive activity. Biochem Biophys Res Commun. 2008;365:439–445. doi: 10.1016/j.bbrc.2007.10.170. [DOI] [PubMed] [Google Scholar]
- Haskell-Luevano C, Cone R D, Monck E K, Wan Y P. Structure activity studies of the melanocortin-4 receptor by in vitro mutagenesis: identification of agouti-related protein (AGRP), melanocortin agonist and synthetic peptide antagonist interaction determinants. Biochemistry. 2001;40:6164–6179. doi: 10.1021/bi010025q. [DOI] [PubMed] [Google Scholar]
- Chen M, Celik A, Georgeson K E, Harmon C M, Yang Y. Molecular basis of melanocortin-4 receptor for AGRP inverse agonism. Regul Pept. 2006;136:40–49. doi: 10.1016/j.regpep.2006.04.010. [DOI] [PubMed] [Google Scholar]
- VanLeeuwen D, Steffey M E, Donahue C, Ho G, MacKenzie R G. Cell surface expression of the melanocortin-4 receptor is dependent on a C-terminal di-isoleucine sequence at codons 316/317. J Biol Chem. 2003;278:15935–15940. doi: 10.1074/jbc.M211546200. [DOI] [PubMed] [Google Scholar]
- Schioth H B, Haitina T, Fridmanis D, Klovins J. Unusual genomic structure: melanocortin receptors in Fugu. Ann N Y Acad Sci. 2005;1040:460–463. doi: 10.1196/annals.1327.090. [DOI] [PubMed] [Google Scholar]
- Yamano Y, Kamon R, Yoshimizu T, Toda Y, Oshida Y, Chaki S, Yoshioka M, Morishima I. The role of the DRY motif of human MC4R for receptor activation. Biosci Biotechnol Biochem. 2004;68:1369–1371. doi: 10.1271/bbb.68.1369. [DOI] [PubMed] [Google Scholar]
- Chai B X, Pogozheva I D, Lai Y M, Li J Y, Neubig R R, Mosberg H I, Gantz I. Receptor-antagonist interactions in the complexes of agouti and agouti-related protein with human melanocortin 1 and 4 receptors. Biochemistry. 2005;44:3418–3431. doi: 10.1021/bi0478704. [DOI] [PubMed] [Google Scholar]
- Yang Y, Dickinson C, Haskell-Luevano C, Gantz I. Molecular basis for the interaction of [Nle4,D-Phe7]melanocyte stimulating hormone with the human melanocortin-1 receptor. J Biol Chem. 1997;272:23000–23010. doi: 10.1074/jbc.272.37.23000. [DOI] [PubMed] [Google Scholar]
- Nickolls S A, Cismowski M I, Wang X, Wolff M, Conlon P J, Maki R A. Molecular determinants of melanocortin 4 receptor ligand binding and MC4/MC3 receptor selectivity. J Pharmacol Exp Ther. 2003;304:1217–1227. doi: 10.1124/jpet.102.044974. [DOI] [PubMed] [Google Scholar]
- Frandberg P A, Doufexis M, Kapas S, Chhajlani V. Cysteine residues are involved in structure and function of melanocortin 1 receptor: substitution of a cysteine residue in transmembrane segment two converts an agonist to antagonist. Biochem Biophys Res Commun. 2001;281:851–857. doi: 10.1006/bbrc.2001.4429. [DOI] [PubMed] [Google Scholar]
- Tao Y X. Inactivating mutations of G protein-coupled receptors and diseases: structure-function insights and therapeutic implications. Pharmacol Ther. 2006;111:949–973. doi: 10.1016/j.pharmthera.2006.02.008. [DOI] [PubMed] [Google Scholar]
- Mossner R, Anders N, Konig I R, Kruger U, Schmidt D, Berking C, Ziegler A, Brockmoller J, Kaiser R, Volkenandt M, Westphal G A, Reich K. Variations of the melanocortin-1 receptor and the glutathione-S transferase T1 and M1 genes in cutaneous malignant melanoma. Arch Dermatol Res. 2007;298:371–379. doi: 10.1007/s00403-006-0708-7. [DOI] [PubMed] [Google Scholar]
- Larsen L H, Echwald S M, Sorensen T I, Andersen T, Wulff B S, Pedersen O. Prevalence of mutations and functional analyses of melanocortin 4 receptor variants identified among 750 men with juvenile-onset obesity. J Clin Endocrinol Metab. 2005;90:219–224. doi: 10.1210/jc.2004-0497. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.