

Abstract
The erythrocyte membrane is a newly appreciated platform for thiol-based circulatory signaling, and it requires robust free thiol maintenance. We sought to define physiological constraints on erythrocyte antioxidant defense. Hemoglobin (Hb) conformation gates glycolytic flux through the hexose monophosphate pathway (HMP), the sole source of nicotinamide adenine dinucleotide phosphate (NADPH) in erythrocytes. We hypothesized elevated intraerythrocytic deoxyHb would limit resilience to oxidative stress. Human erythrocytes were subjected to controlled oxidant (superoxide) loading following independent manipulation of oxygen tension, Hb conformation, and glycolytic pathway dominance. Sufficiency of antioxidant defense was determined by serial quantification of GSH, NADPH, NADH redox couples. Hypoxic erythrocytes demonstrated greater loss of reduction potential [Δ GSH Ehc (mV): 123.4±9.7 vs. 57.2±11.1] and reduced membrane thiol (47.7±5.7 vs. 20.1±4.3%) (hypoxia vs. normoxia, respectively; P<0.01), a finding mimicked in normoxic erythrocytes after HMP blockade. Rebalancing HMP flux during hypoxia restored resilience to oxidative stress at all stages of the system. Cell-free studies assured oxidative loading was not altered by oxygen tension, heme ligation, or the inhibitors employed. These data indicate that Hb conformation controls coupled glucose and thiol metabolism in erythrocytes, and implicate hypoxemia in the pathobiology of erythrocyte-based vascular signaling.—Rogers, S. C., Said, A., Corcuera, D., McLaughlin, D., Kell, P., Doctor, A. Hypoxia limits antioxidant capacity in red blood cells by altering glycolytic pathway dominance.
Keywords: band 3 protein, hemoglobin, glucose metabolism, thiol, reactive oxygen species, glycolysis
Robust antioxidant systems are required by red blood cells (RBCs) to confront the significant oxidative load encountered during circulatory transit; this is particularly true during physiological stress. Hemoglobin (Hb) deoxygenation results in methemoglobin and superoxide production (1); this is amplified as oxygen extraction from RBCs increases, such as in the setting of reduced blood flow or hypoxia. Moreover, substrate limiting availability of molecular oxygen provokes the production of reactive oxygen and nitrogen species (ROS and RNS) in tissue (2). Normally, these and related free radicals feed back into vascular signaling devoted to oxygen (O2) delivery homeostasis (2, 3). In the setting of sustained hypoxia, however, ROS cascades may exceed antioxidant capacity, leaving the biochemical footprint of hypoxia-induced oxidative stress. This chemistry results in maladaptive modifications to proteins, to which membrane thiols are particularly vulnerable (4).
Erythrocytic membrane thiols are newly appreciated to play a central role in RBC-based vascular signaling, by serving as a platform for context-responsive processing of both ROS and RNS (5, 6). Erythrocytes also play a critical role in free radical metabolism, serving as an ROS sink during oxidative stress (7, 8). Notably, both ROS scavenging and vascular signaling by RBCs require regeneration of oxidized thiols for ongoing functionality (9,10,11). In states of severe oxidative stress, significant vascular dysfunction is commonly observed. Under such conditions, the RBC thiol pool is often depleted. In prior work (12,13,14), we have linked RBC membrane redox events to abnormal vascular signaling. Here, we sought to define the physiological constraints on erythrocyte antioxidant defense, to better understand the events leading to the pathobiology of abnormal RBC-based vascular signaling.
Although unexplored to date, the influence of hypoxia on antioxidant defense in RBCs is suggested by known O2-responsive cycling in RBC glucose metabolism (15,16,17,18,19), which is linked to reducing equivalent recycling (Fig. 1). In RBCs, glucose is metabolized via two pathways: the Embden-Meyerhof pathway (EMP), and the hexose monophosphate pathway (HMP; also referred to as the pentose shunt) (20). Although the EMP generates adenosine triphosphate (ATP), NADH, and 2,3-diphosphoglycerate (2,3 DPG); the HMP is the erythrocytes’ sole source of NADPH. The relationship between O2 gradients and the flux through each glycolytic pathway arises from the influence of Hb conformation on protein complex assembly on the cytoplasmic domain of the Band 3 membrane protein (cdB3) (21). Specifically, competition exists between deoxyHb and key EMP enzymes for binding to cdB3 (22, 23). Because binding to cdB3 inactivates the EMP enzymes (24), the glycolytic pathway flux is gated as a function of Hb O2 saturation (HbSO2) (15) (Fig. 1). Therefore, the route of glucose metabolism in RBCs varies as O2 gradients are crossed in the course of circulatory transit, with glycolytic product ratios expected to shift between arteries and veins (25). We speculate that in patients with significant hypoxemia, the persistence of deoxyHb in arterial blood inappropriately maintains masking of cdB3 binding sites and prevents sequestration of key EMP enzymes. Hypoxia may thereby introduce continuous substrate competition between the EMP and HMP, and constrain the increase in glucose flux through the HMP that should occur during arterial transit. As a result, hypoxic RBCs may lose their otherwise considerable capacity to regenerate NADPH, the key reducing equivalent for GSH recycling, which is central to thiol-based antioxidant defenses (26) (Fig. 1).
Figure 1.

Relation between oxygen tension and reducing capacity in RBCs. A simplified scheme of glucose metabolism in the RBC (A), which proceeds through either the Embden-Meyerhof pathway (EMP, orange arrows), or the hexose monophosphate pathway (HMP, blue arrows). Both share glucose-6-phosphate (G6P) as an initial substrate. HMP is the sole source of NADPH in RBCs and generates fructose-6-phosphate (F6P) or glyceraldehyde-3-phosphate (G3P), which rejoin the EMP prior to glyceraldehyde-3-phosphate dehydrogenase (G3PD), a key regulatory point in the EMP. The EMP generates NADH (utilized by metHb reductase), as well as ATP (to drive ion pumps) and 2,3-DPG (to modulate hemoglobin p50) (neither ATP or 2,3 DPG shown). Relative flux through the EMP and the HMP is modulated by O2-linked transitions in Hb conformation due to competitive binding for the cytoplasmic domain of Band 3 (cdB3) between deoxyHb and key EMP enzymes (PFK, Aldo, G3PD, PK, and LDH). In fully oxygenated RBCs (B), these EMP enzymes are inactivated by sequestration on cdB3, resulting in glucose being channelled through the HMP, maximizing NADPH (and thus GSH) recycling capacity. In deoxygenated RBCs (C), EMP enzymes are dispersed by deoxyHb binding to Band 3, creating competition for G6P as a substrate and thereby constraining recycling capacity for NADPH and GSH and limiting resilience to attack by ROS. Hydrogen peroxide (H2O2) and superoxide (O2−) are the principal endogenous ROS encountered by RBCs. Both ROS may be generated internally (not shown); however, only H2O2 can cross the membrane directly. O2− enters RBCs through Band 3 (anion exchange protein 1, or AE-1). H2O2 and O2− are ultimately reduced to water by catalase or glutathione peroxidase (GPx). CAT, catalase; GSH, glutathione; GR, glutathione reductase; NADPH, nicotinamide adenine dinucleotide phosphate; PFK, phosphofructokinase; Aldo, aldolase; PK, pyruvate kinase; LDH, lactate dehydrogenase.
Here, we report on the influence of intraerythrocytic deoxyHb on RBC defense of membrane protein thiols during graded exposure to oxidative stress. We exposed RBCs to ROS after independently manipulating pO2, Hb conformation (and thus Hb-cdB3 binding), and the route of glucose metabolism. Our aims were to test the influence of hypoxemia on RBC resilience to oxidative stress, as quantified by loss of membrane thiol; link alteration in antioxidant defense to a specific disruption in reducing equivalent recycling and RBC energy metabolism; and determine whether this alteration was transduced by Hb conformation or pO2 per se.
MATERIALS AND METHODS
Reagents and antibodies
Unless otherwise indicated, all chemicals and reagents were purchased from Sigma-Aldrich (St. Louis, MO, USA). Koningic acid (KA) was kindly provided by Dr. Keiji Hasumi (Tokyo Noko University, Tokyo, Japan). Antibodies to Band 3 and stomatin were obtained from Santa Cruz Biotechnology (Santa Cruz, CA, USA); antibodies to GLUT-1, β-spectrin, β-actin, and G3PD were purchased from Abcam (Cambridge, MA, USA); HRP linked antibody to mouse IgG was obtained from Cell Signaling Technology (Danvers, MA, USA).
Blood sampling and generation of RBC suspensions
This project was approved by the human research protection office at Washington University, and written informed consent was obtained from all participants. Venous blood (30 ml) was drawn from healthy volunteers into a heparinized syringe; blood was centrifuged (2000 g, 10 min, 4°C), and the plasma and buffy coat were removed. The RBCs were washed (3×) in ice-cold phosphate buffered saline (PBS) with diethylenetriaminepentaacetic acid (DTPA; 500 μM), then resuspended to a hematocrit of ∼50% in Krebs buffer containing 2 g/L glucose (pH 7.4) for further experiments (referred to as washed RBCs).
RBC treatments
For manipulation of hemoglobin conformation, washed RBCs (20–25 ml) were injected into a rotating glass tonometer (in a water bath, 37°C), allowing controlled RBC oxygenation or deoxygenation, as well as sample extraction without atmospheric exposure (12). The tonometer chamber was joined via a vacuum-tight, motorized couple (Rotavapor R110; Buchi, Flawil, Switzerland) to a gas source and purge ports to permit simultaneous chamber rotation and flushing by gas blended from O2, N2, and CO2 tanks (OxyStreamer; BioSpherix, Lacona, NY, USA). Washed RBCs were exposed for 25 min to either oxygenating conditions (21% O2, 5–9% CO2, balance N2), yielding >97% HbSO2, or to deoxygenating conditions (0% O2, 5–9% CO2, balance N2), yielding <10% HbSO2 (to fully deoxygenate the washed RBC sample took 25 min, as determined from prior experiments). CO2 content in the flushing gas was adjusted in all experiments to provide physiological pCO2 and pH in the Krebs buffer. In control experiments, Hb conformation was locked in R state during RBC deoxygenation by preexposure to 100% carbon monoxide (CO) [5 min, room temperature (RT), forming >97% carboxyHb]. All deoxygenated samples were processed in an O2-controlled glove box (5% O2, 95% N2) (Coy Labs, Grass Lake, MI, USA), utilizing degassed buffers and reagents. All samples were analyzed for pO2, pCO2, pH, total [Hb], and percentages of oxyHb, deoxyHb, carboxyHb, and metHb (Bayer Rapidlab 800 blood gas analyzer and co-oximeter; Bayer Corp., East Walpole, MA, USA). For inhibition of superoxide entry into RBCs, washed RBCs (∼10% Hct) were incubated with 4,4′-diisothiocyanostilbene-2,2′- disulfonic acid (DIDS) (500 μM, 1 h, 37°C), then washed and resuspended in Krebs (as above). DIDS inhibits superoxide entry into RBCs, which occurs through Band 3 (27). For glucose depletion, washed RBCs (∼50% Hct) were incubated in glucose-free Krebs buffer (1 h, 37°C) (28). For inhibition of glucose utilization through the HMP, washed RBCs (∼10% Hct) were incubated with dehydroepiandrosterone (DHEA) (5 μM, 15 min, 37°C), a selective inhibitor of glucose-6-phosphate dehydrogenase (G6PD) (29). For inhibition of glucose utilization through the EMP, washed RBCs (∼10% Hct) were incubated with KA (15 μM, 15 min, 37°C), a selective inhibitor of glyceraldehyde-3-phosphate dehydrogenase (G3PD) (30).
Preparation of RBC ghosts
Washed RBCs were lysed in 30 vol of Tris-HCl (10 mM, pH 7.4) containing 500 μM DTPA and 200 μM phenylmethanesulfonyl fluoride (PMSF). The lysate was centrifuged (25,000 g, 20 min, 4°C) to pellet the membrane fraction, which was washed (4–5×) in Tris-HCl (10 mM, pH 7.4) (40,000 g, 20 min, 4°C). One milliliter of washed RBC membranes was incubated (20 min, RT) in 5 ml of 5 mM sodium phosphate containing 1 mM magnesium sulfate, and then centrifuged (22,000 g, 20 min, 4°C) to yield a pellet of resealed RBC ghosts (31). Intact ghost formation was confirmed by phase-contrast microscopy.
Preparation of purified hemoglobin
Washed RBCs were exposed to 100% CO (5 min, RT) to prevent heme autooxidation during processing, then lysed by freezing. After thawing, all processing was conducted at 4°C. The membrane fraction was separated by centrifugation (20,000 g, 15 min); the remaining lysate was dialyzed overnight against buffer A (20 mM Tris-HCl, pH 8.2), then loaded onto a Q-sepharose HP anion exchange column (Hi-Load 16/10; GE Healthcare, New York, NY, USA). A linear gradient elution was performed from 100% buffer A to 75% buffer B (20 mM Tris-HCl and 0.2 M NaCl, pH 8.2) in 5 CVs (Äkta FPLC system; GE Healthcare) (32). Protein elution was monitored at 280 nm; fractions were collected and assessed by native PAGE against an HbAO standard (Sigma). Fractions containing pure Hb tetramers were pooled, concentrated (Amicon Ultra 30-kDa MWCO; Millipore, Billerica, MA, USA), and redialyzed (PBS, pH 7.4) to restore physiological pH and salt concentration. Before study, carboxyHb was converted back to oxyHb by exposure to 100% O2. [Hb] was adjusted to 500 μM, and 2,3-diphosphoglycerate was added (equimolar to [Hb]) to facilitate subsequent deoxygenation (HbSO2 ∼20% at pO2 5 Torr, 37°C).
In vitro model of oxidative stress
A hypoxanthine/xanthine oxidase (HX/XO) system was used to generate steady, gradable production of superoxide (33). Either RBC ghosts or intact washed RBCs (in Krebs) were portioned into aliquots and incubated with 1.5 mM HX and escalating doses of XO (0, 0.2, 0.4, or 0.8 U/ml) in a shaking heater block (37°C) for 2, 5, 10, 20, or 45 min, at which time 1 mM allopurinol was added to the sample to arrest superoxide production. Following HX/XO exposure, ghosts and RBCs were washed (3× in PBS) and assayed for quantification and distribution of reduced (free) thiol density in membrane proteins (samples measured only at 45 min), and ratios of the GSH, NADH, and NADPH redox couples were measured. To isolate the effect of heme occupancy on GSH conversion to glutathione disulfide (GSSG) by superoxide, we employed a cell-free system composed of 500 μM purified Hb tetramers, 500 μM 2,3-DPG, and 500 μM GSH. These components were suspended in Krebs buffer (pH 7.4, 37°C), exposed to either oxygenating or deoxygenating conditions (as above), and then incubated (as above) with 1.5 mM HX and 0.2 U/ml XO.
RBC membrane protein fractionation and determination of free thiol density
Washed RBC membranes were prepared from RBC lysates as above. To isolate intrinsic membrane proteins, washed membranes were incubated (30 min, 37°C) in a stripping buffer (15 mM NaOH, 2 mM DTPA, and 0.2 mM PMSF, pH 12), then centrifuged (25,000 g, 20 min, 4°C). The supernatant was decanted, extrinsic proteins were recovered following acetone precipitation, and the remaining membrane pellet (intrinsic proteins) was resolubilized. Protein content in the fractions was measured with the bicinchoninic acid assay (Pierce Protein Research Products; Thermo Scientific, Rockford, IL, USA). Aliquots from membrane fractions containing equal amounts of protein were separated by SDS-PAGE and either visualized by Coomassie blue staining or transferred to a nitrocellulose membrane. Blotting for proteins known to be associated with each fraction was performed using specific antibodies and Western blot chemiluminescence detection (Pierce). We probed for the intrinsic (membrane spanning) proteins Band 3, GLUT-1, and stomatin, as well as the extrinsic (skeletal, anchoring, or membrane-associated) proteins β-spectrin, β-actin, and G3PD. The reduced (free) thiol concentration [SH] in the protein preparations was determined via a modification of a method previously described (33). Protein fractions were incubated (PBS, 15 min, 37°C) with 10 mM 5,5′ dithiobis(2-nitrobenzoic acid) (DTNB) in the presence of 8 M guanidine; absorbance (412 nm) was measured and compared to a standard curve generated from GSH (0–2 μM). Protein concentration in each fraction was determined (ADV01 assay; Cytoskeleton, Denver, CO), and the density of reduced thiol was expressed as a ratio of moles reduced thiol to protein mass (μM/mg).
Determination of GSH and GSSG
Following exposure to HX/XO, RBCs were washed (3×), resuspended in PBS (50% Hct), and split into samples paired for quantification of the GSSG alone and for total RBC glutathione content (GSH+GSSG). The samples for analysis of GSSG alone were incubated with the GSH scavenger 1-methyl-2-vinylpyridinium trifluoromethanesulfonate (M2VP) (2 min, 100 μl resuspended RBCs plus 10 μl of 3 mM M2VP). After preparation, all samples were stored (−80°C) for batch assay with the Bioxytech GSH/GSSG-412 assay (OxisResearch, Foster City, CA). After thawing (25°C, 10 min), 290 μl ice-cold 5% metaphosphoric acid (MPA) was added to the GSSG samples and 390 μl to the GSH + GSSG samples, which were then centrifuged (16,000 g, 30 s, RT). Fifty microliters of the MPA supernatant was removed and added to either 700 μl or 3 ml of sodium phosphate (NaPO4) with EDTA (GSSG or total GSH samples, respectively). Fifty microliters of sample was then added to a 96-well plate along with 50 μl of chromagen (DTNB in NaPO4 with EDTA and ethanol) and 50 μl of enzyme (glutathione reductase in NaPO4 with EDTA). After a 5-min incubation (RT), 50 μl of NADPH was added. The change in absorbance (412 nm) was measured over 3 min and compared to a standard curve generated from GSSG (0–1.5 μM). Values are reported as a redox ratio (GSSG/GSHtotal) × 100.
Redox potential for glutathione
Glutathione represents the most abundant low-molecular-weight thiol (34) and the principle thiol redox buffer in RBCs (35, 36). We calculated the half-cell reduction potential (Ehc) for intraerythrocytic glutathione in our model. The half-cell reaction for the GSSG/2GSH redox pair is
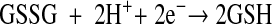 |
Thus, the Nerst equation for the reduction potential of the GSSG/2GSH half cell at 37°C, pH 7.4, has the form (37):
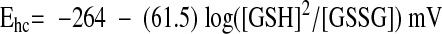 |
Determination of nicotinamide adenine dinucleotides: NADH and NADPH
RBCs were washed (3×) with 10 mM PBS (pH 7.4), then snap frozen in liquid nitrogen in an alkaline extraction solution containing 20 mM nicotinamide, 20 mM sodium bicarbonate (NaHCO3), and 100 mM sodium carbonate (Na2CO3). Reduced and total pyridine nucleotides were quantified by monitoring the change in absorbance (570 nm) during enzyme cycling reactions (38, 39). After thawing (25°C, 10 min), insoluble proteins and membranes were removed from lysates (16,000 g, 30 s, RT). The supernatants were immediately placed on ice and protected from light. Selective measurement of reduced nucleotides (NADH and NADPH) was performed from heated aliquots of the RBC extract (500 μl of lysate was heated at 60°C for 30 min). NADtotal or NADPtotal was determined in unheated aliquots of the same RBC extract. Oxidized nucleotide concentrations (NAD+ and NADP+) were calculated by subtracting the reduced from total values.
Cycling reaction for determination of NADtotal/NADH
One hundred microliters of RBC lysate (unheated/heated) was added to 800 μl of freshly prepared NAD cycling buffer containing 100 mM Tris (pH 8), 0.5 ml thiazolyl blue (MTT), 1 ml phenazine ethosulfate (PES), 0.2 mg/ml alcohol dehydrogenase (ADH) and 1% BSA. Following incubation (5 min, dark, 37°C), 100 μl of ethanol was added to the sample. The sample was mixed, and then centrifuged (16,000 g, 30 s, RT). The change in absorbance (570 nm) was measured over 2.5 min.
Cycling reaction for determination of NADPtotal/NADPH
One hundred microliters of RBC lysate (unheated/heated) was added to 800 μl of freshly prepared NADP cycling buffer containing 100 mM Tris (pH 8), 0.5 mM thiazolyl blue (MTT), 2 mM phenazine ethosulfate (PES), and 1.3 IU/ml glucose-6-phosphate dehydrogenase (G6PD). Following incubation (5 min, dark, 37°C), 100 μl of 10 mM glucose-6-phosphate was added to the sample. The sample was mixed, and the change in absorbance (570 nm) was measured over 2.5 min.
Statistical comparisons
The density of reduced membrane thiol, glutathione redox ratios, and the pyridine nucleotide redox ratios were compared using a 2-way analysis of variance, with Hb oxygenation as the main effect, and either HX/XO dose or HX/XO exposure time as the within-subject factor. Differences in means between groups were also compared by independent samples t test or 1-way analysis of variance (ANOVA), where appropriate. Bonferroni’s post hoc test was used for multiple comparisons. All data are reported as means ± se. Two-tailed values of P < 0.05 were considered significant throughout (GraphPad Prism 5; GraphPad, La Jolla, CA, USA).
RESULTS
Resilience of membrane proteins to oxidative stress
RBC ghosts
Membrane proteins were processed from ghosts into total, intrinsic, and extrinsic fractions. Qualitative analysis of Western blots for proteins known to be associated with these fractions confirmed appropriate protein partitioning (Fig. 2A, B). Minor (ns) difference was observed in the density of reduced protein thiols between ghost membrane fractions prepared from oxygenated or deoxygenated RBCs (Table 1). Following exposure to graded oxidative stress, the density of reduced protein thiols were quantified and indexed to baseline values (Fig. 3A, B). At the highest exposure, 26.6 ± 3.1 vs. 31.8 ± 3.8% of reduced membrane thiol was lost (ns) in oxygenated and deoxygenated ghost preparations, respectively. Thiol losses appeared to be evenly proportioned between the intrinsic and extrinsic protein fractions (Fig. 3A, B). In all ghost preparations, thiol loss was attenuated by limiting XO substrate.
Figure 2.

Partitioning of RBC membrane proteins into intrinsic and extrinsic fractions. Membrane proteins from oxygenated and deoxygenated RBC lysates (total) were processed into intrinsic (membrane spanning) and extrinsic (skeletal, anchoring, or membrane-associated) proteins. Aliquots from membrane fractions containing equal amounts of protein were separated by SDS-PAGE and either visualized by Coomassie blue staining (A), or transferred to a nitrocellulose membrane for immunoblotting (B). Note appropriate partitioning of proteins known to be associated with each fraction. Relative to signal from the unfractionated (total) preparation, partitioning led to enhancement of individual proteins within their respective fractions, precluding quantitative comparisons.
TABLE 1.
Absolute density of reduced thiol in RBC membrane fractions following oxidative stress
| RBC Preparation | Total | Intrinsic | Extrinsic |
|---|---|---|---|
| Normoxic RBCs | |||
| Control | 13.11 ± 2.19 | 6.27 ± 0.55 | 16.65 ± 5.31 |
| HX/XO (1.5 mM/0.8 U·ml−1) | 9.13 ± 1.28 | 5.70 ± 0.32 | 9.76 ± 1.31 |
| Ghosts from Normoxic RBCs | |||
| Control | 9.77 ± 1.39 | 5.23 ± 0.54 | 12.52 ± 2.61 |
| HX/XO (1.5 mM/0.8 U·ml−1) | 7.35 ± 0.60 | 3.75 ± 0.95 | 10.45 ± 1.51 |
| Hypoxic RBCs | |||
| Control | 11.50 ± 1.40 | 6.06 ± 0.63 | 17.01 ± 4.13 |
| HX/XO (1.5 mM/0.8 U·ml−1) | 5.98 ± 0.38* | 5.58 ± 0.53 | 5.74 ± 1.01* |
| Ghosts from Hypoxic RBCs | |||
| Control | 8.73 ± 1.02 | 5.60 ± 0.18 | 11.86 ± 0.18 |
| HX/XO (1.5 mM/0.8 U·ml−1) | 5.96 ± 0.33* | 4.19 ± 0.08 | 7.72 ± 0.74* |
Data are expressed as micromolar free thiol per milligram membrane protein.
P < 0.05 vs. corresponding normoxic value.
Figure 3.

Conservation of reduced RBC membrane thiol in ghost and intact RBCs preparations exposed to graded oxidative stress. Percentage of reduced membrane thiol remaining (indexed to membrane protein) following graded oxidative stress (45-min exposure). Protein fractions in ghosts prepared from oxygenated RBCs (A) and deoxygenated RBCs (B) were compared to fractions from intact oxygenated RBCs (C) and intact deoxygenated RBCs (D). Data are plotted as means ± se; n = 5–11. *P < 0.05, **P < 0.01 for intact deoxygenated RBC protein fractions vs. corresponding intact oxygenated RBCs. ƒP < 0.05 for intact deoxygenated RBC protein fractions vs. corresponding ghosts from deoxygenated RBCs.
Intact RBCs: effect of hypoxia
The density of reduced thiols in control membrane protein fractions differed slightly between preparations from ghosts and intact RBCs and between preparations from oxygenated and deoxygenated RBCs (ns), likely reflecting O2-dependent variation in the composition of extrinsic membrane proteins (Table 1 and Fig. 1) (15, 22). As with the ghost preparations, the loss of membrane thiols from intact RBCs progressed with the severity of oxidative insult (Fig. 3C, D), and thiol loss was blocked by allopurinol, an XO inhibitor. The extent of thiol oxidation following the highest oxidative exposure in intact, oxygenated RBCs was similar to that in ghosts from oxygenated RBCs [20.13±4.3 vs. 26.6±3.1% reduced (free) thiol lost, respectively; ns] and to that in ghosts from deoxygenated RBCs (20.1±4.3 vs. 31.8±3.8%, reduced thiol lost, respectively; ns). However, with increasing oxidative stress, the loss of reduced thiol was significantly greater in hypoxic RBCs than in ghosts from hypoxic RBCs (P<0.05), as well as more severe in hypoxic RBCs than in oxygenated RBCs (P<0.05) (at the highest dose of HX/XO, the percentage of reduced thiol lost was: 47.7±5.7 vs. 20.1±4.3% in hypoxic RBCs and normoxic RBCs, respectively; P<0.01) (Fig. 3C, D). Moreover, the pattern of oxidative injury between ghosts and intact RBCs differed, with losses strictly limited to extrinsic proteins in the intact cells (Table 1, Fig. 3).
Effect of hypoxia on recycling during oxidative stress
GSH recycling
The ability of RBCs to maintain intracellular glutathione in a reduced state during a steady oxidative challenge was determined by serial quantification of redox ratios for the GSH:GSSG couple and calculation of the half-cell reduction potential Ehc. At each level of oxidative stress, hypoxia impaired the ability of RBCs to recycle oxidized glutathione (P<0.05) (Fig. 4G, H), thereby significantly reducing Ehc (P<0.01) (Fig. 4A, B). This effect was blocked by allopurinol, an inhibitor of XO (data not shown). In control experiments, the following cell-free preparations were analyzed under high (pO2∼500 Torr) and low (pO2∼10 Torr) oxygen conditions, (where indicated, we added 1.5 mM HX and 0.2 U/ml XO, the lowest oxidant exposure in our model): GSH alone (data not shown); GSH with HX alone (data not shown); GSH with XO alone (data not shown); GSH with HX/XO; GSH with HX/XO and either DIDS, DHEA, or KA at the concentration used in the RBC exposures; GSH with Hb and HX (and equimolar 2,3-DPG, to approximate p50 of intra-RBC Hb); and GSH with HX/XO and Hb (and equimolar 2,3-DPG) (Fig. 5). We found no significant oxidation of GSH with GSH alone, GSH with HX alone, or GSH with XO alone. In the presence of HX/XO, buffer oxygen content (pO2∼500 or 10 Torr) did not significantly influence the rate of GSH oxidation to GSSG (Fig. 5A). DIDS, DHEA, or KA with HX/XO did not affect the rate of GSH oxidation to GSSG under either high or low oxygen conditions (Fig. 5B, C). oxygenated (HbSO2 88%) or deoxygenated Hb (HbSO2 24%) (with 12 or 9% metHb, respectively), in the presence of HX alone, did not result in the oxidation of GSH to GSSG. Hb, in the presence of HX/XO, significantly accelerated the oxidation of GSH to GSSG (P<0.05); this effect was less pronounced following Hb deoxygenation (HbSO2 88 or 24%). In the presence of either oxygenated or deoxygenated Hb, GSH in the system was brought to full oxidation at 10 min (Fig. 5A).
Figure 4.

Serial glutathione redox state of deoxygenated and oxygenated RBCs exposed to graded oxidative stress. Measurement of total glutathione (GSH+GSSG) and GSSG alone and calculation of GSH (from the difference) in oxygenated or deoxygenated washed RBCs exposed to HX/XO. Glutathione-related parameters, including half-cell reduction potential Ehc (A, B), total GSH + GSSG (C, D), GSH alone (E, F), and GSSG alone (G, H) are shown for oxygenated RBCs (top panels) and deoxygenated RBCs (bottom panels). Data are presented as means ± se; n = 4–6. *P < 0.05, **P < 0.01 for deoxygenated RBCs vs. corresponding oxygenated RBC sample. Note that P < 0.01 for all 0.4 and 0.8 U/ml XO deoxygenated RBC Ehc values from 5 min onward, vs. corresponding oxygenated RBC Ehc (not presented graphically).
Figure 5.

Effect of oxygen tension, heme occupancy, and presence of inhibitors on oxidation of GSH by HX/XO. Cell-free system to isolate effect of oxygen tension, heme occupancy, and presence of inhibitors on oxidant production from HX/XO, as measured by GSH conversion to GSSG. HX (1.5 mM) and XO (0.2 U/ml) alone, or purified Hb tetramers (500 μM) with 2,3-DPG (500 μM), HX (1.5 mM) and XO (0.2 U/ml) were incubated with GSH (500 μM), in Krebs buffer (pH 7.4, 37°C) under oxygenated or deoxygenated conditions (A). In addition, experiments were performed under oxygenated (B) and deoxygenated (C) conditions with GSH (500 μM), HX (1.5 mM), and XO (0.2 U/ml) in the presence of either DIDS (500 μM), DHEA (5 μM), or KA (15 μM). GSSG/GSH redox ratios were calculated from the measurement of total and oxidized glutathione . In the presence of HX/XO, buffer oxygen content (pO2∼500 or 10 Torr) did not significantly influence the rate of GSH oxidation to GSSG (A). Hb, in the presence of HX/XO, significantly accelerated the oxidation of GSH to GSSG (P<0.05); this effect was less pronounced following Hb deoxygenation (HbSO2 88 or 24%) (A). DIDS, DHEA, or KA with HX/XO did not affect the rate of GSH oxidation to GSSG under either high- or low-oxygen conditions (B, C). Data are plotted as means ± se; n = 3–6. *P < 0.05 vs. corresponding low oxygen tension sample.
NADH recycling
As above, for oxygenated and deoxygenated RBCs, the ability to maintain the dinucleotides NADH and NADPH in a reduced state was determined by serial quantification of the redox ratios for the NADPH:NADP+ (Fig. 6A, B) and the NADH:NAD+ (Fig. 6C, D) couples. In control samples not subjected to oxidative stress, both redox couples were stable over time in oxygenated RBCs; however, deoxygenation alone affected both NADPH and NADH redox ratios (depleting NADH moreso). With graded oxidative stress, the ability to recycle NADPH was significantly impaired in hypoxic RBCs, when compared to normoxic cells (P<0.05); however, NADH depletion was immediate and sustained in both normoxic and hypoxic RBCs (Fig. 6).
Figure 6.

Serial NADPH and NADH redox ratios from oxygenated and deoxygenated RBCs exposed to graded oxidative stress. NADPH/NADP (A, B) and NAD/NADH (C, D) redox ratios calculated from the measurement of total and reduced pyridine nucleotides. Change in the (NADPH/NADPTotal) × 100 ratio over time during steady oxidant production reflects recycling efficiency for the NADPH-reducing systems in RBCs. Data are presented from oxygenated (A, C; left panel) and deoxygenated (B, D; right panel) RBCs. Data are plotted as means ± se; n = 3–6. *P < 0.05, **P < 0.01 for deoxygenated RBC samples vs. corresponding oxygenated sample.
Reducing equivalent recycling
Inhibition of superoxide entry into RBCs
Recycling capacity for the coupled redox pairs NADPH/NADP+ and GSSG/GSH was determined by serial quantification of the appropriate redox ratios, as above. Oxygenated RBCs were incubated with the Band 3 inhibitor DIDS prior to deoxygenation (deoxyHb 91.8%, pO2 9.2 Torr) and then were subjected to the most severe oxidative insult in our model (HX, 1.5 mM; XO, 0.8 U/ml). DIDS exposure prevented the loss in GSH and NADPH recycling capacity that was evident in deoxygenated RBC suspensions (in which superoxide entry into RBCs was not inhibited) (P<0.05) (Fig. 7A, B).
Figure 7.

Reducing equivalent recycling: effect of Hb conformation and the inhibition of superoxide entry into RBCs. Paired NADPH and GSH redox ratios from oxygenated and deoxygenated RBCs exposed to HX/XO (1.5 mM and 0.8 U/ml, respectively), plotted with data from deoxygenated RBCs pretreated with Band 3 inhibitor DIDS (500 μM, 1 h, 37°C; to block superoxide entry into RBCs) (A, B), or deoxygenated RBCs pretreated with 100% CO (5 min, RT, forming >97% carboxyHb; to lock Hb conformation in R state and prevent release of EMP enzymes from Band 3; see Fig. 1) (C, D). Glutathione redox ratio is calculated as (GSSG/GSHTotal) × 100. NADPH redox ratio is calculated as (NADPH/NADPTotal) × 100. Recycling capacity in deoxygenated RBCs was preserved by preventing superoxide entry into deoxygenated RBCs (DIDS), or by preventing hypoxia-induced transition in Hb conformation (CO treatment). Data are plotted as means ± se; n = 3–4. *P < 0.05, **P < 0.01 for CO or DIDS values vs. deoxygenated values.
Effect of Hb conformation
In other control experiments, RBCs were exposed to pure CO gas prior to deoxygenation, and then subjected to the most severe oxidative insult in our model (HX, 1.5 mM; XO, 0.8 U/ml). CO exposure stabilized Hb conformation (in R state) during suspension deoxygenation (COHb 95.5±2.7% at pO2 6.8±2.1 Torr) and prevented the loss of GSH and NADPH recycling capacity that was evident in control deoxygenated suspensions (in which Hb transition to T state was permitted, deoxyHb 93.7±2.1%, pO2 10±2.7 Torr) (P<0.05) (Fig. 7C, D).
Effect of glycolytic pathway dominance
Recycling capacity for GSH and NADPH was completely lost following glucose depletion of RBC suspensions (HX, 1.5 mM; XO, 0.8 U/ml) (note that a GSH redox ratio of 50% represents complete conversion to GSSG) (P<0.01) (Fig. 8A, B). Preventing glucose metabolism through the HMP, with the G6PD inhibitor DHEA, significantly limited the ability of oxygenated red blood cells to recycle GSH and NADPH (P<0.05) (Fig. 8C, D), whereas inhibition of the EMP, with the selective G3PD inhibitor KA, restored reducing equivalent recycling capacity in deoxygenated RBC suspensions (P<0.05) (Fig. 8E, F).
Figure 8.

Reducing equivalent recycling: effect of glycolytic pathway dominance. Paired NADPH and GSH redox ratios from oxygenated and deoxygenated RBCs exposed to HX/XO (1.5 mM and 0.8 U/ml, respectively), plotted with data from glucose depleted oxygenated RBC (A, B); oxygenated RBCs pretreated with the G6PD inhibitor DHEA (5 μM, 15 min, 37°C; to block the HMP) (C, D), or deoxygenated RBCs pretreated with the G3PD inhibitor KA (15 μM, 15 min, 37°C; to block the EMP) (E, F). Glutathione redox ratio is calculated as (GSSG/GSHTotal) × 100. NADPH redox ratio is calculated as (NADPH/NADPTotal) × 100. Reducing equivalent recycling capacity was lost in oxygenated RBCs following glucose depletion (A, B) and HMP blockade at G6PD (C, D); reducing equivalent recycling capacity was restored in hypoxic RBCs following EMP blockade (E, F). Data are plotted as means ± se; n = 3–4. *P < 0.05, **P < 0.01 for glucose-depleted or DHEA values vs. oxygenated values or KA values vs. deoxygenated values.
RBC membrane integrity: effect of hypoxia and reducing equivalent recycling
Washed RBCs were treated with DHEA, CO gas, DIDS, or KA (as described above). These RBCs and controls were placed into our tonometer under normoxic or hypoxic conditions and then exposed to oxidative stress (HX, 1.5 mM; XO, 0.8 U/ml; 45 min). As above, the density of reduced protein thiols was quantified and indexed to baseline values. The extent of reduced (free) membrane thiol loss was more severe in hypoxic RBCs than oxygenated RBCs (47.7±5.7 vs. 20.1±4.3% in hypoxic and normoxic RBCs, respectively; P<0.01). Blocking glucose metabolism via the HMP with the G6PD inhibitor DHEA increased membrane thiol losses from normoxic RBCs (20.1±4.3 vs. 33.0±2.6% in normoxic and normoxic DHEA-treated RBCs, respectively; P<0.05). Membrane thiol losses in hypoxic RBCs were diminished by inhibiting superoxide entry into hypoxic RBCs with DIDS (47.7±5.7 vs. 20.6±9.6% hypoxic and DIDS pretreated hypoxic RBCs, respectively; P<0.05); stabilizing Hb conformation with CO pretreatment (47.7±5.7 vs. 16.5±3.4% in hypoxic and CO pretreated hypoxic RBCs, respectively; P<0.05); and blocking glucose metabolism via the EMP with the G3PD inhibitor KA (47.7±5.7 vs. 21.6±3.0% hypoxic and KA pretreated hypoxic RBCs, respectively; P<0.05) (Fig. 9).
Figure 9.

Conservation of reduced RBC membrane thiol in RBCs confronted with oxidative stress. Percentage of reduced membrane thiol remaining (indexed to membrane protein) in RBCs exposed to HX/XO (1.5 mM and 0.8 U/ml, respectively) for 45 min. Following either oxygenation or deoxygenation in the tonometer, washed RBCs (in Krebs) were removed, portioned into aliquots, and incubated with HX/XO in a shaking heater block (37°C). As observed for measures of recycling capacity for reducing equivalents (Figs. 7 and 8), oxygenated RBCs lost the ability to defend membrane thiols following HMP blockade with DHEA, while hypoxic RBCs were rescued by stabilizing Hb conformation (CO), preventing superoxide entry into RBCs (DIDS), or following EMP blockade (KA). Data are plotted as means ± se; n = 4–8. *P < 0.05.
DISCUSSION
The principal findings of these studies were that hypoxic RBCs demonstrated limited ability to recycle the linked reducing agents NADPH and GSH; consequently, these RBCs lost the ability to defend membrane thiols from oxidative attack. These defects were recapitulated in normoxic RBCs following glucose depletion, or by blocking glucose flux through the hexose monophosphate pathway (HMP) (the sole source of NADPH in RBCs); moreover, hypoxic RBCs were rescued by inhibiting superoxide entry into RBCs, stabilizing Hb conformation in R state (preventing Hb-cdB3 association), and by redirecting glucose flux through the HMP (i.e., by blocking the EMP). These data are consistent with the model of RBC metabolism, in which the mode of glucose utilization is modulated by cdB3 and O2-linked Hb transitions (16, 21, 40). Specifically, our findings indicate that hypoxia imposes constraint on flux through the HMP in RBCs, with a resultant inability to regenerate NADPH and GSH during periods of oxidative loading (Fig. 1).
We chose the native oxidant, superoxide, for our model for several reasons. RBCs are specifically equipped to deal with superoxide (Fig. 1), as it is generated internally in the course of normal arteriovenous transit (1); furthermore, superoxide is a major oxidative product of activated endothelium and leukocytes, and a significant source of oxidative stress in the human microcirculation (41). The HX/XO system that we employed to generate superoxide is very well characterized (42) and commonly used in RBC studies (33, 43), and the degree of superoxide generation by the HX/XO ratios that we chose is in the physiologically relevant range produced by activated vascular NADPH oxidase (44, 45).
To characterize the effect of constrained flux through the HMP, quantitative assessment was made of the relation between oxidative load and RBC reducing capacity. This was accomplished by tracking redox ratios for GSH and NADPH in the face of a steady, gradable superoxide production, as afforded by the HX/XO system. Controls were performed in a cell-free system to assure that the severity of oxidative stress presented to normoxic and hypoxic RBCs was equivalent. These controls (Fig. 5) demonstrated that 1) in the presence of HX/XO, buffer oxygen content (pO2 ∼500 or 10 Torr) did not significantly influence the rate of GSH oxidation to GSSG (Fig. 5A); 2) DIDS, DHEA, or KA with HX/XO did not affect the rate of GSH oxidation to GSSG under either high- or low-oxygen conditions (Fig. 5B, C); 3) oxygenated (HbSO2 88%) or deoxygenated Hb (HbSO2 24%) (with 12 or 9% metHb, respectively), in the presence of HX alone, did not result in the oxidation of GSH to GSSG; and 4) Hb, in the presence of HX/XO, significantly accelerated the oxidation of GSH to GSSG by HX/XO (metHb was briskly produced from both oxyHb and deoxyHb, likely from reduction of O2·− to hydrogen peroxide, which decomposes to the hydroxyl radical, ·OH−); this effect did not differ substantially between oxyHb and deoxyHb. These observations reassure that the severity of oxidant stress presented to normoxic and hypoxic RBCs was not significantly altered by the differences in pO2, HbSO2, or by the presence of the inhibitors used.
The data in our cell-free system, however, demonstrated that Hb amplified the oxidative stress from HX/XO, as evidenced by accelerating conversion of GSH to GSSG. This finding suggests that intact RBCs were subjected to greater oxidative stress than were the ghost preparations. Moreover, this observation explains the dramatic reduction in NADH observed in both oxy and deoxy RBCs, since metHb would have been generated at an overwhelming rate in both groups. It is also telling that membrane thiol losses from oxygenated RBCs were similar to those from ghosts (despite the greater stress), while membrane thiol losses from deoxygenated RBCs were significantly greater than in ghosts (reflecting a profound loss in resilience to oxidative stress). Our findings demonstrate that constrained flux through the HMP in deoxygenated RBCs is the cause of limited antioxidant defense in deoxygenated RBCs.
We fully tested the salient features of our RBC oxidant model. As a negative control, we utilized the Band 3 inhibitor DIDS to prevent HX/XO superoxide entry into RBCs (27). This averted the loss of GSH and NADPH recycling in hypoxic RBC and restored the maintenance of reduced membrane thiol. Additionally, we tested other control points of RBCs thiol-based antioxidant defense. This included the manipulation of Hb-cdB3 interaction during hypoxia by stabilizing R state Hb conformation with CO, and also manipulating glycolytic pathway dominance with specific enzyme inhibitors of the EMP and HMP.
It is worth noting the striking similarity between our characterization of hypoxic RBCs in the setting of oxidative stress and previous characterization of RBCs from patients with enzymopathies in the HMP (most notably, G6PD deficiency), all of which are associated with hemolytic anemia and intolerance for oxidative stress (46). This commonality suggests the influence of functional polymorphisms in glycolytic enzymes on phenotype in diseases characterized by hypoxemia or oxidative stress. Moreover, our findings have implications for several classes of clinical problems; specifically, conditions with arterial hypoxemia (e.g., respiratory failure, cyanotic congenital heart disease, altitude illness, obstructive sleep apnea, etc.), conditions in which Hb p50 is altered (hemoglobinopathies, diabetes mellitus, uremia), and for transfusion medicine. Notably, each of the conditions above has been associated with oxidative stress overall and specifically, with oxidative injury to RBCs (particularly to membrane thiols). In addition, these data suggest therapeutic strategies to address oxidative stress by altering interaction between cdB3 and Hb or rebalancing glycolytic flux in hypoxic RBCs.
Overall, our data are consistent with an emerging model of RBC metabolism, in which diverse metabolic streams and signaling pathways articulate with, and are regulated by, O2-linked transitions in Hb conformation and Hb conformation-dependent interactions with cdB3 (15, 16, 20, 22,23,24,25). Diverse RBC functions are coordinated in this fashion and oscillate with O2 gradients during ateriovenous transit; these include ion and amino acid transport (47), glycolysis (15,16,17,18,19), and cytoskeleton-membrane interactions (48), as well as processing of vasoactive effectors in plasma by RBC (5, 6, 12, 13, 49,50,51). Such coupling between regional O2 gradients and RBC metabolism and signaling functions reinforces integration of metabolic demand and the regulation of regional blood flow. This paradigm extends the implications of our findings. While hypoxia may render RBCs vulnerable to oxidative injury, with the direct consequences of altered rheology, hemolysis (20), accelerated clearance (43, 52), and eryptosis (53), oxidative injury may further disrupt O2 delivery through maladaptive alterations in RBC signaling in the microcirculation, which normally maintains coupling between regional blood flow and metabolic demand.
Acknowledgments
We greatly appreciate the gift of KA from Dr. Keiji Hasumi, (Tokyo Noko University, Tokyo, Japan). We thank Dr. Alan Schwartz and Dr. Jonathan Gitlin for thoughtful comments. This work was supported by U.S. National Institutes of Health grant 1K08GM069977 and by an interdisciplinary research initiative from the Children’s Discovery Institute (both to A.D.).
References
- Balagopalakrishna C, Manoharan P T, Abugo O O, Rifkind J M. Production of superoxide from hemoglobin-bound oxygen under hypoxic conditions. Biochemistry. 1996;35:6393–6398. doi: 10.1021/bi952875+. [DOI] [PubMed] [Google Scholar]
- Droge W. Free radicals in the physiological control of cell function. Physiol Rev. 2002;82:47–95. doi: 10.1152/physrev.00018.2001. [DOI] [PubMed] [Google Scholar]
- Gonzalez C, Sanz-Alfayate G, Agapito M T, Gomez-Nino A, Rocher A, Obeso A. Significance of ROS in oxygen sensing in cell systems with sensitivity to physiological hypoxia. Respir Physiol Neurobiol. 2002;132:17–41. doi: 10.1016/s1569-9048(02)00047-2. [DOI] [PubMed] [Google Scholar]
- Kemp M, Go Y M, Jones D P. Nonequilibrium thermodynamics of thiol/disulfide redox systems: a perspective on redox systems biology. Free Rad Biol Med. 2008;44:921–937. doi: 10.1016/j.freeradbiomed.2007.11.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buehler P W, Alayash A I. Oxygen sensing in the circulation: “cross talk” between red blood cells and the vasculature. Antioxidants Redox Signal. 2004;6:1000–1010. doi: 10.1089/ars.2004.6.1000. [DOI] [PubMed] [Google Scholar]
- Singel D J, Stamler J S. Chemical physiology of blood flow regulation by red blood cells: the role of nitric oxide and S-nitrosohemoglobin. Annu Rev Physiol. 2005;67:99–145. doi: 10.1146/annurev.physiol.67.060603.090918. [DOI] [PubMed] [Google Scholar]
- Buehler P W, Alayash A I. Redox biology of blood revisited: the role of red blood cells in maintaining circulatory reductive capacity. Antioxidants Redox Signal. 2005;7:1755–1760. doi: 10.1089/ars.2005.7.1755. [DOI] [PubMed] [Google Scholar]
- Richards R S, Roberts T K, McGregor N R, Dunstan R H, Butt H L. The role of erythrocytes in the inactivation of free radicals. Med Hypotheses. 1998;50:363–367. doi: 10.1016/s0306-9877(98)90206-7. [DOI] [PubMed] [Google Scholar]
- Forman H J, Fukuto J M, Torres M. Redox signaling: thiol chemistry defines which reactive oxygen and nitrogen species can act as second messengers. Am J Physiol Cell Physiol. 2004;287:C246–C256. doi: 10.1152/ajpcell.00516.2003. [DOI] [PubMed] [Google Scholar]
- Mingone C J, Gupte S A, Ali N, Oeckler R A, Wolin M S. Thiol oxidation inhibits nitric oxide-mediated pulmonary artery relaxation and guanylate cyclase stimulation. Am J Physiol Lung Cell Mol Physiol. 2006;290:L549–L557. doi: 10.1152/ajplung.00331.2005. [DOI] [PubMed] [Google Scholar]
- Minetti M, Agati L, Malorni W. The microenvironment can shift erythrocytes from a friendly to a harmful behavior: pathogenetic implications for vascular diseases. Cardiovasc Res. 2007;75:21–28. doi: 10.1016/j.cardiores.2007.03.007. [DOI] [PubMed] [Google Scholar]
- Doctor A, Platt R, Sheram M L, Eischeid A, McMahon T, Maxey T, Doherty J, Axelrod M, Kline J, Gurka M, Gow A, Gaston B. Hemoglobin conformation couples erythrocyte S-nitrosothiol content to O2 gradients. Proc Natl Acad Sci U S A. 2005;102:5709–5714. doi: 10.1073/pnas.0407490102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Palmer L A, Doctor A, Chhabra P, Sheram M L, Laubach V E, Karlinsey M Z, Forbes M S, Macdonald T, Gaston B. S-nitrosothiols signal hypoxia-mimetic vascular pathology. J Clin Invest. 2007;117:2592–2601. doi: 10.1172/JCI29444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bennett-Guerrero E, Veldman T H, Doctor A, Telen M J, Ortel T L, Reid T S, Mulherin M A, Zhu H, Buck R D, Califf R M, McMahon T J. Evolution of adverse changes in stored RBCs. Proc Natl Acad Sci U S A. 2007;104:17063–17068. doi: 10.1073/pnas.0708160104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Messana I, Orlando M, Cassiano L, Pennacchietti L, Zuppi C, Castagnola M, Giardina B. Human erythrocyte metabolism is modulated by the O2-linked transition of hemoglobin. FEBS Lett. 1996;390:25–28. doi: 10.1016/0014-5793(96)00624-2. [DOI] [PubMed] [Google Scholar]
- Barvitenko N N, Adragna N C, Weber R E. Erythrocyte signal transduction pathways, their oxygenation dependence and functional significance. Cell Physiol Biochem. 2005;15:1–18. doi: 10.1159/000083634. [DOI] [PubMed] [Google Scholar]
- Murphy J R. Erythrocyte metabolism. II. Glucose metabolism and pathways. J Lab Clin Med. 1960;55:286–302. [PubMed] [Google Scholar]
- Hamasaki N, Asakura T, Minakami S. Effect of oxygen tension on glycolysis in human erythrocytes. J Biochem. 1970;68:157–161. doi: 10.1093/oxfordjournals.jbchem.a129341. [DOI] [PubMed] [Google Scholar]
- Rapoprot I, Berger H, Rapoport S M, Elsner R, Gerber G. Response of the glycolysis of human erythrocytes to the transition from the oxygenated to the deoxygenated state at constant intracellular pH. Biochim Biophys Acta. 1976;428:193–204. doi: 10.1016/0304-4165(76)90120-3. [DOI] [PubMed] [Google Scholar]
- Bossi D, Giardina B. Red cell physiology. Molec Aspects Med. 1996;17:117–128. doi: 10.1016/0098-2997(96)88343-9. [DOI] [PubMed] [Google Scholar]
- Chu H, Breite A, Ciraolo P, Franco R S, Low P S. Characterization of the deoxyhemoglobin binding site on human erythrocyte band 3: implications for O2 regulation of erythrocyte properties. Blood. 2008;111:932–938. doi: 10.1182/blood-2007-07-100180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Campanella M E, Chu H, Low P S. Assembly and regulation of a glycolytic enzyme complex on the human erythrocyte membrane. Proc Natl Acad Sci U S A. 2005;102:2402–2407. doi: 10.1073/pnas.0409741102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Campanella M E, Chu H, Wandersee N J, Peters L L, Mohandas N, Gilligan D M, Low P S. Characterization of glycolytic enzyme interactions with murine erythrocyte membranes in wild type and membrane protein knockout mice. Blood. 2008;112:3900–3906. doi: 10.1182/blood-2008-03-146159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Low P S, Rathinavelu P, Harrison M L. Regulation of glycolysis via reversible enzyme binding to the membrane protein, band 3. J Biol Chem. 1993;268:14627–14631. [PubMed] [Google Scholar]
- Kinoshita A, Tsukada K, Soga T, Hishiki T, Ueno Y, Nakayama Y, Tomita M, Suematsu M. Roles of hemoglobin Allostery in hypoxia-induced metabolic alterations in erythrocytes: simulation and its verification by metabolome analysis. J Biol Chem. 2007;282:10731–10741. doi: 10.1074/jbc.M610717200. [DOI] [PubMed] [Google Scholar]
- Cimen M Y. Free radical metabolism in human erythrocytes. Clin Chim Acta. 2008;390:1–11. doi: 10.1016/j.cca.2007.12.025. [DOI] [PubMed] [Google Scholar]
- Lynch R E, Fridovich I. Permeation of the erythrocyte stroma by superoxide radical. J Biol Chem. 1978;253:4697–4699. [PubMed] [Google Scholar]
- Feig S A, Segel G B, Shohet S B, Nathan D G. Energy metabolism in human erythrocytes. II. Effects of glucose depletion. J Clin Invest. 1972;51:1547–1554. doi: 10.1172/JCI106951. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Niort G, Boccuzzi G, Brignardello E, Bonino L, Bosia A. Effect of dehydroepiandrosterone on human erythrocytes redox metabolism: inhibition of glucose-6-phosphate dehydrogenase activity in vivo and in vitro. J Steroid Biochem. 1985;23:657–661. doi: 10.1016/0022-4731(85)90018-4. [DOI] [PubMed] [Google Scholar]
- Endo A, Hasumi K, Sakai K, Kanbe T. Specific inhibition of glyceraldehyde-3-phosphate dehydrogenase by koningic acid (heptelidic acid) J Antibiotics. 1985;38:920–925. doi: 10.7164/antibiotics.38.920. [DOI] [PubMed] [Google Scholar]
- Kondo T. Preparation of microcapsules from human erythrocytes: use in transport experiments of glutathione and its S-conjugate. Methods Enzymol. 1989;171:217–225. doi: 10.1016/s0076-6879(89)71013-2. [DOI] [PubMed] [Google Scholar]
- Sun G, Palmer A F. Preparation of ultrapure bovine and human hemoglobin by anion exchange chromatography. J Chromatogr. 2008;867:1–7. doi: 10.1016/j.jchromb.2008.02.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang X, Wu Z, Song G, Wang H, Long M, Cai S. Effects of oxidative damage of membrane protein thiol groups on erythrocyte membrane viscoelasticities. Clin Hemorheol Microcirc. 1999;21:137–146. [PubMed] [Google Scholar]
- Griffith O W. Biologic and pharmacologic regulation of mammalian glutathione synthesis. Free Rad Biol Med. 1999;27:922–935. doi: 10.1016/s0891-5849(99)00176-8. [DOI] [PubMed] [Google Scholar]
- Griffith O W. Glutathione turnover in human erythrocytes. Inhibition by buthionine sulfoximine and incorporation of glycine by exchange. J Biol Chem. 1981;256:4900–4904. [PubMed] [Google Scholar]
- Lunn G, Dale G L, Beutler E. Transport accounts for glutathione turnover in human erythrocytes. Blood. 1979;54:238–244. [PubMed] [Google Scholar]
- Schafer F Q, Buettner G R. Redox environment of the cell as viewed through the redox state of the glutathione disulfide/glutathione couple. Free Rad Biol Med. 2001;30:1191–1212. doi: 10.1016/s0891-5849(01)00480-4. [DOI] [PubMed] [Google Scholar]
- Zerez C R, Lee S J, Tanaka K R. Spectrophotometric determination of oxidized and reduced pyridine nucleotides in erythrocytes using a single extraction procedure. Anal Biochem. 1987;164:367–373. doi: 10.1016/0003-2697(87)90506-9. [DOI] [PubMed] [Google Scholar]
- Wagner T C, Scott M D. Single extraction method for the spectrophotometric quantification of oxidized and reduced pyridine nucleotides in erythrocytes. Anal Biochem. 1994;222:417–426. doi: 10.1006/abio.1994.1511. [DOI] [PubMed] [Google Scholar]
- Messner K R, Imlay J A. In vitro quantitation of biological superoxide and hydrogen peroxide generation. Methods Enzymol. 2002;349:354–361. doi: 10.1016/s0076-6879(02)49351-2. [DOI] [PubMed] [Google Scholar]
- Crimi E, Ignarro L J, Napoli C. Microcirculation and oxidative stress. Free Rad Res. 2007;41:1364–1375. doi: 10.1080/10715760701732830. [DOI] [PubMed] [Google Scholar]
- Fridovich I. Quantitative aspects of the production of superoxide anion radical by milk xanthine oxidase. J Biol Chem. 1970;245:4053–4057. [PubMed] [Google Scholar]
- Baskurt O K, Temiz A, Meiselman H J. Effect of superoxide anions on red blood cell rheologic properties. Free Rad Biol Med. 1998;24:102–110. doi: 10.1016/s0891-5849(97)00169-x. [DOI] [PubMed] [Google Scholar]
- Corbisier P, Houbion A, Remacle J. A new technique for highly sensitive detection of superoxide dismutase activity by chemiluminescence. Anal Biochem. 1987;164:240–247. doi: 10.1016/0003-2697(87)90392-7. [DOI] [PubMed] [Google Scholar]
- Souza H P, Liu X, Samouilov A, Kuppusamy P, Laurindo F R, Zweier J L. Quantitation of superoxide generation and substrate utilization by vascular NAD(P)H oxidase. Am J Physiol Heart Circ Physiol. 2002;282:H466–H474. doi: 10.1152/ajpheart.00482.2001. [DOI] [PubMed] [Google Scholar]
- Valentine W N, Tanaka K R, Paglia D E. Hemolytic anemias and erythrocyte enzymopathies. Ann Intern Med. 1985;103:245–257. doi: 10.7326/0003-4819-103-2-245. [DOI] [PubMed] [Google Scholar]
- Gibson J S, Cossins A R, Ellory J C. Oxygen-sensitive membrane transporters in vertebrate red cells. J Exp Biol. 2000;203:1395–1407. doi: 10.1242/jeb.203.9.1395. [DOI] [PubMed] [Google Scholar]
- Chang S H, Low P S. Regulation of the glycophorin C-protein 4.1 membrane-to-skeleton bridge and evaluation of its contribution to erythrocyte membrane stability. J Biol Chem. 2001;276:22223–22230. doi: 10.1074/jbc.M100604200. [DOI] [PubMed] [Google Scholar]
- McMahon T J, Moon R E, Luschinger B P, Carraway M S, Stone A E, Stolp B W, Gow A J, Pawloski J R, Watke P, Singel D J, Piantadosi C A, Stamler J S. Nitric oxide in the human respiratory cycle. Nat Med. 2002;8:711–717. doi: 10.1038/nm718. [DOI] [PubMed] [Google Scholar]
- Pawloski J R, Hess D T, Stamler J S. Export by red blood cells of nitric oxide bioactivity. Nature. 2001;409:622–626. doi: 10.1038/35054560. [DOI] [PubMed] [Google Scholar]
- Dietrich H H, Ellsworth M L, Sprague R S, Dacey R G., Jr Red blood cell regulation of microvascular tone through adenosine triphosphate. Am J Physiol Heart Circ Physiol. 2000;278:H1294–H1298. doi: 10.1152/ajpheart.2000.278.4.H1294. [DOI] [PubMed] [Google Scholar]
- Uyesaka N, Hasegawa S, Ishioka N, Ishioka R, Shio H, Schechter A N. Effects of superoxide anions on red cell deformability and membrane proteins. Biorheology. 1992;29:217–229. doi: 10.3233/bir-1992-292-303. [DOI] [PubMed] [Google Scholar]
- Lang K S, Lang P A, Bauer C, Duranton C, Wieder T, Huber S M, Lang F. Mechanisms of suicidal erythrocyte death. Cell Physiol Biochem. 2005;15:195–202. doi: 10.1159/000086406. [DOI] [PubMed] [Google Scholar]