

Abstract
We recently generated an HT-1080-derived cell line called HT-AR1 that responds to dihydrotestosterone (DHT) treatment by undergoing cell growth arrest in association with cytoskeletal reorganization and induction of neuroendocrine-like cell differentiation. In this report, we show that DHT induces a dose-dependent increase in G0/G1 growth-arrested cells using physiological levels of hormone. The arrested cells increase in cell size and contain a dramatic redistribution of desmoplakin, keratin 5, and chromogranin A proteins. DHT-induced cytoskeletal changes were also apparent from time lapse video microscopy that showed that androgen treatment resulted in the rapid appearance of neuronal-like membrane extensions. Expression profiling analysis using RNA isolated from DHT-treated HT-AR1 cells revealed that androgen receptor activation leads to the coordinate expression of numerous cell signaling genes including RhoB, PTGF-β, caveolin-2, Egr-1, myosin 1B, and EHM2. Because RhoB has been shown to have a role in tumor suppression and neuronal differentiation in other cell types, we investigated RhoB signaling functions in the HT-AR1 steroid response. We found that steroid induction of RhoB was DHT-specific and that newly synthesized RhoB protein was post-translationally modified and localized to endocytic vesicles. Moreover, treatment with a farnesyl transferase inhibitor reduced DHT-dependent growth arrest, suggesting that prenylated RhoB might function to inhibit HT-AR1 cell proliferation. This was directly shown by transfecting HT-AR1 cells with RhoB coding sequences containing activating or dominant negative mutations.
Steroid signaling controls numerous cellular processes in development that require cytoskeletal reorganization to facilitate cell migration during embryogenesis (1, 2). In addition, neuronal precursor cells are sensitive to estrogen and androgen treatment that induces cytoskeletal changes resulting in increased neurite outgrowth (3). These steroid-regulated cytoskeletal responses are poorly understood and may be related to steroid effects on cancer cell migration and tumor metastasis (4, 5).
Recently, LNCaP cells (6, 7) and the mouse fibroblast cell line NIH3T3 (8) have been shown to respond to androgen signaling by inducing rapid cytoskeletal changes that appear to reflect nongenomic signaling mechanisms (9–11). Because both LNCaP and NIH3T3 cells express endogenous androgen receptor (AR), 1 the use of AR mutants to investigate genomic and nongenomic mechanisms involved in cytoskeletal reorganization in these cell lines is not feasible. Therefore, we recently developed an alternative cell model to study androgen control of cytoskeletal organization by taking advantage of the well characterized human cell line HT-1080 (12). This fibrosarcoma cell line responds to glucocorticoid treatment by undergoing cytoskeletal changes that are associated with increased fibronectin expression (13–16). The HT-1080 cell line was established in 1974 from a tumor biopsy taken from the acetabulum of a 35-year-old male who had not received chemotherapy and died of metastatic disease without an autopsy 3 months after diagnosis (12). It has been shown that HT-1080 cells express functional glucocorticoid receptor, but lack AR, progesterone receptor, and mineralocorticoid receptor (17). Because androgen and glucocorticoid responses are partially overlapping in a variety of cell types (18–20), we reasoned that ectopic expression of human AR in HT-1080 cells might recapitulate some or all of the steroid-induced cytoskeletal changes seen with glucocorticoid receptor, and moreover, provide a null genetic background to investigate molecular determinants of AR signaling.
As described in Chauhan et al. (21), stable transfection of HT-1080 cells with a puromycin-resistant expression vector encoding full-length human AR led to the isolation of several subclones, including HT-AR1, which was shown to express normal levels of functional AR protein. Bourgeois and colleagues (13, 14) had shown that dexamethasone (Dex) treatment of HT-1080 cells induced fibronectin expression without altering cell proliferation. Similarly we found that DHT treatment induced fibronectin protein expression, however, unlike Dex, DHT treatment also led to pronounced HT-AR1 cell growth arrest and increased expression of chromogranin A, neuron-specific enolase, and the recently discovered FERM domain encoding gene EHM2 (21). The androgen response of HT-AR1 cells was shown to be AR-dependent because a puromycin-resistant HT-1080 subclone containing only the expression vector (HT-VC1) was insensitive to DHT treatment.
In this report, we describe results of experiments aimed at identifying key downstream signaling events that are required for the AR-dependent response of HT-AR1 cells. Cell biological studies and expression profiling demonstrated that androgen signaling induces transcriptional reprogramming of HT-AR1 cells resulting in cell cycle arrest, cytoskeletal reorganization, and coordinate expression of numerous cell signaling genes. One of these differentially expressed genes was RhoB, a small GTPase that is post-translationally modified by prenylation and functions to regulate cell growth (22–24), cytoskeletal reorganization (23, 25, 26), and cell differentiation (27–29). Although much is known about the upstream and downstream effectors of RhoA, Rac1, and Cdc42, the best characterized members of this family of small GTPases, regulation of RhoB activity, and identification of RhoB targets are not well understood (30). To investigate RhoB signaling in fibrosarcoma cells, we use a combination of experimental approaches to demonstrate that DHT-induced expression of RhoB in HT-AR1 cells contributes to the androgen response phenotype by mediating growth arrest, a result that is consistent with the proposed tumor suppressing activities of RhoB (22–24).
EXPERIMENTAL PROCEDURES
Cell Cycle Analysis
Puromycin-resistant HT-1080 subclones expressing AR protein were isolated in selection media as previously described (21). Selection media was made by combining Dulbecco’s modified Eagle’s media with defined calf bovine serum (CBS) (Hyclone, Logan, UT) to 10% (v/v), streptomycin (Sigma) to 0.1 mg/ml, penicillin G (Sigma) to 100 units/ml, 0.3 μg/ml puromycin (Sigma), and filter sterilized in the final step with a 0.22 μM filter membrane. Five days prior to an experiment, HT-AR1 cells were grown in selection media to reselect cells containing the AR expression vector (pDC-hAR-PAC (21)). All experiments were performed in culture media (selection media without puromycin) containing 10% CBS or 5% heat-inactivated charcoal-stripped (C/S) serum as described in the text. Cell cycle analysis of HT-AR1 cells was performed after growth for 3 days in culture media with or without 10 nM DHT. The cells were harvested by trypsinization, washed with phosphate-buffered saline, and fixed in methanol at 4 °C for 15 min. The cells were then incubated in modified Kirshan buffer (20 μg/ml RNase A, 3.4 mM sodium citrate, 0.3% Nonidet P-40) containing 50 μg/ml propidium iodide. Propidium iodide fluorescence of nuclei was determined using FACScan (BD Biosciences) and analyzed with CELLQuest ModFit LT 2.0.
Immunostaining
Detection of protein expression in HT-AR1 cells by immunostaining was performed using antibodies specific for the androgen receptor (Santa Cruz SC-815, 1:10 dilution), desmoplakin (Progen antibodies 2.15, 2.17, 2.20, undiluted mixture), cytokeratin 5 (BD Pharmingen RCK 103, 1:10 dilution), chromogranin A (Zymed Laboratories Inc. 18-0094, 1:60 dilution), RhoB (Santa Cruz SC-180, 1:10 dilution), and hemagglutinin (Roche 3F10, 1:10 dilution). Cells were grown in culture media containing 5% C/S CBS on 12-mm coverslips coated with human fibronectin, allowed to adhere overnight, and then supplemented with fresh media ± DHT (10 nM). After 4 days, the coverslips were harvested, dipped sequentially in phosphate-buffered saline, chilled in 100% methanol (10 min) and cold acetone (dipped six times), and then air dried. Diluted primary antibodies were applied to cells for 30 min at room temperature and then the coverslips were washed three times in phosphate-buffered saline for 5 min each. Primary antibodies were detected with either anti-rabbit Alexa 568 or anti-mouse Alexa 568 (Molecular Probes) using 1:400 dilution. For some experiments, nuclei were stained with Hoechst 33258 (Sigma) using a stock of 2 mg/ml diluted to 1:100 for use, rinsed twice in phosphate-buffered saline for 2–3 min each, and post-fixed in 100% ethanol and mounted using Mowial (Sigma). Stained cells were visualized using appropriate filters on a Zeiss Axiophot 200 microscope and imaged using Axiovision software.
Time Lapse Video Microscopy
Cells were seeded on a glass Delta T dish (0.15 mm; Bioptechs, Inc. Butler, PA) at 40,000/dish and grown overnight in Dulbecco’s modified Eagle’s media selection containing 10% CBS at 37 °C. The following morning, fresh culture media with 10% CBS was added with or without DHT and the cells were incubated for 1 h at 37 °C before beginning the time lapse video. Images were captured using a grayscale CCD camera (ORCA-100, Hamamatsu, Japan) mounted on an inverted Olympus IMT2 microscope (Olympus America, Melville, NY) equipped with a BiopTechs Delta T live cell system (Bioptechs, Inc. Butler, PA) under a humidified (5% CO2 balanced with ultrazero air) atmosphere. Data analysis was performed using SimplePCI 4.0 software (Compix Imaging Inc., Cranberry Township, PA). Representative video images obtained from HT-AR1 cells grown in ±DHT media during the first 15 min are presented in Fig. 3 and 5 h of the 16-h time lapse video are contained in the Supplemental Materials (available at http://www.jbc.org).
Fig. 3. Time lapse video microscopy of HT-AR1 cells shows rapid changes in membrane morphology in DHT cultures.

Cells were seeded on a glass Delta T dish and grown overnight in Dulbecco’s modified Eagle’s selection media and then changed to culture media the next day with or without DHT. Images were captured every 5 min beginning with T0, which was 1 h after adding culture media ±DHT. Representative fields from the two cultures are shown of the first 15 min of video microscopy. The arrows identify individual cells in the same field over the time course. QuickTime videos of these two cultures out to 5 h are included as Supplemental Materials (available at http://www.jbc.org).
Fig. 5. Western blot of selected proteins expressed in HT-AR1 cells.
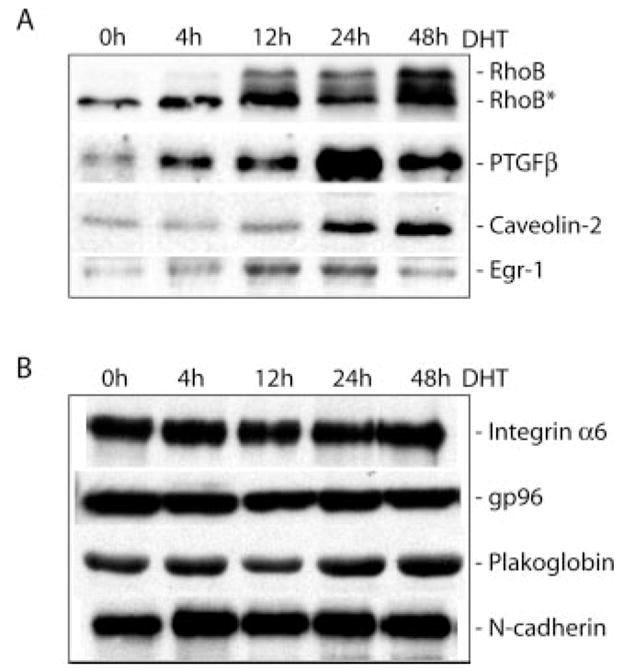
Whole cell protein extracts were prepared from HT-AR1 cells grown in DHT-containing media for the indicated times and separated by 7.5 or 12.5% SDS-PAGE. A, differential expression of RhoB, PTGFβ, caveolin-2, and Egr-1 proteins in DHT-treated HT-AR1 cells. The RhoB antibody detects both the unmodified and post-translationally modified (RhoB*) forms of the protein (50, 51). B, expression levels of integrin α6, gp96, plakoglobin, and N-cadherin proteins are not altered by DHT treatment in HT-AR1 cells under these same conditions.
Expression Profiling
Total RNA was isolated from untreated and DHT-treated HT-AR1 cells using the RNA-Bee™ RNA isolation reagent (Tel-test, Inc., Friendswood, TX). Fluorescently labeled cDNA was prepared from 30 μg of total RNA using Micromax direct cDNA microarray system (PerkinElmer Life Sciences) and Cy3 (control) or Cy5 (experimental) fluorescent dyes. Labeled cDNA was purified using Qiagen QIAquick PCR purification kit (Qiagen, Valencia, CA), combined into one tube, and precipitated at −20 °C overnight using 3 M sodium acetate (pH 5.2) and isopropyl alcohol. The human cDNA microarray slides containing 5,300 human gene sequences were obtained from the Microarray Core Service of the Arizona Cancer Center.2 Slide hybridization, washing, and processing were performed as previously described (21). Slides were scanned for fluorescent emission using Gene Pix 4000A microarray Scanner (Axon Instruments, Inc., Foster City, CA). GenePix Pro 4.0 software (Axon Instruments, Inc.) was used to analyze the fluorescence intensities and to format the intensity data as log-transformed ratio values. The data were filtered by eliminating genes in which no hybridization signal was detected at two or more time points. All genes used for expression profiling analysis were validated by replica hybridizations and by visual inspection of the primary data using GenePix 4.0 analysis software. The Arizona Cancer Center BioResource for Gene Array analysis (BioRag) was used for initial screening of differentially expressed genes to identify candidates most likely involved in cell signaling. This resource can be accessed online.3 A complete list of DHT-regulated genes in HT-AR1 cells can be obtained on request.
Northern Blots
Total RNA was isolated from untreated and DHT-treated HT-AR1 cells as described above and equal amounts of RNA (20 μg) were run on a denaturing formaldehyde gel and transferred to a Duralon-UV™ membrane (Stratagene, La Jolla, CA) by standard capillary methods using 10× SSC. Subsequently, RNA was UV cross-linked to the nylon membrane using UV Stratalinker (Stratagene) and Northern blotting was performed using radioactively labeled cDNA probes as described (21). The GenBank™ sequences used to generate cDNA probes from ATCC clones were RhoB (X06820), PTGF-β (AA450062), CAV2 (T89391), Egr-1 (AA486628), Myo1B (N95358), EHM2 (AA948217), FKBP51 (W86653), and urokinase plasminogen activator (AA284668). Sequential hybridization and rehybridization was performed with each of the eight cDNA probes using the same Duralon-UV membrane after stripping the blot twice with hot (92 °C) 0.02% SDS in double distilled H2O after each hybridization round.
Western Blots
For most blots, cells were lysed on the tissue culture dish with RIPA buffer (150 mM NaCl, 50 mM Tris, 5 mM EDTA, 1% (v/v) Triton X-100, 1% (w/v) deoxycholate, 0.1% (w/v) SDS, pH 7.5) plus protease inhibitors (phenylmethylsulfonyl fluoride, 2 mM; leupeptin and aprotinin, 1 μg/ml). The lysate was briefly sonicated on ice and protein concentration was determined by the BCA assay (Pierce). Protein samples for SDS-PAGE were prepared by adding 20 μg of protein with equal volumes of 2× reducing sample buffer. Samples were boiled for 5 min and after a quick chill on ice they were loaded onto 7.5, 12.5, or 4 –20% gradient SDS-PAGE gels. Following electrophoresis, proteins were electrotransferred to Millipore Immobilon-P polyvinylidene fluoride membrane (Millipore, Bedford, MA), and incubated with antibodies specific for α-tubulin (Sigma B512), RhoB (Santa Cruz SC-180), PTGFβ (Santa Cruz SC-10603), caveolin-2 (BD Bioscience 610684), EGR-1 (Santa Cruz SC-110), integrin α6 (AA6A previously described (31)), gp96 (SPE850, clone 9G10, Stressgen Biotechnologies, Victoria, British Columbia), plakoglobin (Chemicon International, MAB2083), anti-pan-cadherin (as described in Ref. 32), fibronectin (Santa Cruz SC-9068), or hemagglutinin (Roche 3F10). Secondary antibodies used in Western blotting were conjugated to horseradish peroxidase and visualized by chemiluminescence (ECL Western blotting Detection System, Amersham Biosciences).
DNA Transfections
Transient transfections were performed in 6- or 12-well plates using 1 μg of plasmid DNA and Polyfect transfection reagent according the manufacturer’s recommendations (Qiagen, Valencia, CA). DHT was added after 4 h of Polyfect treatment and cells were harvested 16 –24 h later for cell counting and immunostaining with anti-HA antibody. The human RhoB pCDNA3.1 plasmids were obtained from the Guthrie Institute (Sayer, PA) and contained a 3× hemagglutinin (HA) epitope tag at the N terminus of either an activating RhoB mutation (G14V) called HA-RhoBact, or a dominant negative RhoB mutation (T19N) called HA-RhoBdn. The HT-AR1 stable cell lines were generated by recloning the HA-RhoBact and HA-RhoBdn constructs into the tetracycline-regulated expression vector (pCDNA4/TO) (T-Rex expression system, Invitrogen, Inc.). The pCDNA4/TO RhoB plasmids were stably co-transfected into HT-AR1 cells with the tetracycline repressor expressing plasmid pCDNA6/TR at a molar ratio of 1:6 using Avanti DOTAP:DOPE lipofection reagent (Alabaster, AL). Both the TO and TR vectors were linearized before transfection with the enzyme Bst1107, which cleaves once in the vector backbone. Colonies were isolated and expanded in selection media containing 10% tetracycline-free serum (Hyclone, Logan, UT), 1.0 μg/ml zeocin, and 0.6 μg/ml blasticidin according to the manufacturer’s recommendations (Invitrogen Inc.). Candidate HT-AR1 T-Rex subclones were screened for tetracycline-regulated expression by treating cells with 1.0 μg/ml doxycycline (Dox) for 16 h and preparing protein extracts for analysis by anti-HA Western blots. The efficiency of obtaining HT-AR1 subclones that exhibited low basal level expression of HA-RhoB protein with a 10–50-fold Dox induction was >70%.4
RESULTS
Physiological Levels of DHT Induce G0/G1 Growth Arrest
Treatment of HT-AR1 cells with 100 nM DHT was shown by Chauhan et al. (21) to induce growth arrest over a 3–7-day period with no evidence of apoptosis. To determine whether the observed DHT-dependent growth inhibition was because of super physiological levels of steroid, we cultured HT-AR1 cells for 4 days with or without the addition of 10 pM to 1 μM DHT. As shown in Fig. 1A, HT-AR1 cell growth was found to be inhibited up to 90% by DHT concentrations ranging from 1 nM to 1 μM, with a small but reproducible growth stimulatory effect at 100 pM DHT.
Fig. 1. Growth inhibition and cell cycle arrest in DHT-treated HT-AR1 cells.

A, dose response profile of HT-AR1 cells cultured for 5 days in media containing carrier alone (ETOH) as compared with media containing DHT at concentrations between 10−11 and 10−6 M. Values plotted are mean numbers ± S.E. of viable cells relative to the untreated control. B, cell cycle parameters of HT-AR1 cell cultures maintained in media with or without DHT for 3 days as determined by propidium iodide fluorescence using FACS. Percentage of cells in G0/G1, S, and G2/M phases are shown. The inset shows a plot of FACS forward and side scatter data as an indication of cell size under each culture condition.
To better understand the nature of this growth arrest phenotype, we performed cell cycle analysis using fluorescent activated cell sorting (FACS) to determine whether growth inhibition was because of cell cycle arrest. The data shown in Fig. 1B revealed that following 3 days of culture in DHT, there was a 28% increase in the number of HT-AR1 cells in the G0/G1 phase of the cell cycle relative to untreated cells. Moreover, we observed a concomitant decrease in the number of HT-AR1 cells in the S and G2/M phases of the cell cycle, showing only 62 (S) and 83% (G2/M) of the cells in DHT-treated cultures as compared with untreated controls. In addition, cell size determinations based on forward and side scatter measurements showed that DHT-treated HT-AR1 cells were consistently larger than untreated cells (Fig. 1B, inset).
Cytoskeletal Reorganization in DHT-treated HT-AR1 Cells
Based on the finding that DHT treatment of HT-AR1 cells induced a growth arrest phenotype that was accompanied by an increase in cell size (Fig. 1B), we used immunostaining to determine whether the expression level and cellular distribution of desmoplakin, keratin 5, and chromogranin A proteins was altered by DHT treatment as might be expected for differentiating cells. As shown in Fig. 2A, HT-AR1 cells displayed a spindle cell shape and grew to high cell densities when cultured in C/S CBS media on fibronectin-coated coverslips for 4 days. However, when DHT was added to the media for the same time period, the cells flattened out and cell boundaries were greatly extended. Immunostaining of these cells with AR antibody revealed that DHT treatment resulted in predominant nuclear localization of AR that would be consistent with the induction of a AR-mediated genomic response in HT-AR1 cells (33).
Fig. 2. DHT treatment of HT-AR1 cells induces cytoskeletal reorganization and neuroendocrine-like differentiation.

A, cell morphology and AR protein expression in HT-AR1 cells cultured for 3 days on fibronectin-coated coverslips in Dulbecco’s modified Eagle’s media containing 5% C/S CBS serum with or without 10 nM DHT. AR protein was detected by immunostaining using an anti-AR antibody. The same field of cells is shown by phase-contrast or fluorescence microscopy. B, HT-AR1 cells cultured the same as in A and stained with antibodies that specifically recognize desmoplakin, keratin 5, or chromogranin A. The cells in the keratin 5 immunostaining were counter-stained with Hoechst dye to visualize the nuclei.
Using the same growth conditions, the data in Fig. 2B show that untreated HT-AR1 cells contained diffuse desmoplakin staining, whereas DHT-treated HT-AR1 cells displayed a clear condensation of desmoplakin along cell boundaries (see arrowheads). Desmoplakin polarization at cell boundaries is characteristic of differentiated cells undergoing desmosome production (34). Consistent with our earlier results (21), immunostaining of DHT-treated HT-AR1 cells using an anti-keratin 5 antibody (RCK103), revealed increased levels of highly organized keratin 5 protein that extended into intracellular networks. The expression of cytokeratin 5 in DHT-treated HT-AR1 cells suggests that HT-1080 cells can acquire epithelial cell-like qualities upon activation of androgen signaling. We have previously confirmed that the HT-AR1 subclone and HT-1080 cell line are isogenic by comparing the HT-AR1 and HT-1080 genotypes at 11 independent single nucleotide polymorphisms (21).
Western blotting had showed that DHT-treated HT-AR1 cells have elevated expression of the neuroendocrine marker proteins chromogranin A and neuron-specific enolase (21). To determine whether DHT treatment causes characteristic redistribution of chromogranin A into secretory granules on the surface of HT-AR1 cells, we used immunostaining to visualize chromogranin A protein expression. The results shown in Fig. 2B revealed that in the absence of DHT, HT-AR1 cells were faintly positive for chromogranin A staining. However, following DHT treatment, there was a dramatic increase in chromogranin A expression manifested as a pattern of secretory granules. These results are consistent with our chromogranin A immunoblotting results (21) and suggest that AR signaling activates an intracellular signaling pathway in HT-AR1 cells that leads to the induction of a neuroendocrine-like differentiation program.
One of the striking morphological changes we had observed in DHT-treated HT-AR1 cells was the appearance of neuronal-like membrane extensions (21). Based on the recent report of Castoria et al. (8) showing that androgen signaling in mouse NIH3T3 cells caused AR-dependent changes in membrane structure within minutes of hormone addition, we used time lapse video imaging to monitor HT-AR1 cell morphology and motility in the absence and presence of DHT. For these experiments, HT-AR1 cells were plated in glass Delta T culture dishes at 40,000 cells/well and digital images were captured every 5 min for 16 h. Representative screen images of HT-AR1 cell cultures at 5-min intervals over a course of 15 min are presented in Fig. 3 where it can be seen that DHT treatment caused significant alterations in the size and location of membrane extensions (see arrows). In contrast, cells cultured in the absence of DHT exhibited very little movement or change in cell morphology over the same 15-min time period. The first 5 h of the 16-h experiment are included as a video movie under Supplemental Materials (available at http://www.jbc.org).
Transcriptional Control of HT-AR1 Cell Proliferation and Differentiation
To determine whether AR-mediated growth arrest and differentiation was accompanied by significant changes in the HT-AR1 cell transcriptome, we used expression profiling to identify differentially expressed genes during the first 48 h of DHT treatment. For these experiments, we used a 5.3k human cDNA array obtained from the Arizona Cancer Center and co-hybridized Cy3-labeled cDNA produced with RNA isolated from untreated HT-AR1 cells and Cy5-labeled cDNA using RNA from HT-AR1 cells treated with DHT for 4, 12, 24, or 48 h. Data analysis was performed with GenePix 4.0 software as described under “Experimental Procedures.” Genes were considered differentially expressed if reproducible fluorescence signals were observed at multiple time points and there was at least a 3-fold difference in expression level (induced or repressed). Based on these criteria, we identified 148 differentially expressed genes of a total of 2355 that displayed a detectable level of expression in HT-AR1 cells. Of these 148 genes, 34 encoded proteins in the GenBank™ data base that were broadly defined as regulatory proteins. The complete list of 148 differentially expressed genes in HT-AR1 cells can be obtained on request.3
Transcriptional changes identified by expression profiling were confirmed by Northern blotting using cDNA probes specific for eight of the most highly regulated cell signaling genes. For these experiments, sequential hybridization, stripping, and rehybridization of the same Duralon-UV nylon filter membrane was used to analyze the expression of the RhoB, PTGF-β, CAV2, Egr-1, Myo1B, EHM2, FKBP51, and urokinase genes in DHT-treated HT-AR1 cells. As shown in Fig. 4, these Northern blot analyses confirmed that RhoB, EHM2, and FKBP51 were early androgen response genes based on DHT-induced expression within the first 4 h of treatment. RhoB is a small GTPase involved in intracellular signaling and tumor suppression (30, 35), whereas, EHM2 is a FERM domain encoding gene that may be involved in cytoskeletal reorganization (21). The FKBP51 gene has been shown to be steroid-regulated in a number of cell types (36, 37) and the FKBP51 protein appears to function in protein folding as a component of the steroid receptor-chaperone complex (38, 39).
Fig. 4. Northern blot of differentially expressed genes in DHT-treated HT-AR1 cells.
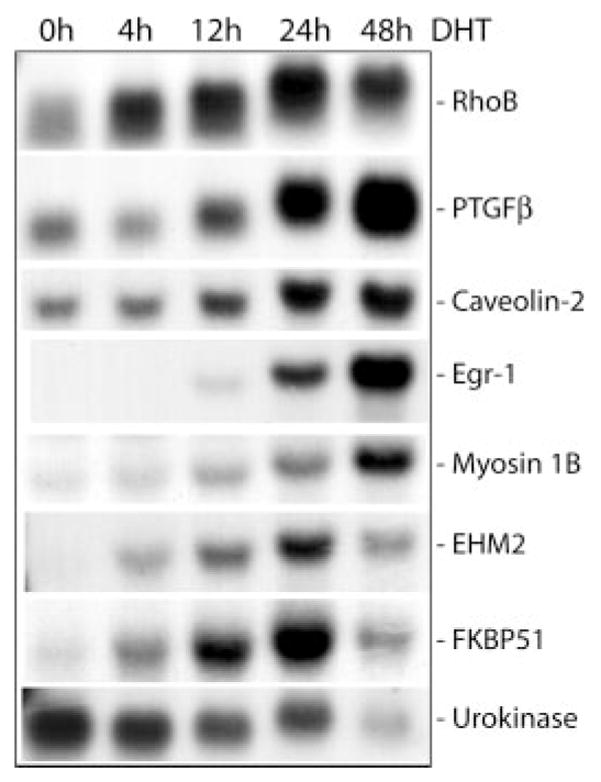
Total RNA was isolated from HT-AR1 cells grown in DHT-containing media for the indicated times and electrophoresed under denaturing conditions. The Northern blot was sequentially hybridized with 32P-labeled cDNA probes specific for RhoB, PTGF-β, CAV2, Egr-1, Myo1B, EHM2, FKBP51, and urokinase as described under “Experimental Procedures.” The size of each gene transcript was verified using RNA molecular weight markers (data not shown).
Fig. 4 also shows the DHT-induced expression of the placental TGFβ (PTGF-β) gene that encodes a TGFβ-related peptide hormone that is expressed at high levels in placental tissue (40) and has been shown to be androgen-regulated in rat ventral prostate cells (41). Other names for PTGF-β are NAG-1 (42), MIC-1 (43), and GDF-15 (44). Three additional DHT-induced genes we identified in HT-AR1 cells were CAV2 that may be involved in membrane signaling (45) and has been shown to be induced 25-fold during adipocyte differentiation (46); Egr-1, an early response gene encoding a zinc finger transcription factor that controls the expression of numerous cell growth and differentiation genes (47); and Myo1B, a member of the myosin superfamily that plays a role in membrane dynamics and transport (48). Last, the data in Fig. 4 reveal that DHT treatment of HT-AR1 cells caused significant down-regulation of the urokinase plasminogen activator gene, a well characterized serine protease that is also down-regulated in HT-1080 cells by glucocorticoid treatment (49).
Fig. 5 shows the results of Western blots using antibodies directed against some of these protein products. In addition to RhoB, it can be seen that PTGFβ, caveolin-2, and EGR-1 protein levels are all increased relative to that of integrin α6, gp96, plakoglobin, and N-cadherin proteins. EGR-1 protein levels were increased transiently between 12 and 24 h, whereas, PTGF-β and caveolin-2 protein levels were not significantly increased until 24 –48 h post-treatment. Note that the RhoB protein is post-translationally modified by farnesyl (C15) or geranylgeranyl (C20) at the carboxyl-terminal CAAX sequence (22), and these modifications increase the electrophoretic mobility of RhoB protein in polyacrylamide gels as shown by RhoB* (50, 51). Consistent with this interpretation, nascent RhoB protein is unmodified (slower migrating protein), however, over time, the majority of this species is prenylated and accumulates as the modified form. These protein expression data confirm the Northern blotting results and indicate that androgen signaling in HT-AR1 cells involves numerous changes at both the RNA and protein levels of select cell signaling genes.
Activation of RhoB Signaling Induces Growth Arrest in HT-AR1 Cells
Among the genes we characterized at the RNA and protein levels, we were especially interested in RhoB because of its potential to be a key mediator in HT-AR1 growth arrest (22–24), cytoskeletal reorganization (23, 25, 26), and cell differentiation (27–29). As shown in Fig. 6A, RhoB immunostaining of HT-AR1 cells grown on fibronectin-coated coverslips in the presence or absence of 10 nM DHT resulted in the appearance of a punctuate cytoplasmic staining pattern that is indicative of RhoB localization to endocytic vesicles (26, 35). Fig. 6B shows a Northern blot in which it can be seen that RhoB transcription is induced by DHT but not Dex treatment, suggesting that RhoB expression might be involved in mediating the HT-AR1 growth arrest because DHT treatment induces both growth arrest and RhoB expression, whereas, Dex treatment has no effect on cell proliferation (21).
Fig. 6. Characterization of RhoB expression in DHT-treated HT-AR1 cells.
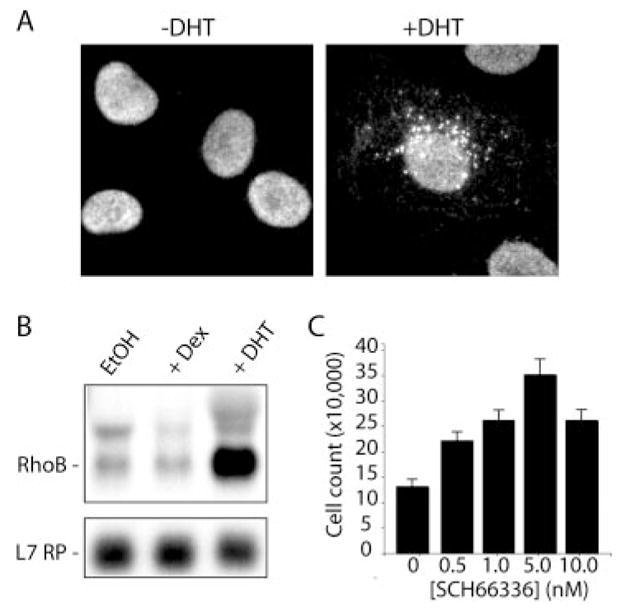
A, HT-AR1 cells were treated with DHT for 3 days and immunostained with anti-RhoB antibody. Characteristic punctate pattern of RhoB protein in DHT-treated HT-AR1 cells is consistent with RhoB localization to endocytic vesicles as seen in other cell types (26, 35). B, Northern blot showing androgen-specific induction of RhoB expression in HT-AR1 cells after 48 h of treatment with 10 nM DHT or 100 nM Dex. The L7 ribosomal protein gene was used as an RNA loading control. C, treatment of HT-AR1 cells with the farnesyl transferase inhibitor SCH66336 partially blocks DHT-induced growth arrest. Cells were grown in 10 nM DHT for 3 days with or without the indicated concentrations of SCH66336 and viable cells were counted by trypan blue exclusion. Mean values are shown ± S.E.
To follow-up on the RhoB expression studies, we took advantage of the finding that some fraction of prenylated RhoB protein in cells is farnesylated (22), unlike RhoA, Rac, and cdc42, which are only geranylgeranylated (30). Farnesyl transferase inhibitors such as SCH66336 have been used to block RhoB signaling in cells by inhibiting farnesylation and membrane localization that is required for RhoB activity (51). As shown in Fig. 6C, when adding SCH66336 to media containing 10 nM DHT, we observed a 3-fold increase in HT-AR1 cell growth as compared with growth in DHT media lacking SCH66336. Importantly, this range of SCH66336 concentrations (1–10 nM) had no effect on HT-AR1 cell viability or proliferation in the absence of DHT (data not shown), and was in the concentration range needed to inhibit Ras activity (3–6 nM) (52). Whereas SCH66336 treatment did not completely reverse the growth arrest phenotype, these data do suggest that DHT induction of RhoB expression, and lipid modification of RhoB protein by the enzyme farnesyl transferase, contribute to the response.
A second more direct experiment was performed using RhoB expression vectors encoding either a constitutively active variant of RhoB (RhoBact) containing a point mutation in the GTPase domain (G14V), or a dominant negative mutant (RhoBdn) that is defective in nucleotide binding (T19N). These human RhoB cDNA constructs contained three copies of the hemagglutinin epitope tag (HA3) and were cloned into the pcDNA3.1 + eukaryotic expression vector (Guthrie Institute). To determine whether ectopic expression of RhoB affected HT-AR1 cell proliferation or cytoskeletal reorganization, we transiently transfected HT-AR1 cells with the two RhoB constructs, or with vector alone and cultured the cells in ±DHT media for 48 h. As shown in Fig. 7A, transfection with the constitutively active RhoBact variant caused growth arrest independent of DHT treatment, whereas, cells transfected with the dominant negative RhoBdn construct were partially protected from DHT-induced growth arrest. Immunostaining with anti-HA antibody (Fig. 7B) showed that DHT induced increase in cell size still occurred in RhoBact- or RhoBdn-transfected cells, suggesting that under these conditions, RhoB signaling stimulated growth arrest but not cytoskeletal reorganization.
Fig. 7. Transient transfection of HT-AR1 cells with a constitutively active form of RhoB induces growth arrest.
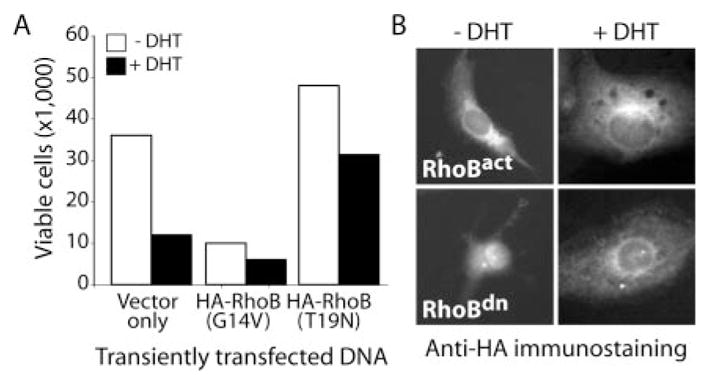
A, transient transfection of HT-AR1 cells with the pCDNA3.1 vector alone, or the same vector encoding HA-tagged activated HA-RhoBact (G14), or dominant negative HA-RhoBdn (T19N) variant, alters cell proliferation. Transfected cells were cultured in the presence or absence of 10 nM DHT for 3 days and viable cells were counted by trypan blue exclusion. B, representative results of HA immunostaining of transiently transfected HT-AR1 cells from the same experiment as in A. Results show that DHT-induced cytoskeletal reorganization was not affected by ectopic expression of RhoBact or RhoBdn. The HA immunostaining showed that >60% of the HT-AR1 cells were HA positive 3 days after transfection (data not shown).
To better control the level of ectopic RhoB protein in transfected HT-AR1 cells, we recloned the HA-RhoBact and HA-RhoBdn constructs into a tetracycline-regulated expression vector (pCDNA4/TO) that could be stably co-transfected into cells with the tetracycline repressor (TetR) expressing plasmid pCDNA6/TR (Invitrogen Inc.). Because the two plasmids carry different antibiotic resistance genes (blasticidin and zeocin), subclones containing both the target plasmid and the TetR plasmid can be selected in a single round of transfection (53). For these experiments, we used the pCDNA4/TO plasmid encoding the HA-RhoBact or HA-RhoBdn genes, or empty vector, and co-transfected HT-AR1 cells with linearized pCDNA6/TR plasmid at a ratio of 1:6, TO:TR. Double antibiotic resistant colonies were isolated after 10 –14 days and protein extracts from Dox-treated cells were analyzed by Western blotting using an anti-HA antibody. Based on the results of this screening, three representative stable cell lines from each transfection were chosen for further study.
The results in Fig. 8A show that Dox-induced expression of RhoBact and RhoBdn with either 0.1 or 1.0 μg/ml Dox for 16 h had no effect on fibronectin protein levels, consistent with the lack of phenotypic change in cytoskeleton organization of these cells (data not shown). However, we did observe a dramatic reduction in cell growth in the Dox-treated HT-RhoBact (G14V) cell line independent of DHT treatment (Fig. 8B). In these experiments, Dox treatment did not alter cell growth of either the vector control cell line or the RhoBdn mutant stable cell line (T19N), even though DHT treatment induced growth arrest as expected. One reason why expression of the dominant negative RhoBdn protein in this T-Rex stable cell line did not inhibit the DHT response in Dox + DHT media may be that the level of HA-RhoBdn protein may not have been sufficient to recapitulate the transient transfection assays (Fig. 7).
Fig. 8. Tet-regulated expression of HA-RhoBact induces growth arrest in HT-AR1 T-Rex stable cell lines.

A, Western blot using anti-HA antibody to analyze protein extracts prepared from selected HT-AR1 T-Rex stable cells cultured in 0, 0.1, or 1.0 μg/ml Dox for 16 h. The blot was stripped and reprobed with anti-fibronectin antibody. B, relative cell growth of the same HT-AR1 T-Rex stable cell lines as in A cultured with or without 1 μg/ml Dox in the presence or absence of 10 nM DHT for 48 h. Mean ± S.E. ere calculated using untreated cells as the control.
DISCUSSION
Androgen regulation of cytoskeletal reorganization and neuroendocrine-like differentiation in the HT-1080 human fibrosarcoma cell model provides a powerful approach to investigate steroid signaling mechanisms in the context of a cancer cell phenotype. Extending our initial observations (21), we have now characterized the HT-AR1 response to androgen treatment and found that growth arrest occurs at physiological levels of DHT (1–10 nM) and involves G0/G1 cell cycle arrest. Moreover, redistribution of desmoplakin, keratin 5, and chromogranin A proteins support our conclusion that HT-AR1 cells undergo a neuroendocrine-like cell differentiation. Time lapse video microscopy analysis indicated that both rapid and delayed changes in cell attachment properties are a hallmark of the HT-AR1 phenotype.
Based on the time course of most of these cell biological responses (2–5 days), and their dependence on expression of functional AR protein (21), we used expression profiling to identify genes that function downstream of AR activation to determine what cell signaling pathways might be contributing to the overall response. One of the early response genes we identified by expression profiling was RhoB. We found that DHT, but not Dex, treatment led to the rapid induction of RhoB gene expression and the synthesis of unmodified and modified RhoB protein that accumulated in the plasma membrane. RhoB signaling was shown to be important in mediating the HT-AR1 growth arrest response based on results using the farnesyl transferase inhibitor SCH66336 and by ectopic expression of the RhoB variants RhoBact and RhoBdn.
RhoB is a unique member of the Rho family for a number of reasons. First, unlike RhoA, which is expressed at high levels in most cells and primarily associated with the assembly of filamentous actin (25), RhoB expression is tightly regulated (54, 55), and shown to be important to the differentiation of neuronal cells (27, 28). Moreover, RhoA activation promotes malignant transformation and cell proliferation (29), whereas, RhoB functions as a tumor suppressor and shows elevated expression in quiescent cells (24, 56). Another distinguishing feature of RhoB is that it is localized to membranes, both the plasma membrane and endosomes even when it is inactive (35). RhoA is cytosolic in a complex and only moves to the plasma membrane upon activation and dissociation of RhoGDI (57). Interestingly, RhoGDI does not bind to RhoB, which is consistent with the finding that RhoB is always in association with membranes (57). Last, only RhoB has been shown to inhibit NFκB signaling through stabilization of the IκB complex (58), and to repress the transcription of TGFβ type II receptors by a mechanism involving AP-1 (59). The RhoB signaling pathways in HT-AR1 cells are not known, but they could involve any number of Rho effector proteins that have been identified in other cells. For example, the Ser/Thr kinases ROCK and PRK, and the scaffold proteins Rhotekin and Dia (30). Other Rho effector proteins include the AR transcriptional coactivator protein FHL2 (60), G protein βγ subunits (61), and phospholipase C isozymes (62).
Signaling by Rho proteins has been shown to activate the function of FERM domain proteins (63, 64), and in some cases, Rho signaling can be modulated by this same family of proteins (65). Because EHM2 encodes a FERM domain protein (21, 66), which is likely involved in epithelial cell morphogenesis (67), we find it intriguing that DHT treatment co-induces the expression of RhoB and EHM2 genes in HT-AR1 cells (see Fig. 4). This could be indicative of an interdependent function because both genes are expressed at very low levels in the absence of DHT. Results from expression profiling studies investigating the transcriptional response of cells to developmental (68), metabolic (69), and oncogenic (70) stimuli, have shown that entire sets of genes encoding proteins with interdependent functions are often co-regulated. In many cases, co-regulated genes share a common set of cis-elements in the 5′ gene regulatory regions, suggesting that they are members of an evolutionarily conserved “gene network” (71). Similar to what has recently been proposed by Nelson et al. (72) to explain coordinate expression of androgen-regulated genes in the LNCaP prostate cancer cell line, it is possible that the RhoB and EHM2 genes might each contain one or more functional androgen response elements, linking them together in the same gene network. Alternatively, DHT-induced expression of RhoB and EHM2 could be the result of AR-dependent activation of Src and other cytoplasmic kinase pathways that lead to stimulation of Elk-1 or STAT5 functions, two transcription factors that have been shown to be activated by nongenomic steroid signaling (73–75).
In addition to EHM2 and RhoB, we also identified the transcription factor Egr-1 as an androgen response gene (Fig. 4). Mouse knockout studies by Milbrandt and colleagues (76) have demonstrated that Egr-1, along with other EGR family members, plays a critical role in mediating steroidogenesis in mammalian reproductive tissues. In fact, Egr-1 has been shown to stimulate AR transcriptional regulatory activity through direct receptor binding (77), and could therefore function as a feed forward regulatory loop in HT-AR1 cells. Recently, Svaren et al. (78) used a transient expression system to identify Egr-1 target genes in human prostate cells by expression profiling. Relevant to our findings with HT-AR1 cells, they found that among the Egr-1-induced genes, five of the most highly regulated genes were associated with neuroendocrine cells (neurogranin, neuron-specific enolase, neurosin, neuro-d4, and ICAM-5). Moreover, Raychowdhury et al. (79), reported that Egr-1 regulates the expression of chromogranin A in gastric epithelial cells that could explain the co-induction of Egr-1 and chromogranin A in HT-AR1 cells. With regard to steroid regulation of Egr-1 expression in HT-AR1 cells, it is interesting that Egr-1 transcripts are extremely low in the absence of DHT and that Egr-1 expression does not peak until 48 h after DHT treatment (Fig. 4). Because Egr-1 transcription has been shown to be regulated by both MEK/ERK and the cAMP-mediated signaling pathway in other cell types (80), it is possible that the delay in Egr-1 expression following DHT treatment may be the result of AR-mediated nongenomic signaling events that require activation of downstream transcription factors (33).
In summary, we found that induced expression of RhoB in HT-AR1 cells was responsible for the growth arrest phenotype but not for cytoskeletal reorganization, indicating that RhoB signaling accounts for only a subset of the androgen responses in this fibrosarcoma cell model. The observed coordinate expression of numerous cell signaling genes (RhoB, PTGF-β, CAV2, Egr-1, Myo1B, and EHM2), suggests that DHT treatment activates downstream targets through a combination of genomic and nongenomic signaling mechanisms. By exploiting the HT-AR1 model using functional genomics, proteomics, and molecular genetic approaches, it should be possible to dissect the relevant contributions of cytoplasmic and nuclear AR functions to gain a better understanding of signaling pathways that modulate steroid responses in human cancer cells.
Acknowledgments
We thank Jeff Way for help in establishing the HT-AR1 cell line, Norma Seaver (The Arizona Cancer Center) for help in FACS analysis, Dr. Bernie Futscher and Skip Vaught (The Arizona Cancer Center Microarray Facility) for advice using the 5k Human Gene Set 1 array, the University of Arizona Department of Cell Biology & Anatomy Core Service for help with the time lapse video microscopy, Dr. Ritu Pandey of the Arizona Cancer Center Bioinformatics core service for help with development of the steroid-regulated gene expression (SRGE) data base, and Dr. Alan List of the Arizona Cancer Center for the SCH66336.
Footnotes
This work was supported by National Institutes of Health Grant CA 23074, Arizona Cancer Center Core Grants PO1 CA 56666 and CA 75152 (to A. E. C.), and the generous support of the Jack Findlay Doyle II Charitable Fund (to R. L. M.).
The on-line version of this article (available at http://www.jbc.org) contains two video movies of HT-AR1 cells cultured in media ±DHT for 5 h as separate ~5 MB QuickTime media files (HTAR1-Control.mov and HTAR1-DHT.mov).
The abbreviations used are: AR, androgen receptor; C/S, charcoal-stripped; Dex, dexamethasone; DHT, dihydrotestosterone; Dox, doxycycline; FACS, fluorescence-activated cell sorting; HA, hemagglutinin; TGF, transforming growth factor; MEK/ERK, mitogen-activated protein kinase/extracellular signal-regulated kinase kinase.
azcc-microarray.arl.arizona.edu.
K. Davis and R. L. Miesfeld, unpublished data.
References
- 1.Perlman WR, Ramachandran B, Arnold AP. J Comp Neurol. 2003;455:513–530. doi: 10.1002/cne.10510. [DOI] [PubMed] [Google Scholar]
- 2.Morley SD, Viard I, Chung BC, Ikeda Y, Parker KL, Mullins JJ. Mol Endocrinol. 1996;10:585–598. doi: 10.1210/mend.10.5.8732689. [DOI] [PubMed] [Google Scholar]
- 3.Simerly RB. Annu Rev Neurosci. 2002;25:507–536. doi: 10.1146/annurev.neuro.25.112701.142745. [DOI] [PubMed] [Google Scholar]
- 4.Lokeshwar VB, Lokeshwar BL, Pham HT, Block NL. Cancer Res. 1996;56:651–657. [PubMed] [Google Scholar]
- 5.Xing RH, Mazar A, Henkin J, Rabbani SA. Cancer Res. 1997;57:3585–3593. [PubMed] [Google Scholar]
- 6.Papakonstanti EA, Kampa M, Castanas E, Stournaras C. Mol Endocrinol. 2003;17:870 –881. doi: 10.1210/me.2002-0253. [DOI] [PubMed] [Google Scholar]
- 7.Kampa M, Papakonstanti EA, Hatzoglou A, Stathopoulos EN, Stournaras C, Castanas E. FASEB J. 2002;16:1429 –1431. doi: 10.1096/fj.02-0131fje. [DOI] [PubMed] [Google Scholar]
- 8.Castoria G, Lombardi M, Barone MV, Bilancio A, Di Domenico M, Bottero D, Vitale F, Migliaccio A, Auricchio F. J Cell Biol. 2003;161:547–556. doi: 10.1083/jcb.200211099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Simoncini T, Genazzani AR. Eur J Endocrinol. 2003;148:281–292. doi: 10.1530/eje.0.1480281. [DOI] [PubMed] [Google Scholar]
- 10.Losel R, Wehling M. Nat Rev Mol Cell Biol. 2003;4:46 –56. doi: 10.1038/nrm1009. [DOI] [PubMed] [Google Scholar]
- 11.Heinlein CA, Chang C. Mol Endocrinol. 2002;16:2181–2187. doi: 10.1210/me.2002-0070. [DOI] [PubMed] [Google Scholar]
- 12.Rasheed S, Nelson-Rees WA, Toth EM, Arnstein P, Gardner MB. Cancer. 1974;33:1027–1033. doi: 10.1002/1097-0142(197404)33:4<1027::aid-cncr2820330419>3.0.co;2-z. [DOI] [PubMed] [Google Scholar]
- 13.Oliver N, Newby RF, Furcht LT, Bourgeois S. Cell. 1983;33:287–296. doi: 10.1016/0092-8674(83)90357-4. [DOI] [PubMed] [Google Scholar]
- 14.Dean DC, Newby RF, Bourgeois S. J Cell Biol. 1988;106:2159 –2170. doi: 10.1083/jcb.106.6.2159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Brenner KA, Corbett SA, Schwarzbauer JE. Oncogene. 2000;19:3156 –3163. doi: 10.1038/sj.onc.1203626. [DOI] [PubMed] [Google Scholar]
- 16.Kondoh N, Shuda M, Arai M, Oikawa T, Yamamoto M. In Vitro Cell Dev Biol Anim. 2002;38:111–117. doi: 10.1290/1071-2690(2002)038<0111:AOAIGO>2.0.CO;2. [DOI] [PubMed] [Google Scholar]
- 17.Bulens F, Merchiers P, Ibanez-Tallon I, De Vriese A, Nelles L, Claessens F, Belayew A, Collen D. J Biol Chem. 1997;272:663–671. doi: 10.1074/jbc.272.1.663. [DOI] [PubMed] [Google Scholar]
- 18.Rundlett SE, Miesfeld RL. Mol Cell Endocrinol. 1995;109:1–10. doi: 10.1016/0303-7207(95)03477-o. [DOI] [PubMed] [Google Scholar]
- 19.Chapman MS, Askew DJ, Kuscuoglu U, Miesfeld RL. Mol Endocrinol. 1996;10:967–978. doi: 10.1210/mend.10.8.8843413. [DOI] [PubMed] [Google Scholar]
- 20.Whitacre DC, Karnas KJ, Miesfeld RL. Endocrine. 2001;15:111–118. doi: 10.1385/ENDO:15:1:111. [DOI] [PubMed] [Google Scholar]
- 21.Chauhan S, Pandey R, Way J, Sroka TC, Demetriou MC, Kunz S, Cress AE, Mount DW, Miesfeld RL. Biochem Biophys Res Commun. 2003;310:421–432. doi: 10.1016/j.bbrc.2003.08.147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Chen Z, Sun J, Pradines A, Favre G, Adnane J, Sebti SM. J Biol Chem. 2000;275:17974 –17978. doi: 10.1074/jbc.C000145200. [DOI] [PubMed] [Google Scholar]
- 23.Liu AX, Rane N, Liu JP, Prendergast GC. Mol Cell Biol. 2001;21:6906 –6912. doi: 10.1128/MCB.21.20.6906-6912.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Adnane J, Muro-Cacho C, Mathews L, Sebti SM, Munoz-Antonia T. Clin Cancer Res. 2002;8:2225–2232. [PubMed] [Google Scholar]
- 25.Price LS, Collard JG. Semin Cancer Biol. 2001;11:167–173. doi: 10.1006/scbi.2000.0367. [DOI] [PubMed] [Google Scholar]
- 26.Gampel A, Parker PJ, Mellor H. Curr Biol. 1999;9:955–958. doi: 10.1016/s0960-9822(99)80422-9. [DOI] [PubMed] [Google Scholar]
- 27.Laplante I, Paquin J, Beliveau R. Brain Res Dev Brain Res. 2001;129:157–168. doi: 10.1016/s0165-3806(01)00197-3. [DOI] [PubMed] [Google Scholar]
- 28.Liu JP, Jessell TM. Development. 1998;125:5055–5067. doi: 10.1242/dev.125.24.5055. [DOI] [PubMed] [Google Scholar]
- 29.Olenik C, Aktories K, Meyer DK. Brain Res Mol Brain Res. 1999;70:9–17. doi: 10.1016/s0169-328x(99)00121-7. [DOI] [PubMed] [Google Scholar]
- 30.Bishop AL, Hall A. Biochem J. 2000;348:241–255. [PMC free article] [PubMed] [Google Scholar]
- 31.Davis T, Rabinovitz I, Futscher B, Schnölzer M, Buerger F, Liu Y, Kulesz-Martin M, Cress A. J Biol Chem. 2001;276:26099 –26106. doi: 10.1074/jbc.M102811200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Tran NL, Nagle RB, Cress AE, Heimark RL. Am J Pathol. 1999;155:787–798. doi: 10.1016/S0002-9440(10)65177-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Losel RM, Falkenstein E, Feuring M, Schultz A, Tillmann HC, Rossol-Haseroth K, Wehling M. Physiol Rev. 2003;83:965–1016. doi: 10.1152/physrev.00003.2003. [DOI] [PubMed] [Google Scholar]
- 34.Jones JC, Goldman RD. J Cell Biol. 1985;101:506 –517. doi: 10.1083/jcb.101.2.506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Ellis S, Mellor H. Trends Cell Biol. 2000;10:85–88. doi: 10.1016/s0962-8924(99)01710-9. [DOI] [PubMed] [Google Scholar]
- 36.Chauhan S, Leach CH, Kunz S, Bloom JW, Miesfeld RL. J Steroid Biochem Mol Biol. 2003;84:441–452. doi: 10.1016/s0960-0760(03)00065-7. [DOI] [PubMed] [Google Scholar]
- 37.Amler LC, Agus DB, LeDuc C, Sapinoso ML, Fox WD, Kern S, Lee D, Wang V, Leysens M, Higgins B, Martin J, Gerald W, Dracopoli N, Cordon-Cardo C, Scher HI, Hampton GM. Cancer Res. 2000;60:6134 –6141. [PubMed] [Google Scholar]
- 38.Cheung J, Smith DF. Mol Endocrinol. 2000;14:939 –946. doi: 10.1210/mend.14.7.0489. [DOI] [PubMed] [Google Scholar]
- 39.Schiene-Fischer C, Yu C. FEBS Lett. 2001;495:1–6. doi: 10.1016/s0014-5793(01)02326-2. [DOI] [PubMed] [Google Scholar]
- 40.Yokoyama-Kobayashi M, Saeki M, Sekine S, Kato S. J Biochem (Tokyo) 1997;122:622–626. doi: 10.1093/oxfordjournals.jbchem.a021798. [DOI] [PubMed] [Google Scholar]
- 41.Paralkar VM, Vail AL, Grasser WA, Brown TA, Xu H, Vukicevic S, Ke HZ, Qi H, Owen TA, Thompson DD. J Biol Chem. 1998;273:13760 –13767. doi: 10.1074/jbc.273.22.13760. [DOI] [PubMed] [Google Scholar]
- 42.Baek SJ, Horowitz JM, Eling TE. J Biol Chem. 2001;276:33384 –33392. doi: 10.1074/jbc.M101814200. [DOI] [PubMed] [Google Scholar]
- 43.Bootcov MR, Bauskin AR, Valenzuela SM, Moore AG, Bansal M, He XY, Zhang HP, Donnellan M, Mahler S, Pryor K, Walsh BJ, Nicholson RC, Fairlie WD, Por SB, Robbins JM, Breit SN. Proc Natl Acad Sci U S A. 1997;94:11514 –11519. doi: 10.1073/pnas.94.21.11514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Strelau J, Bottner M, Lingor P, Suter-Crazzolara C, Galter D, Jaszai J, Sullivan A, Schober A, Krieglstein K, Unsicker K. J Neural Transm Suppl. 2000;60:273–276. doi: 10.1007/978-3-7091-6301-6_18. [DOI] [PubMed] [Google Scholar]
- 45.Razani B, Wang XB, Engelman JA, Battista M, Lagaud G, Zhang XL, Kneitz B, Hou H, Jr, Christ GJ, Edelmann W, Lisanti MP. Mol Cell Biol. 2002;22:2329 –2344. doi: 10.1128/MCB.22.7.2329-2344.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Scherer PE, Okamoto T, Chun M, Nishimoto I, Lodish HF, Lisanti MP. Proc Natl Acad Sci U S A. 1996;93:131–135. doi: 10.1073/pnas.93.1.131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Adamson ED, Mercola D. Tumor Biol. 2002;23:93–102. doi: 10.1159/000059711. [DOI] [PubMed] [Google Scholar]
- 48.Bement WM, Hasson T, Wirth JA, Cheney RE, Mooseker MS. Proc Natl Acad Sci U S A. 1994;91:6549 –6553. doi: 10.1073/pnas.91.14.6549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Medcalf RL, Richards RI, Crawford RJ, Hamilton JA. EMBO J. 1986;5:2217–2222. doi: 10.1002/j.1460-2075.1986.tb04487.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Engel ME, Datta PK, Moses HL. J Biol Chem. 1998;273:9921–9926. doi: 10.1074/jbc.273.16.9921. [DOI] [PubMed] [Google Scholar]
- 51.Mattingly RR, Gibbs RA, Menard RE, Reiners JJ., Jr J Pharmacol Exp Ther. 2002;303:74 –81. doi: 10.1124/jpet.102.036061. [DOI] [PubMed] [Google Scholar]
- 52.Haluska P, Dy GK, Adjei AA. Eur J Cancer. 2002;38:1685–1700. doi: 10.1016/s0959-8049(02)00166-1. [DOI] [PubMed] [Google Scholar]
- 53.Tate CG, Haase J, Baker C, Boorsma M, Magnani F, Vallis Y, Williams DC. Biochim Biophys Acta. 2003;1610:141–153. doi: 10.1016/s0005-2736(02)00719-8. [DOI] [PubMed] [Google Scholar]
- 54.Fritz G, Kaina B. Nucleic Acids Res. 2001;29:792–798. doi: 10.1093/nar/29.3.792. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Fritz G, Kaina B, Aktories K. J Biol Chem. 1995;270:25172–25177. doi: 10.1074/jbc.270.42.25172. [DOI] [PubMed] [Google Scholar]
- 56.Schwarze SR, DePrimo SE, Grabert LM, Fu VX, Brooks JD, Jarrard DF. J Biol Chem. 2002;277:14877–14883. doi: 10.1074/jbc.M200373200. [DOI] [PubMed] [Google Scholar]
- 57.Michaelson D, Silletti J, Murphy G, D’Eustachio P, Rush M, Philips MR. J Cell Biol. 2001;152:111–126. doi: 10.1083/jcb.152.1.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Fritz G, Kaina B. J Biol Chem. 2001;276:3115–3122. doi: 10.1074/jbc.M005058200. [DOI] [PubMed] [Google Scholar]
- 59.Adnane J, Seijo E, Chen Z, Bizouarn F, Leal M, Sebti SM, Munoz-Antonia T. J Biol Chem. 2002;277:8500 –8507. doi: 10.1074/jbc.M104367200. [DOI] [PubMed] [Google Scholar]
- 60.Muller JM, Metzger E, Greschik H, Bosserhoff AK, Mercep L, Buettner R, Schule R. EMBO J. 2002;21:736 –748. doi: 10.1093/emboj/21.4.736. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Thodeti CK, Massoumi R, Bindslev L, Sjolander A. Biochem J. 2002;365:157–163. doi: 10.1042/BJ20020248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Wing MR, Snyder JT, Sondek J, Harden TK. J Biol Chem. 2003;278:41253–41258. doi: 10.1074/jbc.M306904200. [DOI] [PubMed] [Google Scholar]
- 63.Takahashi K, Sasaki T, Mammoto A, Takaishi K, Kameyama T, Tsukita S, Takai Y. J Biol Chem. 1997;272:23371–23375. doi: 10.1074/jbc.272.37.23371. [DOI] [PubMed] [Google Scholar]
- 64.Yonemura S, Matsui T, Tsukita S. J Cell Sci. 2002;115:2569 –2580. doi: 10.1242/jcs.115.12.2569. [DOI] [PubMed] [Google Scholar]
- 65.Speck O, Hughes SC, Noren NK, Kulikauskas RM, Fehon RG. Nature. 2003;421:83–87. doi: 10.1038/nature01295. [DOI] [PubMed] [Google Scholar]
- 66.Shimizu K, Nagamachi Y, Tani M, Kimura K, Shiroishi T, Wakana S, Yokota J. Genomics. 2000;65:113–120. doi: 10.1006/geno.2000.6154. [DOI] [PubMed] [Google Scholar]
- 67.Hoover KB, Bryant PJ. Dev Genes Evol. 2002;212:230 –238. doi: 10.1007/s00427-002-0231-6. [DOI] [PubMed] [Google Scholar]
- 68.Davidson EH, Rast JP, Oliveri P, Ransick A, Calestani C, Yuh CH, Minokawa T, Amore G, Hinman V, Arenas-Mena C, Otim O, Brown CT, Livi CB, Lee PY, Revilla R, Rust AG, Pan Z, Schilstra MJ, Clarke PJ, Arnone MI, Rowen L, Cameron RA, McClay DR, Hood L, Bolouri H. Science. 2002;295:1669 –1678. doi: 10.1126/science.1069883. [DOI] [PubMed] [Google Scholar]
- 69.Webb GC, Akbar MS, Zhao C, Steiner DF. Proc Natl Acad Sci U S A. 2000;97:5773–5778. doi: 10.1073/pnas.100126597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Khan J, Bittner ML, Saal LH, Teichmann U, Azorsa DO, Gooden GC, Pavan WJ, Trent JM, Meltzer PS. Proc Natl Acad Sci U S A. 1999;96:13264 –13269. doi: 10.1073/pnas.96.23.13264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Davidson EH, Rast JP, Oliveri P, Ransick A, Calestani C, Yuh CH, Minokawa T, Amore G, Hinman V, Arenas-Mena C, Otim O, Brown CT, Livi CB, Lee PY, Revilla R, Schilstra MJ, Clarke PJ, Rust AG, Pan Z, Arnone MI, Rowen L, Cameron RA, McClay DR, Hood L, Bolouri H. Dev Biol. 2002;246:162–190. doi: 10.1006/dbio.2002.0635. [DOI] [PubMed] [Google Scholar]
- 72.Nelson PS, Clegg N, Arnold H, Ferguson C, Bonham M, White J, Hood L, Lin B. Proc Natl Acad Sci U S A. 2002;99:11890 –11895. doi: 10.1073/pnas.182376299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Peterziel H, Mink S, Schonert A, Becker M, Klocker H, Cato AC. Oncogene. 1999;18:6322–6329. doi: 10.1038/sj.onc.1203032. [DOI] [PubMed] [Google Scholar]
- 74.Kinoshita Y, Chen S. Cancer Res. 2003;63:3546 –3555. [PubMed] [Google Scholar]
- 75.Bjornstrom L, Sjoberg M. Mol Endocrinol. 2002;16:2202–2214. doi: 10.1210/me.2002-0072. [DOI] [PubMed] [Google Scholar]
- 76.Lee SL, Tourtellotte LC, Wesselschmidt RL, Milbrandt J. J Biol Chem. 1995;270:9971–9977. doi: 10.1074/jbc.270.17.9971. [DOI] [PubMed] [Google Scholar]
- 77.Yang SZ, Abdulkadir SA. J Biol Chem. 2003;278:39906 –39911. doi: 10.1074/jbc.M307250200. [DOI] [PubMed] [Google Scholar]
- 78.Svaren J, Ehrig T, Abdulkadir SA, Ehrengruber MU, Watson MA, Milbrandt J. J Biol Chem. 2000;275:38524 –38531. doi: 10.1074/jbc.M005220200. [DOI] [PubMed] [Google Scholar]
- 79.Raychowdhury R, Schafer G, Fleming J, Rosewicz S, Wiedenmann B, Wang TC, Hocker M. Mol Endocrinol. 2002;16:2802–2818. doi: 10.1210/me.2001-0292. [DOI] [PubMed] [Google Scholar]
- 80.Thiel G, Cibelli G. J Cell Physiol. 2002;193:287–292. doi: 10.1002/jcp.10178. [DOI] [PubMed] [Google Scholar]