

Abstract
IL-2 has been extensively examined to promote clinical T and NK cell responses. Regulatory T cells (Tregs) have been shown to regulate many aspects of the immune system, including natural killer (NK) cell-mediated responses. In our study, we demonstrated that in vivo administration of IL-2 led to activation and expansion of both NK cells and immunosuppressive Tregs. Therefore, we attempted to augment NK cell anti-tumor effects by concurrently depleting Tregs using anti-CD25. Increased NK cell activation by IL-2 correlated with an increase in classical, short-term NK cell in vitro killing assays regardless of depletion of Tregs. However, when splenocytes of the treated mice were used in long-term tumor outgrowth experiments, we observed that prior depletion of Tregs with IL-2 administration led to improved anti-tumor effects compared to either treatment alone. Importantly, these in vitro data correlated with subsequent in vivo survival of leukemia-bearing mice in which co-treatment of IL-2 with anti-CD25 led to significantly improved survival compared to mice treated with either IL-2 alone or Treg depletion. Prior depletion of NK1.1+ cells, but not CD8+ cells, completely abrogated all anti-tumor effects mediated by IL-2 and anti-CD25 combination therapy. These findings demonstrate that superior NK cell-mediated anti-leukemic effects can be achieved with IL-2 administration and concurrent depletion of CD25+ cells.
Introduction
Natural killer (NK) cells are members of the innate immune system and are known to mediate MHC unrestricted cytotoxicity against virally infected and neoplastic cells (1, 2). Besides their ability to kill target cells directly, NK cells are also known to be potent immune modulators with the ability to produce abundant cytokines capable of altering immune responses (2, 3). NK cells can promote T helper type-1 (Th1) responses (2, 4, 5) and can participate in dendritic cell (DC) maturation (6) and in the generation of cytotoxic T lymphocytes and tumor-specific memory T cells against various tumors (7–9). These studies demonstrated the broad range of methods by which NK cells can control immune responses, but none have adequately explained how NK cell function is modulated by other cells.
CD4+CD25+ regulatory T (Treg) cells are critical immunomodulatory cells that are responsible for maintaining immune tolerance (10). Treg cells make up approximately 10% of the entire CD4+ T cell pool. They exclusively express the transcription factor Foxp3 and are known to prevent organ-specific autoimmune diseases (10). The role of Tregs has been demonstrated in tolerance during infections (11) and in allogeneic transplantation (12).
The inhibition of NK cells by Tregs has been demonstrated previously. Recent studies demonstrated that adoptive transfer of Treg cells into C57BL/6 athymic mice resulted in a down regulation of NKG2D on NK cells (13). This transfer of Treg cells correlated with a significant increase in lung metastases of the NKG2D-sensitive tumor cell line B16-Rae, suggesting that Treg cells were capable of suppressing the clearance of NKG2D-ligand expressing cells. Subsequent studies have shown the cytotoxic ability of purified NK cells in short-term killing assays could be inhibited by the addition of purified Tregs (14). This inhibition worked against Rae-1-transfected cell lines but not with non-Rae1-transfected control tumor cells. The investigators also demonstrated that treatment of B16-Rae1-bearing mice with anti-CD25 led to decreased lung metastases. The treatment of mice injected with non-transfected cells did not result in any difference in the number of lung metastases. In these studies, only tumor cell lines transfected with ligands to the NKG2D receptor were affected by the manipulation of Tregs.
We have recently demonstrated that Tregs affect NK cell function in a murine bone marrow allograft transplant model in which Tregs inhibit NK cell-mediated rejection of bone marrow allografts (15). These studies further support the unique link between NK cells and Tregs, and suggest that future studies designed to enhance NK cell function should consider removing Tregs as part of the therapeutic regimen.
Many immunotherapies currently explored for treating leukemia revolve around NK cell activation. NK cells are typically activated with Th1-inducing cytokines (i.e. IL-2, IL-12, IL-18) or with nucleotide analogues (poly I:C, CpGs) that engage Toll-like receptors (TLRs). Historically, IL-2 stimulation was the primary method through which human NK cell activation has been accomplished with mixed clinical results (16–18). However, studies also demonstrated that this cytokine can also be a potent activator of Treg cells both in vivo and in vitro (10). In this paper, we demonstrated that some of the important immunostimulatory functions of IL-2 are lost via activation of Treg cells, therefore, concurrent Treg cell depletion allows for more thorough NK cell activation which increases the anti-tumor potential of NK cells.
Materials and Methods
Mice
C57BL/6 mice were purchased from the Animal Production Area (National Cancer Institute [NCI] at Frederick, Frederick, MD). Female mice were 2 to 4 months of age at the initiation of experiments. Animals were maintained under specific-pathogen free conditions at the University of Nevada, Reno facility. Animal care was provided in accordance with the procedures outlined by the NIH. Animal studies were performed at the University of Nevada, Reno animal facility according to approved protocols and in accordance with the Animal Care and Use Committees.
Reagents
Recombinant human IL-2 (IL-2; TECIN (Teceleukin)) was provided by the National Cancer Institute. Anti-CD25 (clone PC61.5.3) and anti-NK1.1 (PK136) were grown by National Cell Culture Center (Minneapolis, MN). Purified rat IgG was purchased from Jackson ImmunoResearch Laboratories. The rat antibody to mouse CD8 (clone YTS169.4), was produced in ascites. The antibody concentration was 4.3 mg/mL, and endotoxin level was 3.6 endotoxin units (EU)/mg antibody.
Cytotoxicity Assays
Yac-1 tumor cells were labeled with Na251CrO4 (Perkin Elmer, Waltam, MA) and used in a standard 4-hr 51Cr-release assay as described previously (19). For chromium release assays, 2000 51Cr-labeled Yac-1 target cells were used per well. The percent lysis was calculated as follows: [(experimental lysis - spontaneous lysis)/(maximal lysis - spontaneous lysis)] × 100. Each sample was analyzed in triplicate. These experiments were performed three times with similar results.
Tumor Outgrowth Assay
Spleens from mice undergoing IL-2 (2×105 IU IL-2 on days 1, 3, 5, and 7, i.p.) and anti-CD25 (1mg anti-CD25 on day 1, 4, and 7, i.p.) therapy were removed on day 8, processed into a single cell suspension, and added to 96-well culture plates at indicated effector to target (E:T) ratios in RF10 complete media containing 1000 IU/mL IL-2 (Roche). Target cells were then added at 1000 targets/well in RF10 complete media containing 1000 IU/mL IL-2. For some control wells, tumor cells were left out and instead media containing IL-2 alone was added to measure the proliferation of effector cells alone. After 4 days, 10μL of the cell milieu was passed into new wells containing 90μL of fresh RF10 complete media but lacking IL-2. In some experiments, the passage occurred a second time after an additional 4 days. Three days following passage (day 7), the cells were pulsed with 3H-thymidine (1 μCi per well) (Amersham Pharmacia Life Sciences; Buckinghamshire, UK) 16–18 h before they were harvested and counted in the presence of scintillation fluid on a Wallac β-plate reader (Perkin Elmer). In experiments designed to determine the mechanism of NK cell cytotoxicity against C1498, wildtype or perforin knockout (pfp−/−) splenocytes were depleted of T cells by anti-Thy1.2 and rabbit complement and incubated in complete media containing 1000 IU/mL IL-2 for 6 days. Activated NK cells were then added to 96-well culture plates at the indicated E:T ratios in media containing 1000 IU/mL IL-2. C1498 target cells were added as described above. After 4 days, cell proliferation was determined by an MTT Cell Proliferation Kit (Roche). Tumor outgrowth was determined by subtracting absorbance values of effectors alone from wells containing both tumor and effector cells at indicated E:T ratios. Three individual wells were analyzed per data point.
Flow Cytometry
Splenocytes from C57BL/6 were prepared at 2×107 cells/mL in staining buffer (PBS with 1% FBS). 106 cells were labeled with FITC-anti-CD25 (7D4), R-phycoerythrin (PE) anti-CD25 (PC61), PE-cyanine 5 (PC5) anti-CD4 (GK1.5), PC5-anti-CD3 (145-2C11), FITC or PE anti-NK1.1 (PK136), biotinylated anti-CD122 (TM-β1) plus PC7-Streptavidin (SA) (all from eBiosciences; San Diego, CA) or Fc-block (anti-CD16; BD Pharmingen) and labeled for 15 min at 4°C. For Foxp3 analysis, intracellular staining was performed using the Foxp3 kit (eBiosciences). The two clones of anti-CD25 antibody were used because they recognize non-competing epitopes enabling us to examine CD25+ cells after in vivo treatment with PC61 antibody. Phenotype analysis was performed four times. Cell counts were determined by percent expression of light-scatter cells by cell counts as determined by a Coulter Z1 particle counter (Beckman Coulter; Fullerton, CA). For granzyme B analysis, surface-labeled cells were incubated with Intraprep kit (Beckman Coulter) and labeled with PE-anti-human Granzyme B (GB12 which also reacts with mouse Gzmb) or PE-mouse IgG1 control antibodies (Invitrogen; Carlsbad, CA). Granzyme B analysis was performed two times. All flow cytometry was performed on a BD FACSCAN flow cytometer using CellQuest software (Becton Dickinson) or a 4-color Beckman Coulter XL/MCL using System II software. Listmode flow cytometric data files were analyzed with FlowJo (Tree Star).
Survival Studies
C57BL/6 mice were injected with 2×105 C1498 leukemia cells, i.v., on day 0. Groups of 8 mice were treated with 2×105 IU IL-2 on days 1, 3, 5, and 7 i.p., 1mg anti-CD25, PC61.5.3, on day 1, 4, and 7, i.p., or a combination of the two. In some experiments, NK cells were depleted with 500 μg anti-NK1.1 (clone: PK136), or 300 μg anti-CD8 antibody (clone:YTS169.4) on day 1 and day 5 as performed previously (15). Mice were monitored for survival and euthanized if mice showed morbidity. Each experiment was performed two times.
Statistical Analysis
Statistical analysis was performed using Graphpad Prism 4 software (Graphpad, El Camino, CA). T-tests, Fisher Exact test, one-way ANOVA, or two-way ANOVA, or log-rank test were used where appropriate. A P value of less than 0.05 was considered significant. Means ± standard deviations are shown unless otherwise specified.
Results
IL-2 administration promotes NK cell numbers, function, and anti-tumor effects in vivo
Previous studies have demonstrated a potential anti-tumor effect of IL-2 in cancer, especially renal cancer and melanoma (20, 21). We first examined the effects of IL-2 on NK cell anti-tumor effects ex vivo. C57BL/6 mice were injected with 2×105 IU rhIL-2 on days 0, 2, 4, 6 and the numbers of NK cells in the spleens were quantified (Figure 1A). There was a significant increase in the number of NK cells after IL-2 treatment (P<0.01), from 2×106 cells to over 5×106 cells, and this increase correlated with a significant (P<0.01) increase in killing by splenocytes as determined by a 4hr chromium release assay against the NK cell-sensitive Yac-1 tumor targets (Figure 1B). Finally, the in vivo anti-tumor effects of IL-2 were determined by injecting C57BL/6 mice with 2×105 C1498 leukemia cells, i.v., and then following the same IL-2 regimen as above. IL-2 therapy promoted a slight, but significant (P<0.05), increase in the survival time of C1498-bearing C57BL/6 mice (Figure 1C). These data confirmed that IL-2 promoted modest anti-tumor responses in vivo which correlated with both an increase in NK cell numbers as well as killing by splenocytes.
Figure 1. IL-2 promotes NK cell numbers, function, and anti-tumor effect in vivo.

C57BL/6 mice were left untreated (Control; ■) or given IL-2 (2×105 IU, day 1,3,5,7). Spleens from mice were removed on day 8 and (A.) the number of CD3−NK1.1+ NK cells were determined (n=2) and (B.) splenocytes from indicated treatment mice were used in a 4hr chromium release assay against Yac-1 targets (n=3). (C.) Mice undergoing above treatment were challenged on day 0 with 2×105 C1498 cells, i.v., and analyzed for survival (n=8). * P<0.05 as determined by Log-Rank test. These data are representative of 5 experiments.
IL-2 therapy results in regulatory T cell expansion
Next, we wanted to examine the effects of IL-2 immunotherapy on CD4+CD25+ Treg expansion (Figure 2). Splenocytes from C57BL/6 mice were untreated or treated with the IL-2 regimen listed above were analyzed for the effects on Tregs. Control mice consistently expressed around 10% Foxp3+ cells (Figure 2A), and IL-2 therapy led to an increase in CD4+Foxp3+ Treg percentage (Figure 2B) as well as a significant increase in Treg numbers (Figure 2C; P<0.01). Most Foxp3+ cells expressed CD25 though some Foxp3+ cells were low/negative for CD25 expression. These results demonstrated that IL-2 administration increased both NK cell numbers and function as well as the number of Tregs, which can potentially inhibit NK cell-mediated function.
Figure 2. IL-2 increases Treg percentage and numbers.

C57BL/6 mice were untreated or given IL-2 as in Figure 1. The percentages of Foxp3+ cells out of total CD4+ cells are indicated for untreated mice (A.) or IL-2 treated mice (B.). Total CD4+Foxp3+ cells from control of IL-2 treated mice (n=3) (C.). These data are representative of 3 experiments.
IL-2 receptors on NK cells do not change following IL-2 therapy
Our regulatory T cell depletion strategy was to use the anti-CD25 monoclonal antibody (PC61.5.3) but first we wanted to ensure that murine NK cells did not express CD25. Resting NK cells (CD3−NK1.1+) express the low-affinity IL-2Rβ (CD122) but not the high affinity IL-2Rα (CD25; Figure 3A). Following the earlier IL-2 treatment regimen, while we did see an increase in CD25+ cells (Figure 2B), but there was no difference in the levels of CD25 or CD122 on NK cells (Figure 3B) showing that in vivo IL-2 administration did not stimulate upregulation of the IL-2 receptors. These data demonstrated that co-treatment with IL-2 and anti-CD25 does not result in the depletion of NK cells in vivo.
Figure 3. CD25 is not expressed on resting or IL-2 activated NK cells in vivo.

C57BL/6 mice were untreated or given IL-2 as in Figure 1. Splenocytes were harvested on day 8 and expression of CD25 and CD122 was determined on CD3−NK1.1+ cells from control mice (A.) or IL-2 treated mice (B.) (n=3). These data are representative from 2 experiments.
In vivo CD25 depletion reduces Tregs numbers after IL-2 immunotherapy
Next, we examined the combined effects of IL-2 and anti-CD25 therapy on Tregs. IL-2 was given as before and anti-CD25 (clone: PC61.5.3) was given at 1mg per injection, i.p, on day 1, 4, and 7. Splenocytes were isolated after treatment (day 8) and Tregs were determined by analyzing the number (Figure 4A) or the percentage (Figure 4B) of CD4+Foxp3+ cells. Treating mice with IL-2 consistently resulted in significant expansion of Tregs (Figure 4A, IL-2, P<0.01) whereas treating mice with anti-CD25 resulted in a partial, but significant reduction in CD4+Foxp3+ cells (Figure 4A, anti-CD25, P<0.01). Interestingly, the combination therapy (Figure 4, IL-2 + anti-CD25) resulted in a nearly identical number of cells (Figure 4A) as well as percentage (Figure 4B) compared to anti-CD25 treated mice (Figure 4A), thus any expansion of Treg numbers by IL-2 appeared to have been abrogated by anti-CD25 therapy.
Figure 4. IL-2 therapy increases Treg numbers while anti-CD25 reduces total Tregs.

(A.) Mice undergoing IL-2 therapy as in Figure 1 had spleens removed 1 day following treatment and splenocytes were analyzed for numbers of CD4+Foxp3+ cells. n = 3 mice per group. Analysis by One way ANOVA with Dunnett’s post test. These data are representative of 5 experiments. (B.) Mice undergoing the same treatment as in (A.) but data are shown as percentage of splenocytes that are CD4+ Foxp3+. n=2–3 mice per group. Analysis by One way ANOVA with Bonferroni’s post test. The data are combined from two experiments.
In order to examine the in vivo effect of IL-2 and anti-CD25 on Treg-mediated regulation of NK cells, we developed a model which would allow the simultaneous activation of NK cells and depletion of regulatory T cells. C57BL/6 mice were treated with IL-2 (2×105 IU/0.2mL; i.p., days 1, 3, 5, and 7) and anti-CD25 (PC61.5.3, 1mg/0.2mL, i.p., days 1, 4, 7), either alone or in combination. On day 8, mice were sacrificed and splenocytes were examined in order to determine if IL-2 and/or anti-CD25 had direct effects on NK cell numbers and granzyme B content. As shown above (Figure 1A), IL-2 therapy was capable of generating a large increase in splenic NK cells (CD3−NK1.1+), while anti-CD25 treatment alone did not appear to significantly increase NK cell numbers. The combination of IL-2 and anti-CD25 failed to increase NK cell numbers greater than IL-2 alone (Figure 5A).
Figure 5. Increased NK cell numbers and activation by IL-2 is not enhanced by concurrent CD25 depletion.

Mice undergoing IL-2 and anti-CD25 therapy had spleens removed an analyzed for various parameters. A) Total number of NK cells. (B) Histogram and (C.) MFI of Granzyme B expression in CD3−NK1.1+ cells (n=3). Analysis for A and B by One way ANOVA with Dunnett’s posttest. These data are representative of 2 experiments.
We next examined the granzyme B (GzmB) content to see if NK cells in treated mice were activated following IL-2 and anti-CD25 therapy (Figure 5B–5F). GzmB staining of NK cells was essentially negative in control mice (Figure 5B), as resting murine NK cells generally lack GzmB expression. However, we found GzmB levels of CD3−NK1.1+ increased to a similar level following administration of IL-2 regardless of administration of anti-CD25 antibody (Figure 5C compared to Figure 5E). As expected, treatment of mice with anti-CD25 antibody alone did not result in any increase in GzmB levels (Figure 5D). The data were analyzed statistically by grouping mice together and examining the MFI of GzmB (Figure 5F). Similar data were seen in analysis of other activation markers NKG2D, CD69, and CD90.2, CD27, CD11b, Ly49C/I, and Ly49G2; each of these markers increased following IL-2 administration and no further increase was observed following anti-CD25 treatment (data not shown). Analysis of DX5, which has been shown to be a marker of mature NK cells (22), resulted in equivalent intensity regardless of treatment (data not shown). These results demonstrate that overall NK cell numbers or phenotype did not alter with CD25 depletion.
IL-2 administration combined with CD25 depletion enhances tumor cell killing in long term, but not short term tumor assays
As we did not see any changes in the phenotype or numbers of the NK cells using combination therapy, we tested the NK cells in ex vivo assays to determine their function. Mice treated with IL-2 and/or anti-CD25 were sacrificed and splenocytes were tested in a 4-hr chromium release assay against Yac-1 targets (Figure 6A). While control mice had low cytolytic activity, mice that received IL-2 had enhanced NK cell function (Figure 6A). Mice receiving anti-CD25 did not have enhanced killing compared to control mice, while mice receiving both IL-2 and anti-CD25 were nearly identical to mice receiving IL-2 alone. These data demonstrated that depletion of CD25+ cells does not enhance the cytolytic function of NK cells in short-term assays.
Figure 6. IL-2 and αCD25 promote tumor inhibition in long-term but not short-term assays.

C57BL/6 mice were treated with PBS (control; ■), rhIL-2 (2×105 IU, day 0,2,4,6; ▲), αCD25 (PC61.5.3, 1mg, day 0, 3, 6; ▼;), or rhIL-2 and αCD25 (●) prior to sacrificing the mice and using splenocytes in a chromium release assay (A) or a tumor outgrowth assay (B). (C.) Effector cells used in (B.) were plated in the absence of tumor cells to determine the proliferation of the effector cells. (D.) Mice undergoing IL-2 and anti-CD25 therapy were untreated, or administered anti-NK1.1 or anti-CD8 antibodies and compared in the outgrowth assay. (E.). Activated NK cells from wild type or pfp −/− mice were used in the tumor outgrowth assay. * P<0.01. Statistics performed using a twoway ANOVA with Bonferroni multiple comparison test.
In order to reflect the in vivo situation more accurately, we used a long-term tumor outgrowth assay to assess NK cell anti-tumor function (Figure 6B). In the tumor outgrowth assay, tumor cells were cultured with the indicated number of spleen cells in 96-well plates with media containing 1000 IU/mL IL-2 for up to 4 days, prior to passing 10μL of the cultured cells into 90μL of fresh media lacking IL-2 for an additional 3 days. Following the 3 days of culture lacking IL-2, the cells were pulsed with tritiated thymidine and relative cell numbers were determined by tritiated thymidine incorporation. In the long term assays, the full repertoire of NK cell effector pathways are available as opposed to chromium release assays which generally reflect the short term effects of cytolytic granules. Though redundant cytotoxic mechanisms are available to the NK cell, we have previously determined that the C1498 leukemia cell line is sensitive to perforin, FasL, and TRAIL (23), and we have unpublished data showing that these cells are insensitive to TNF-α and IFN-γ (data not shown). In the tumor outgrowth assay, we observed that mice treated with either IL-2 or anti-CD25 had enhanced anti-tumor functioned compared to control mice (Figure 6B). Importantly, we found that mice treated with IL-2 and anti-CD25 had significantly enhanced anti-tumor function compared to mice receiving IL-2 alone (Figure 6B; P<0.01).
In order to verify that the proliferation of the observed was from tumor cells and not effector splenocytes, we replicated the assay without adding tumor cells to the effector cells (Figure 6C). In these assays we observed relatively little tritium incorporation, suggesting that following passage of the cultured cells into media lacking IL-2, only the tumor cells incorporate substantial amounts of 3H-thymidine. Finally, we verified that NK cells but not CD8+ T cells were responsible for the anti-tumor effects in the outgrowth assay (Figure 6D). In vivo depletion of NK1.1+ cells resulted in abrogation of anti-tumor effector function, while depletion of CD8+ cells did not result in any change in anti-tumor function in the outgrowth assay. The activity of NK cells in the outgrowth assay is predominantly through perforin exocytosis, as verified in Figure 6E, in which activated wild type and perforin-deficient NK cells were used. Though perforin is the predominant mechanism, there is inhibition of tumor outgrowth from perforin-deficient NK cells at increase E:T ratios, demonstrating that NK cells use other mechanisms other than perforin to inhibit tumors in the outgrowth assay. These results indicate that concurrent treatment of IL-2 and anti-CD25 leads to enhanced killing of tumor cells compared to IL-2 alone. Importantly, this effect was only observed in the long-term tumor outgrowth assays, not the short-term killing assays.
Depletion of CD25+ cells combined with NK cell activation led to markedly increased NK cell-mediated anti-tumor effects in vivo
In order to examine the effects of Treg cells on NK cells, we determined the effect of IL-2 and anti-CD25 on NK cell anti-leukemic effects in vivo (Figure 7). Interestingly, depletion of CD25+ cells alone had no affect on the survival of leukemia-bearing mice. Consistent with our previous experiments, mice treated with IL-2 led to a slight, but significant increase in survival compared to control mice (*; P<0.05). However, mice treated with both IL-2 and anti-CD25 antibodies had a significant increase in survival when compared to either control mice (**; P<0.01) or mice treated with IL-2 alone (**; P<0.01). These results demonstrated that in a leukemia-bearing mouse model, removal of Tregs alone was not enough to increase anti-tumor effects significantly. Similarly, the addition of IL-2, which activated NK cells and Tregs, may have countered its own effects due to the increased activation of Tregs that may have masked increases in NK cell activation. However, the combination of IL-2 and anti-CD25 allowed NK cells to be activated while the inhibitory Treg cells were removed. This combined therapy allowed for synergistic increases in survival of the leukemia-bearing mice. If NK cells were depleted from the mice using anti-NK1.1 antibody in addition to the combination therapy, all effects on survival were abrogated (Figure 7, IL-2 + αCD25 + αNK1.1). These data demonstrated that neither activation of NK cells nor removal of Tregs alone were sufficient to generate optimal anti-leukemic responses; however, the combination promoted powerful anti-leukemic responses in functions mediated by NK cells, which is also supported by the in vitro tumor outgrowth data.
Figure 7. IL-2 therapy combined with CD25 depletion results in significant increase in survival compared to IL-2 alone.
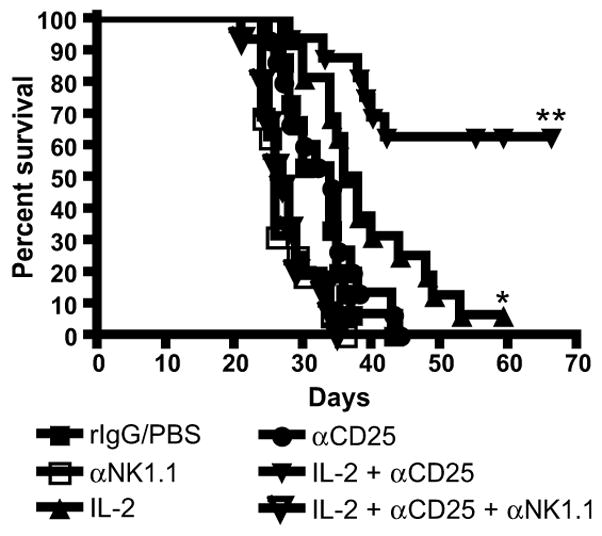
Mice were injected with 2×105 C1498 cells, i.v., on day 0. Mice were either injected with PBS/rIgG controls, with IL-2 (2×105 IU, days 1, 3, 5, 7), with anti-CD25 antibodies (1mg, day 1, 4, 7), with IL-2 and anti-CD25 as above, or with IL-2 and anti-CD25 combined with anti-NK1.1 (500ug, day 1,5). n = 8 mice per group. * P<0.05 compared to Group 1 (rIgG/PBS). ** P<0.01 compared to either rIgG/PBS or IL-2 alone group. These data are combined from 2 experiments.
IL-2 therapy combined with CD25 depletion promotes anti-tumor effects mediated by NK1.1+ but not CD8+ cells
To verify that our effect was mediated by NK cells and not by T cells, we compared the effects of depleting NK and CD8 T cells (Table 1). Since CD8 T cells can acquire cytotoxic activity after IL-2 (24) treatment and can upregulate NK1.1 expression following activation (25), we wanted to ensure that the effect of our depletion was not dependent on the loss of activated CD8+ T cells. Mice either received the IL-2 and anti-CD25 therapy as before or they received the therapy with NK1.1+ depletion or CD8+ depletion. Mice which received the therapy in combination with NK1.1+ depletion had a complete abrogation of effect while depletion of CD8+ cells had no effect on the therapy. Mice depleted of CD8+ T cells had a 98% reduction in the number of splenic CD8+ T cells (data not shown). These data demonstrated that NK cells, and not CD8+ T cells, mediated the anti-tumor effects of IL-2 and anti-CD25 therapy.
Table I.
IL-2 therapy combined with CD25 depletion promotes survival mediated by NK1.1+, but not CD8+ cells1.
| Treatment | Mice Surviving on Day 35 | Mean Survival Time (Mean ± SEM) |
|---|---|---|
| Control (no treatment) | 0/16 | 27.1 ± 0.73 |
| rhIL-2 + anti-CD25 | 8/16* | 39.2 ± 3.4** |
| NK1.1 Depletion | 0/16 | 25.0 ± 0.78 |
| NK1.1 Depletion + rhIL-2 + anti-CD25 | 0/16 | 25.8 ± 0.73 |
| CD8 Depletion | 0/16 | 29.1 ± 0.75 |
| CD8 Depletion + rhIL-2 + anti-CD25 | 8/16* | 43.8 ± 4.9** |
Mice were injected with 2×105 C1498 cells, i.v., on day 0. Mice were either injected with PBS/rIgG controls (Group 1), with rhIL-2 (2×105 IU, days 1, 3, 5, 7) and anti-CD25 antibodies (1mg, day 1, 4, 7)(Group 2), with PK136 (500ug, day 1, 5; Group 3), with IL-2 and anti-CD25 as above plus PK136 (Group 4), with anti-CD8 (300ug, day 1, 5; Group 5) or with IL-2 and anti-CD25 combined with anti-CD8 (Group 6).
P<0.01 by two-tailed Fisher Exact Test compared to specific no treatment controls.
P<0.01 by Oneway ANOVA with Tukey multiple comparison test compared to specific no treatment controls.
Discussion
This study has demonstrated that prior depletion of Treg cells together with IL-2 administration led to synergistic NK cell-mediated anti-leukemic effects which were greater than either therapy alone. The effect of this therapy in vivo was mediated by NK cells and not by CD8+ T cells. Interestingly, classical, short-term NK cell killing assays did not demonstrate increases in NK cell function by combination therapy, and only by using long-term tumor outgrowth assays did we see enhanced function of the splenocytes from the combination treated mice and these effects were correlated with in vivo survival. These data were similar to our previous report which also demonstrated that augmentation of NK cell anti-tumor effects with bortezomib were only seen in long-term assays but not in short-term killing assays which tend to rely on perforin/granzyme lysis (23). The mechanism by which Tregs suppress NK cells has been elusive. It was demonstrated that surface-bound TGF-β on the Tregs was responsible for the inhibition of NK cells (13, 14) while other studies have demonstrated that perforin and granzyme B from Tregs were responsible for inhibiting NK cells (26). Studies performed in our laboratory using purified Tregs and NK cells, regardless of activation status of the two populations, consistently did not result with any suppression using short-term killing assays and equivocal suppression was observed in long-term outgrowth assays (data not shown). It is also possible that Treg-mediated suppression of NK cells may involve an unidentified third party. We also hypothesize that Treg-mediated inhibition of NK cells in vivo is the result of the relative numbers of the two populations. The ratio of Tregs to NK cells in mouse at rest is ~1:1. Administration of IL-2 expands both cell populations, keeping the ratio close while anti-CD25 selectively reduces the Treg population, skewing the ratio toward the NK cells, though the NK cells remain inactivated. Combined administration of IL-2 and anti-CD25 contracts Tregs while expanding and activating NK cells, resulting in significant skewing towards NK cells. These activated NK cells are then capable of mediating anti-tumor effects. Even though the original paper which examined the suppression of NK cells by Tregs was published several years ago (13), we still do not have an understanding of the mechanism of Treg-mediated suppression on NK cells using in vitro assays which may be due to pleiotropic mechanisms of action or assays used for readout.
Recent studies have described a role for Tregs in the suppression of NK cells both in vivo and in vitro (13–15); however, in both of these studies, the mechanism of NK cell inhibition appeared to be through the downregulation of NKG2D on the NK cell. The cytolysis of the C1498 (H2b) tumor cell line was not inhibited by anti-NKG2D antibodies though cytolysis of the renal cell carcinoma RENCA (H2d) was inhibited which is consistent with NK cell recognition of these tumors (data not shown) (27). Therefore, NKG2D downregulation is unlikely to be the mechanism by which Tregs inhibit NK cells in the C1498 leukemia model.
Other reports demonstrated that transfer of wildtype Tregs, but not TGF-β−/− Tregs, into athymic nude mice (which lack T lymphocytes) suppressed NK cell-mediated cytotoxicity (13). Adoptive transfer of activated Tregs into tumor bearing athymic nude mice was correlated with a decrease in lung metastases of the NKG2D-sensitive tumor cell line, B16-Rae, but not to the untransfected cell line B16. This same report showed an increase in proliferation of lymph node resident NK cells, as measured by BrdU incorporation, followed by depletion of CD25+ cells with the PC61 antibody (13).
In contrast, our data did not show any increase in NK cell numbers following depletion of CD25+ cells, even though we used more than 3x more PC61 antibody than was used in that report. Though we did not look at BrdU incorporation or lymph node resident NK cells, mouse colony discrepancies, which may alter NK cell or Treg activation, may be involved with these differences.
In a study which looked at a murine model of colon adenocarcinoma, IL-2 and anti-CD25 immunotherapy was used (28). Using an intra-splenic injection model, which led to tumor growth in the liver, the investigators found that IL-2 and anti-CD25 therapy led to a decrease in liver weight, presumably through the reduction of tumor associated in the liver. The investigators then used an IL-2 transfected cell line and injected these tumors subcutaneously. In these studies, treatment of mice with anti-CD25 reduced tumor growth, but when contrasted with our study, the effect of their treatment was completely abrogated by CD8+ depletion and unaffected by NK1.1+ depletion. This difference may be due to tumor type or route of administration but supports that added benefits can be obtained by combination therapy.
The removal of Tregs has important clinical ramifications. Methods of removal of CD25+ cells, including anti-CD25 monoclonal antibodies daclizumab and basiliximab, are being studied for the removal of Tregs clinically. Denileukin diftitox, which is IL-2 conjugated to diphtheria toxin, has been approved for the depletion of Tregs (29) and has been demonstrated to be remove Tregs in patients with ovarian cancer (30), renal cancer (31), and melanoma (32). A potential problem with using anti-CD25 antibodies clinically is the expression of CD25 on human Tregs does not appear to be as correlative as it is on murine Tregs. Populations of human Tregs have been shown to lack expression of CD25, and thus removal of these cells with anti-CD25 antibodies may be problematic (33, 34). However, the complete removal of Tregs may not be necessary for clinical efficacy, and only partial removal of Tregs, as in this study, may be sufficient to augment immunotherapies.
Depletion of murine Tregs with anti-CD25 is problematic. It was demonstrated that treatment of mice with anti-CD25 resulted in the functional inactivation, but not depletion, of Tregs (35). However, these studies have been controversial. Subsequent studies demonstrated that partial depletion of Foxp3+ Tregs occurred following treatment with PC61, but not with 7D4, as analyzed by flow cytometry (36, 37). Our study, in agreement with previous studies, demonstrates that less than half of splenic Foxp3+ Tregs are depleted by anti-CD25 therapy (37). If the remaining Tregs are functionally inactive, the combined effect may equal to that of a complete depletion.
Previous reports have examined the clinical use of IL-2 to activate NK cells in vivo. Interestingly, a subsequent clinical study demonstrated that IL-2 administration did not express significant levels of CD25 but did upregulate CD122 on NK cells (38). Another clinical study examined the effects of long-term, low-dose IL-2 administration to patients who had undergone either autologous or T-cell-depleted allogeneic bone marrow transplants (39). All patients undergoing IL-2 therapy had increased NK cell numbers and well as cytotoxic function and experienced minimal toxicity. These studies support the use of IL-2 to augment NK cell function in clinical therapies, yet the administration of IL-2 has previously been shown to expand Tregs both clinically (40) and in murine models (41).
The use of IL-15 instead of IL-2 to activate NK cells may also be of interest. Resting Tregs express the high-affinity IL-2 receptor complex (42), and therefore have 100x more affinity for IL-2 (43) than NK cells which generally express the intermediate affinity IL-2 receptor complex (44). Therefore, following IL-2 administration, Tregs may be capable of activating and responding more quickly than other cells. A clinical study in which ultra-low dose IL-2 was administered to patients with HIV demonstrated that Tregs expanded 3x more than NK cells, suggesting that Tregs cells may be more capable of responding to IL-2 (45). The use of IL-15, which is still capable of activating Tregs (46), should reduce the inherent advantage of Tregs to respond to IL-2 greater than cells lacking the high-affinity IL-2 receptor complex.
In our study, we found that Treg depletion with concurrent cytokine-mediated NK cell activation can have powerful anti-leukemic effects. This would suggest that our combined therapy may have broad reaching application and potentially could be used in a variety of tumor therapies. Further studies investigating the mechanisms of Treg inhibition of NK cells are ongoing to understand more completely how to optimize and extend this potential anti-tumor therapy.
Acknowledgments
This publication has been funded all or in part by National Institutes of Health grant R01CA95327-02, R01CA102282-01A1, and P20 RR016464.
The authors would like to acknowledge Kory Alderson, Sean Eller, and Alexandra Liska for reviewing the manuscript. We also thank Rebecca Partain and the UNR Department of Physiology and Cell Biology for sorting the Treg cells.
References
- 1.Cudkowicz G, Bennett M. Peculiar immunobiology of bone marrow allografts. I. Graft rejection by irradiated responder mice. The Journal of experimental medicine. 1971;134:83–102. doi: 10.1084/jem.134.1.83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Herberman RB, Ortaldo JR. Natural killer cells: their roles in defenses against disease. Science. 1981;214:24–30. doi: 10.1126/science.7025208. [DOI] [PubMed] [Google Scholar]
- 3.Trinchieri G. Biology of natural killer cells. Advances in immunology. 1989;47:187–376. doi: 10.1016/S0065-2776(08)60664-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Dredge K, Marriott JB, Todryk SM, Dalgleish AG. Adjuvants and the promotion of Th1-type cytokines in tumour immunotherapy. Cancer Immunol Immunother. 2002;51:521–531. doi: 10.1007/s00262-002-0309-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Trinchieri G, Wysocka M, D’Andrea A, Rengaraju M, Aste-Amezaga M, Kubin M, Valiante NM, Chehimi J. Natural killer cell stimulatory factor (NKSF) or interleukin-12 is a key regulator of immune response and inflammation. Progress in growth factor research. 1992;4:355–368. doi: 10.1016/0955-2235(92)90016-b. [DOI] [PubMed] [Google Scholar]
- 6.Gerosa F, Gobbi A, Zorzi P, Burg S, Briere F, Carra G, Trinchieri G. The reciprocal interaction of NK cells with plasmacytoid or myeloid dendritic cells profoundly affects innate resistance functions. J Immunol. 2005;174:727–734. doi: 10.4049/jimmunol.174.2.727. [DOI] [PubMed] [Google Scholar]
- 7.Geldhof AB, Van Ginderachter JA, Liu Y, Noel W, Raes G, De Baetselier P. Antagonistic effect of NK cells on alternatively activated monocytes: a contribution of NK cells to CTL generation. Blood. 2002;100:4049–4058. doi: 10.1182/blood-2001-11-0106. [DOI] [PubMed] [Google Scholar]
- 8.Kelly JM, Darcy PK, Markby JL, Godfrey DI, Takeda K, Yagita H, Smyth MJ. Induction of tumor-specific T cell memory by NK cell-mediated tumor rejection. Nature immunology. 2002;3:83–90. doi: 10.1038/ni746. [DOI] [PubMed] [Google Scholar]
- 9.Hallett WH, Murphy WJ. Positive and negative regulation of Natural Killer cells: therapeutic implications. Seminars in cancer biology. 2006;16:367–382. doi: 10.1016/j.semcancer.2006.07.003. [DOI] [PubMed] [Google Scholar]
- 10.Shevach EM. Regulatory T cells in autoimmmunity*. Annual review of immunology. 2000;18:423–449. doi: 10.1146/annurev.immunol.18.1.423. [DOI] [PubMed] [Google Scholar]
- 11.Iwashiro M, Messer RJ, Peterson KE, Stromnes IM, Sugie T, Hasenkrug KJ. Immunosuppression by CD4+ regulatory T cells induced by chronic retroviral infection. Proceedings of the National Academy of Sciences of the United States of America. 2001;98:9226–9230. doi: 10.1073/pnas.151174198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Trenado A, Charlotte F, Fisson S, Yagello M, Klatzmann D, Salomon BL, Cohen JL. Recipient-type specific CD4+CD25+ regulatory T cells favor immune reconstitution and control graft-versus-host disease while maintaining graft-versus-leukemia. The Journal of clinical investigation. 2003;112:1688–1696. doi: 10.1172/JCI17702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Ghiringhelli F, Menard C, Terme M, Flament C, Taieb J, Chaput N, Puig PE, Novault S, Escudier B, Vivier E, Lecesne A, Robert C, Blay JY, Bernard J, Caillat-Zucman S, Freitas A, Tursz T, Wagner-Ballon O, Capron C, Vainchencker W, Martin F, Zitvogel L. CD4+CD25+ regulatory T cells inhibit natural killer cell functions in a transforming growth factor-beta-dependent manner. J Exp Med. 2005;202:1075–1085. doi: 10.1084/jem.20051511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Smyth MJ, Teng MW, Swann J, Kyparissoudis K, Godfrey DI, Hayakawa Y. CD4+CD25+ T regulatory cells suppress NK cell-mediated immunotherapy of cancer. J Immunol. 2006;176:1582–1587. doi: 10.4049/jimmunol.176.3.1582. [DOI] [PubMed] [Google Scholar]
- 15.Barao I, Hanash AM, Hallett W, Welniak LA, Sun K, Redelman D, Blazar BR, Levy RB, Murphy WJ. Suppression of natural killer cell-mediated bone marrow cell rejection by CD4+CD25+ regulatory T cells. Proc Natl Acad Sci USA. 2006 doi: 10.1073/pnas.0509249103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Mule JJ, Shu S, Schwarz SL, Rosenberg SA. Adoptive immunotherapy of established pulmonary metastases with LAK cells and recombinant interleukin-2. Science. 1984;225:1487–1489. doi: 10.1126/science.6332379. [DOI] [PubMed] [Google Scholar]
- 17.Rosenberg SA, Mule JJ, Spiess PJ, Reichert CM, Schwarz SL. Regression of established pulmonary metastases and subcutaneous tumor mediated by the systemic administration of high-dose recombinant interleukin 2. J Exp Med. 1985;161:1169–1188. doi: 10.1084/jem.161.5.1169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Antony PA, Restifo NP. CD4+CD25+ T regulatory cells, immunotherapy of cancer, and interleukin-2. J Immunother (1997) 2005;28:120–128. doi: 10.1097/01.cji.0000155049.26787.45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Sayers TJ, Brooks AD, Seki N, Smyth MJ, Yagita H, Blazar BR, Malyguine AM. T cell lysis of murine renal cancer: multiple signaling pathways for cell death via Fas. Journal of leukocyte biology. 2000;68:81–86. [PubMed] [Google Scholar]
- 20.McDermott DF, Atkins MB. Application of IL-2 and other cytokines in renal cancer. Expert opinion on biological therapy. 2004;4:455–468. doi: 10.1517/14712598.4.4.455. [DOI] [PubMed] [Google Scholar]
- 21.Andersen MH, Gehl J, Reker S, Pedersen LO, Becker JC, Geertsen P, thor Straten P. Dynamic changes of specific T cell responses to melanoma correlate with IL-2 administration. Seminars in cancer biology. 2003;13:449–459. doi: 10.1016/j.semcancer.2003.09.009. [DOI] [PubMed] [Google Scholar]
- 22.Rosmaraki EE, Douagi I, Roth C, Colucci F, Cumano A, Di Santo JP. Identification of committed NK cell progenitors in adult murine bone marrow. Eur J Immunol. 2001;31:1900–1909. doi: 10.1002/1521-4141(200106)31:6<1900::aid-immu1900>3.0.co;2-m. [DOI] [PubMed] [Google Scholar]
- 23.Hallett WH, Ames E, Motarjemi M, Barao I, Shanker A, Tamang DL, Sayers TJ, Hudig D, Murphy WJ. Sensitization of tumor cells to NK cell-mediated killing by proteasome inhibition. J Immunol. 2008;180:163–170. doi: 10.4049/jimmunol.180.1.163. [DOI] [PubMed] [Google Scholar]
- 24.Tamang DL, Redelman D, Alves BN, Vollger L, Bethley C, Hudig D. Induction of granzyme B and T cell cytotoxic capacity by IL-2 or IL-15 without antigens: multiclonal responses that are extremely lytic if triggered and short-lived after cytokine withdrawal. Cytokine. 2006;36:148–159. doi: 10.1016/j.cyto.2006.11.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Assarsson E, Kambayashi T, Sandberg JK, Hong S, Taniguchi M, Van Kaer L, Ljunggren HG, Chambers BJ. CD8+ T cells rapidly acquire NK1.1 and NK cell-associated molecules upon stimulation in vitro and in vivo. J Immunol. 2000;165:3673–3679. doi: 10.4049/jimmunol.165.7.3673. [DOI] [PubMed] [Google Scholar]
- 26.Fehniger TA, Cai SF, Cao X, Bredemeyer AJ, Presti RM, French AR, Ley TJ. Acquisition of murine NK cell cytotoxicity requires the translation of a pre-existing pool of granzyme B and perforin mRNAs. Immunity. 2007;26:798–811. doi: 10.1016/j.immuni.2007.04.010. [DOI] [PubMed] [Google Scholar]
- 27.Abdool K, Cretney E, Brooks AD, Kelly JM, Swann J, Shanker A, Bere EW, Jr, Yokoyama WM, Ortaldo JR, Smyth MJ, Sayers TJ. NK cells use NKG2D to recognize a mouse renal cancer (Renca), yet require intercellular adhesion molecule-1 expression on the tumor cells for optimal perforin-dependent effector function. J Immunol. 2006;177:2575–2583. doi: 10.4049/jimmunol.177.4.2575. [DOI] [PubMed] [Google Scholar]
- 28.Imai H, Saio M, Nonaka K, Suwa T, Umemura N, Ouyang GF, Nakagawa J, Tomita H, Osada S, Sugiyama Y, Adachi Y, Takami T. Depletion of CD4+CD25+ regulatory T cells enhances interleukin-2-induced antitumor immunity in a mouse model of colon adenocarcinoma. Cancer science. 2007;98:416–423. doi: 10.1111/j.1349-7006.2006.00385.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Foss F. Clinical experience with denileukin diftitox (ONTAK) Semin Oncol. 2006;33:S11–16. doi: 10.1053/j.seminoncol.2005.12.017. [DOI] [PubMed] [Google Scholar]
- 30.Barnett B, Kryczek I, Cheng P, Zou W, Curiel TJ. Regulatory T cells in ovarian cancer: biology and therapeutic potential. Am J Reprod Immunol. 2005;54:369–377. doi: 10.1111/j.1600-0897.2005.00330.x. [DOI] [PubMed] [Google Scholar]
- 31.Dannull J, Su Z, Rizzieri D, Yang BK, Coleman D, Yancey D, Zhang A, Dahm P, Chao N, Gilboa E, Vieweg J. Enhancement of vaccine-mediated antitumor immunity in cancer patients after depletion of regulatory T cells. J Clin Invest. 2005;115:3623–3633. doi: 10.1172/JCI25947. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Mahnke K, Schonfeld K, Fondel S, Ring S, Karakhanova S, Wiedemeyer K, Bedke T, Johnson TS, Storn V, Schallenberg S, Enk AH. Depletion of CD4+CD25+ human regulatory T cells in vivo: kinetics of Treg depletion and alterations in immune functions in vivo and in vitro. Int J Cancer. 2007;120:2723–2733. doi: 10.1002/ijc.22617. [DOI] [PubMed] [Google Scholar]
- 33.Stephens LA, Mason D. CD25 is a marker for CD4+ thymocytes that prevent autoimmune diabetes in rats, but peripheral T cells with this function are found in both CD25+ and CD25- subpopulations. J Immunol. 2000;165:3105–3110. doi: 10.4049/jimmunol.165.6.3105. [DOI] [PubMed] [Google Scholar]
- 34.Apostolou I, Sarukhan A, Klein L, von Boehmer H. Origin of regulatory T cells with known specificity for antigen. Nat Immunol. 2002;3:756–763. doi: 10.1038/ni816. [DOI] [PubMed] [Google Scholar]
- 35.Kohm AP, McMahon JS, Podojil JR, Begolka WS, DeGutes M, Kasprowicz DJ, Ziegler SF, Miller SD. Cutting Edge: Anti-CD25 monoclonal antibody injection results in the functional inactivation, not depletion, of CD4+CD25+ T regulatory cells. J Immunol. 2006;176:3301–3305. doi: 10.4049/jimmunol.176.6.3301. [DOI] [PubMed] [Google Scholar]
- 36.Zelenay S, Demengeot J. Comment on “Cutting edge: anti-CD25 monoclonal antibody injection results in the functional inactivation, not depletion, of CD4+CD25+ T regulatory cells”. J Immunol. 2006;177:2036–2037. doi: 10.4049/jimmunol.177.4.2036-a. author reply 2037–2038. [DOI] [PubMed] [Google Scholar]
- 37.Stephens LA, Anderton SM. Comment on “Cutting edge: anti-CD25 monoclonal antibody injection results in the functional inactivation, not depletion, of CD4+CD25+ T regulatory cells”. J Immunol. 2006;177:2036. doi: 10.4049/jimmunol.177.4.2036-a. author reply 2037–2038. [DOI] [PubMed] [Google Scholar]
- 38.Voss SD, Robb RJ, Weil-Hillman G, Hank JA, Sugamura K, Tsudo M, Sondel PM. Increased expression of the interleukin 2 (IL-2) receptor beta chain (p70) on CD56+ natural killer cells after in vivo IL-2 therapy: p70 expression does not alone predict the level of intermediate affinity IL-2 binding. J Exp Med. 1990;172:1101–1114. doi: 10.1084/jem.172.4.1101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Soiffer RJ, Murray C, Cochran K, Cameron C, Wang E, Schow PW, Daley JF, Ritz J. Clinical and immunologic effects of prolonged infusion of low-dose recombinant interleukin-2 after autologous and T-cell-depleted allogeneic bone marrow transplantation. Blood. 1992;79:517–526. [PubMed] [Google Scholar]
- 40.Ahmadzadeh M, Rosenberg SA. IL-2 administration increases CD4+ CD25(hi) Foxp3+ regulatory T cells in cancer patients. Blood. 2006;107:2409–2414. doi: 10.1182/blood-2005-06-2399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Fontenot JD, Rasmussen JP, Gavin MA, Rudensky AY. A function for interleukin 2 in Foxp3-expressing regulatory T cells. Nat Immunol. 2005;6:1142–1151. doi: 10.1038/ni1263. [DOI] [PubMed] [Google Scholar]
- 42.Sakaguchi S, Ono M, Setoguchi R, Yagi H, Hori S, Fehervari Z, Shimizu J, Takahashi T, Nomura T. Foxp3+ CD25+ CD4+ natural regulatory T cells in dominant self-tolerance and autoimmune disease. Immunol Rev. 2006;212:8–27. doi: 10.1111/j.0105-2896.2006.00427.x. [DOI] [PubMed] [Google Scholar]
- 43.Taniguchi T, Minami Y. The IL-2/IL-2 receptor system: a current overview. Cell. 1993;73:5–8. doi: 10.1016/0092-8674(93)90152-g. [DOI] [PubMed] [Google Scholar]
- 44.Voss SD, Sondel PM, Robb RJ. Characterization of the interleukin 2 receptors (IL-2R) expressed on human natural killer cells activated in vivo by IL-2: association of the p64 IL-2R gamma chain with the IL-2R beta chain in functional intermediate-affinity IL-2R. J Exp Med. 1992;176:531–541. doi: 10.1084/jem.176.2.531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Shah MH, Freud AG, Benson DM, Jr, Ferkitich AK, Dezube BJ, Bernstein ZP, Caligiuri MA. A phase I study of ultra low dose interleukin-2 and stem cell factor in patients with HIV infection or HIV and cancer. Clin Cancer Res. 2006;12:3993–3996. doi: 10.1158/1078-0432.CCR-06-0268. [DOI] [PubMed] [Google Scholar]
- 46.Yates J, Rovis F, Mitchell P, Afzali B, Tsang JY, Garin M, Lechler RI, Lombardi G, Garden OA. The maintenance of human CD4+ CD25+ regulatory T cell function: IL-2, IL-4, IL-7 and IL-15 preserve optimal suppressive potency in vitro. Int Immunol. 2007;19:785–799. doi: 10.1093/intimm/dxm047. [DOI] [PubMed] [Google Scholar]