

Abstract
Skeletal muscle weakness is a common finding in patients with chronic heart failure (CHF). This functional deficit cannot be accounted for by muscle atrophy alone, suggesting that the syndrome of heart failure induces a myopathy in the skeletal musculature. To determine whether decrements in muscle performance are related to alterations in contractile protein function, biopsies were obtained from the vastus lateralis muscle of four CHF patients and four control patients. CHF patients exhibited reduced peak aerobic capacity and knee extensor muscle strength. Decrements in whole muscle strength persisted after statistical control for muscle size. Thin filaments and myosin were isolated from biopsies and mechanically assessed using the in vitro motility assay. Isolated skeletal muscle thin-filament function, however, did not differ between CHF patients and controls with respect to unloaded shortening velocity, calcium sensitivity, or maximal force. Similarly, no difference in maximal force or unloaded shortening velocity of isolated myosin was observed between CHF patients and controls. From these results, we conclude that skeletal contractile protein function is unaltered in CHF patients. Other factors, such as a decrease in total muscle myosin content, are likely contributors to the skeletal muscle strength deficit of heart failure.
Keywords: thin filament, myosin, fiber type
Exercise intolerance is a hallmark symptom of chronic heart failure (CHF). Although cardiac pump dysfunction can limit exercise capacity, research conducted over the last two decades has highlighted the importance of changes in the skeletal musculature in heart failure (10, 21, 37). Recent studies have shown that skeletal muscle strength is reduced in heart failure and that this decrement cannot be solely accounted for by a reduction in muscle mass (9, 28), suggesting a fundamental defect in skeletal muscle contractile function. Recent studies have confirmed this at the cellular level. In chemically skinned, single skeletal muscle fibers (26), isometric force production and ATPase were profoundly reduced in heart failure patients compared with controls. These data suggest that the contractile machinery directly contributes to the functional deficit of skeletal muscle in heart failure. The mechanism whereby CHF affects the contractile properties of myofilament proteins, however, has not been studied.
In the failing human myocardium, we have demonstrated that changes at the thin-filament level account for a 35% depression in contractile force, a defect that is mediated via the phosphorylation of troponin (19). Phosphorylation of specific myofilament proteins is thought to affect human myocardial contractile function (19, 34). Such processes are mediated through signaling pathways activated by stimulation of specific neurohormone receptors, such as angiotensin II type 1, endothelin, and α-adrenergic (7). Since circulating neurohormone levels are markedly elevated in heart failure, we hypothesized that a similar defect in contractile protein function may exist in the skeletal musculature of patients with heart failure. To test this hypothesis, we examined skeletal muscle thin-filament function from biopsies of the vastus lateralis muscle in CHF patients and controls using an in vitro motility assay. Vastus lateralis muscle is comprised of slow- and fast-twitch fibers (31) that contain different contractile protein isoforms, most notably myosin heavy chain (MHC), myosin light chain, troponin C, troponin I, and troponin T. Although differences in fast- and slow-twitch fiber function related to MHC isoform expression are well described (15), it is not known whether these fiber types exhibit differences in thin-filament function. Thus we initially performed experiments to examine the effect of fiber type on thin-filament function by comparing the function of skeletal muscle thin filaments isolated from rat soleus (predominantly slow-twitch, MHC I-containing fibers) and extensor digitorum longus (predominantly fast-twitch, MHC IIB- and IIX-containing fibers) muscle fibers. Finally, to determine whether the skeletal myopathy of heart failure is associated with an alteration in myosin function, we measured the contractile properties of isolated myosin using the in vitro motility assay.
METHODS
Subjects
Four men with chronic heart failure were recruited from the Heart Failure Clinic of the Cardiology Unit at the University of Vermont. Four male volunteers served as controls. Two of the controls were healthy and free of disease, had no signs or symptoms of heart disease, and had normal rest and exercise electrocardiograms. The other two control volunteers had stable coronary artery disease with normal global and regional left ventricular function, resting electrocardiograms, and no evidence of exertional ischemia, as demonstrated by a normal maximal electrocardiographic stress test without angina. The latter two volunteers were treated with aspirin, one with a Ca2+ channel blocker and the other with an 3-hydroxy-3-methyl-glutaryl-CoA reductase inhibitor. All heart failure patients were taking angiotensin-converting enzyme inhibitors or angiotensin receptor blockers and diuretics, 75% were taking β-blockers, and 50% were taking digoxin. The research protocol was approved by Committees on Human Research at the University of Vermont. Written, informed consent was obtained from each research subject. Data on these volunteers regarding the effect of heart failure on skeletal muscle protein expression and whole muscle function have been published previously (27, 28).
Left ventricular ejection fraction was determined by quantitative echocardiography. Peak oxygen consumption (peak V̇O2) was determined through treadmill stress testing by standard protocol. Isokinetic and isometric leg strength was assessed in the right leg of subjects using a multijoint dynamometer (Loredan Biomedical, Sacramento, CA). New York Heart Association heart failure functional class was determined by the patient’s attending cardiologist.
Skeletal muscle biopsies were taken from the vastus lateralis muscle under local lidocaine anesthesia as previously described (31). Biopsies weighing between 100 and 150 mg were snap frozen in liquid nitrogen after adipose and connective tissues were removed and subsequently stored at −70°C.
Animals
Rats were obtained (Taconic, Germantown, NY) at 6 wk of age and were housed singly in wire-bottom cages. All rats were maintained on a 12:12-h light-dark cycle in a temperature-controlled room. Tap water and a low-salt (0.6%) chow were available ad libitum throughout the study. All procedures were approved by the Institutional Animal Care and Use Committee of the University of Vermont. At 20 wk of age, soleus and extensor digitorum longus (EDL) muscle tissues were harvested under pentobarbital sodium (90 mg/kg ip) anesthesia. Tissue was frozen in liquid nitrogen and stored at −80°C until analysis.
Myofibrillar protein content and isoform distribution
Actin and MHC content were determined from tissue homogenates using gel electrophoresis, as described previously (31). Actin and MHC content were determined as a function of protein load. In addition, the relative MHC isoform content was determined on homogenates from a separate piece of tissue using gel electrophoresis, as described previously (31). MHC isoforms were expressed as a fraction of total MHC protein content determined by densitometry.
Contractile protein preparation
Native thin filaments were isolated from frozen muscle tissue as described previously (35). In brief, myofibrils were isolated from frozen muscle tissue. Myofibrils were then homogenized in 10 mM sodium phosphate (pH 6.5), 100 mM NaCl, 5 mM MgCl2, 1 mM NaN3, 1 mM EGTA, 5 mM ATP, and 2 μg/ml leupeptin at a ratio of 40 ml/g of myofibril. Debris and thick filament were then pelleted with centrifugation at 137,000 g for 20 min. Native thin filaments were then collected with centrifugation (137,000 g for 180 min) with the pellet being raised in a low-salt buffer (25 mM KCl, 25 mM imidazole, 1 mM EGTA, 5 mM MgCl2, 10 mM DTT). Myosin was isolated by homogenization of frozen muscle tissue in 0.3 M KCl, 0.15 M KH2PO4, 10 mM Na4P2O7, 5 mM MgCl2, 2 mM ATP, pH to 6.8, 5 mM DTT. After 60 min on ice, the homogenate was clarified with centrifugation at 157,000 g for 60 min. The supernatant was then diluted 10-fold with 2 mM DTT and left to sit on ice for 60 min. Myosin was then collected at 38,500 g for 20 min. The pellet was then raised in 0.3 M KCl, 10 mM imidazole, 2 mM DTT, stored on ice, and used in the motility assay within 24 h. The purity of the isolations was confirmed by SDS-PAGE. Protein concentration was determined for native thin filaments and myosin with a protein assay (Bio-Rad) using bovine serum albumin (Sigma) as the standard. In the experiments with native thin filaments, chicken pectoralis myosin was used. Similarly, in the experiments using isolated human skeletal myosin, chicken pectoralis actin was used. Native thin filaments and chicken pectoralis actin were labeled with rhodamine-phalloidin (1:1 molar ratio) before use in the motility assay. α-Actinin (Sigma) was dialyzed in (in mM) 25 KCl, 25 Imidazole (pH 7.4), 5 MgCl2, 2 EGTA, 1 NaN3, and 1 DTT. The α-actinin concentration was subsequently determined using a protein assay (Bio-Rad).
Motility assay
The details of the motility assay have previously been described (35). In brief, myosin (100 μg/ml, unless otherwise noted) was applied for 1 min to a nitrocellulose coated coverslip in (in mM) 300 KCl, 25 imidazole, 5 MgCl2, 2 m EGTA, and 10 DTT. The surface was then washed with bovine serum albumin (0.5 mg/ml) in low-salt buffer. Employing epifluorescent microscopy, rhodamine-labeled thin filaments were observed moving across the myosin-coated surface in the presence of ATP (2 mM) and as a function of the free calcium in the motility solutions (25 mM KCl, 25 mM imidazole, 5 mM MgCl2, 2 mM EGTA and 10 mM DTT, 0.1 mg/ml glucose oxidase, 1.8 μg/ml catalase, 2.3 mg/ml glucose, and 0.38% methyl-cellulose). Free calcium in the motility solution was adjusted as described (3). Relative force was determined by adding increasingly higher concentrations of α-actinin to the motility surface until thin-filament motility was arrested (35). Since α-actinin avidly binds actin, it functions to load the movement of actin filaments in a concentration-dependent manner. Thus the concentration of α-actinin required to completely arrest thin-filament motility is a relative measure of isometric force. All experiments were performed at 30°C
Statistics
All data are reported as means ± SE, unless otherwise noted. An unpaired Student’s t-test was used to compare groups. Analysis of covariance was used to compare whole muscle strength data between groups after differences in leg muscle mass were adjusted for. All analyses were conducted with SPSS software 13 (SPSS, Chicago, IL). All values are means ± SE unless otherwise specified.
RESULTS
Clinical characteristics
The CHF and control group were similar in age and weight (Table 1). The etiology of heart failure in the CHF group was ischemic in two patients and idiopathic in two patients. All heart failure patients had severe left ventricular systolic dysfunction, as assessed by two-dimensional echocardiography (mean left ventricular ejection fraction of 18 ± 3%) and impaired functionality as indicated by a mean New York Heart Association class of 2.8 ± 0.5. Consistent with the severity of heart failure was the marked reduction in peak V̇O2 (Table 1; P = 0.02). Leg muscle mass was mildly reduced in the heart failure group, but the differences did not reach significance. Both isokinetic (P = 0.04) and isometric (P < 0.01) muscle strength were reduced in CHF patients (Table 1). The difference in leg strength persisted between the two groups for isometric (P < 0.05) after accounting for differences in muscle mass using covariance analysis, whereas the difference in isokinetic strength was diminished but not statistically significant (P = 0.174).
Table 1.
Clinical characteristics of research subjects
| CHF | Control | P Value | |
|---|---|---|---|
| Mean age, yr | 60.3±5.4 | 64.5±8.3 | 0.68 |
| Mean body weight, kg | 76.3±8.1 | 89.3±11.9 | 0.40 |
| Mean peak V̇O2, ml·kg−1·min−1 | 16.7±1.4 | 34.0±4.3 | 0.02 |
| Leg muscle mass, kg | 18.1±0.7 | 21.2±1.7 | 0.19 |
| Leg force isokinetic, N ·m | 76.7±12.8 | 141.0±20.2 | 0.04 |
| Leg force isometric, N ·m | 148.5±9.6 | 211.8±11.2 | <0.01 |
Values are means ± SE. CHF, chronic heart failure subjects; V̇O2, oxygen consumption.
Myofibrillar protein content and MHC isoform composition
Actin protein content did not differ between heart failure patients and controls (CHF, 167 ± 19 vs. control, 189 ± 22 arbitrary units (AU)/μg protein; P = 0.47). In contrast, there was a strong trend toward reduced (42%) MHC protein content in the heart failure group (CHF, 128 ± 10 vs. control, 222 ± 40 AU/μg protein; P = 0.06). We found no difference in the relative distribution of MHC I (CHF, 63 ± 8 vs. control, 63 ± 7%; P = 0.93), MHC IIa (CHF, 36 ± 8 vs. control, 35 ± 5%; P = 0.97), or MHC IIx (CHF, 1.1 ± 0.4 vs. control, 2.4 ± 1.1%; P = 0.33).
Characterization of thin-filament function from different fiber types
Native thin filaments were isolated from fast-twitch fibers, rat EDL, slow-twitch fibers, and rat soleus. Using a motility substrate of chicken pectoralis myosin in a calcium-regulated system, no difference was found between EDL- and soleus-isolated native thin-filament function (Table 2, Fig. 1A) with regard to maximal unloaded shortening, calcium sensitivity (pCa50), or cooperativity of thin-filament activation (Hill coefficient). Furthermore, using the α-actinin force assay, no difference in force generation as a function of calcium was observed between thin filament isolated from the different fiber types (Table 2, Fig. 1B).
Table 2.
Rat skeletal native thin-filament studies
| EDL | Soleus | P Value | |
|---|---|---|---|
| Unloaded shortening velocity | |||
| Maximal velocity, μm/s | 5.03±0.29 | 5.35±0.25 | 0.21 |
| pCa50 | 6.51±0.04 | 6.41±0.04 | 0.96 |
| Hill coefficient | 2.85±0.67 | 2.74±0.54 | 0.90 |
| Relative isometric force | |||
| Maximal force | 1.00±0.01 | 0.96±0.06 | 0.64 |
| pCa50 | 6.59±0.05 | 6.60±0.05 | 0.46 |
| Hill coefficient | 2.66±0.71 | 3.88±1.64 | 0.53 |
Values are means ± SE. EDL, extensor digitorum longus; maximal velocity, maximal native thin-filament velocity propelled by chicken pectoralis myosin; pCa50, negative log of the calcium concentration at which velocity or force is half maximal; Hill coefficient, slope of the activation curve and a measure of thin-filament cooperativity; maximal force, maximal native thin-filament force using chicken pectoralis myosin.
Fig. 1.

Velocity-pCa (A) and force-pCa (B) relations for native thin filaments isolated from rat fast-twitch muscle (extensor digitorum longus: ●, broken regression) compared with rat slow-twitch muscle (soleus: ○, solid regression) using chicken pectoralis myosin as the motility substrate.
Skeletal thin filament and myosin function in human subjects
As shown in Fig. 2 and Table 3, native thin filaments isolated from the vastus lateralis of CHF patients exhibit similar calcium-activated unloaded shortening velocity compared with healthy volunteers. Furthermore, no difference in maximal isometric force was demonstrated for thin filaments isolated from the two groups performed at pCa 5 (Fig. 3).
Fig. 2.
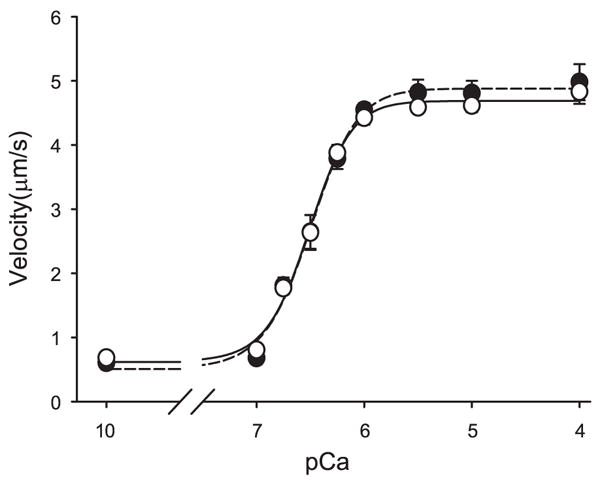
Velocity-pCa relation for isolated native thin filaments from the vastus lateralis muscle of research subjects with chronic heart failure (CHF; ○, solid regression) compared with controls (●, broken regression). Chicken pectoralis was used as the motility substrate.
Table 3.
Human skeletal native thin-filament studies
| CHF | Control | P Value | |
|---|---|---|---|
| Unloaded shortening velocity | |||
| Maximal velocity, μm/s | 4.7±0.2 | 4.9±0.2 | 0.77 |
| pCa50 | 6.51±0.03 | 6.50±0.03 | 0.37 |
| Hill coefficient | 2.16±0.25 | 1.98±0.24 | 0.62 |
| Relative maximal isometric force | 1.00±0.13 | 0.99±0.10 | 0.39 |
Values are means ± SE. Relative maximal force is the relative maximal native thin-filament force for CHF and control subjects using chicken pectoralis myosin.
Fig. 3.
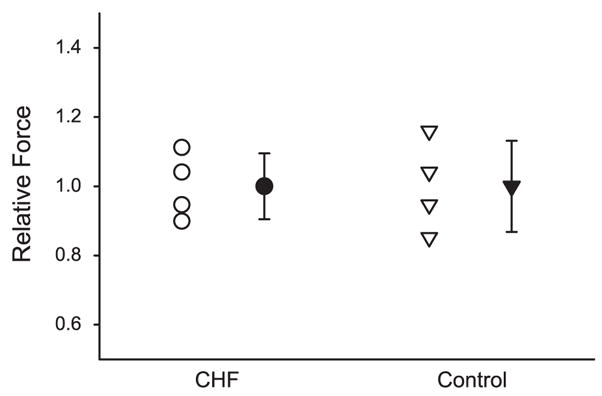
Maximal isometric force (pCa 5) of native thin filaments isolated from the vastus lateralis muscle of research subjects with CHF (circles) compared with controls (triangles). The open data points represent the data from individual research subjects in each group, whereas the filled data points represent the mean and the SE of the CHF and control groups.
Finally, to determine whether an alteration in myosin function was present in the skeletal musculature of heart failure patients, myosin was isolated and characterized in the in vitro motility assay using unregulated actin isolated from chicken pectoralis muscle. The velocity at which myosin translocated actin filaments across a myosin-coated surface as a measure of unloaded shortening was not different between CHF patients and controls (P = 0.94; Fig. 4A). These velocity values are similar to that reported previously for human skeletal myosin (5). As previously shown (35), isometric force demonstrates a linear dependence on myosin surface concentration as demonstrated in Fig. 4B. No difference in force was seen for myosin isolated from CHF and control group skeletal muscle (P = 0.42).
Fig. 4.

A: the velocity of actin filaments propelled by myosin isolated from the vastus lateralis muscle of CHF and control research subjects. Actin is isolated from chicken pectoralis. B: relative isometric force as a function of skeletal myosin concentration in the loading buffers for CHF (squares, solid regression) and control (circles, dashed regression) research subjects. Force increased as a function of myosin concentration with no difference demonstrated between the two groups.
DISCUSSION
Studies have shown that CHF is associated with reduced skeletal muscle strength per unit muscle size (9, 28). This contractile deficit at the whole muscle level has been observed in isolated muscle tissue in animal models (16, 21) and in chemically skinned, single muscle fibers in humans (26), suggesting that the heart failure syndrome promotes contractile dysfunction at the level of the myofilaments. In the present study, we investigated the role of individual myofilament components in the decreased skeletal muscle function of heart failure patients. We hypothesized, similar to our findings in human myocardium (19), that CHF impairs skeletal muscle contractile function at the myofilament level via impaired thin-filament function. In contrast to this hypothesis, and despite the fact that we observed reduced whole skeletal muscle strength in heart failure patients, no alterations in the functional properties of skeletal muscle thin filaments or myosin were observed. These results are discussed in the context of recent observations of altered muscle function and contractile protein expression in heart failure patients.
We originally hypothesized that contractile function would be impaired in skeletal muscle in a manner similar to that observed in the myocardium (18, 19, 25, 38), where impaired thin-filament function has been implicated as a predominant defect. The logic for this hypothesis was based on the fact that skeletal muscle is exposed to the same circulating hormonal milieu that likely incites the contractile abnormalities in the myocardium, most notably, increased neurohormones and cytokines (17). In the myocardium, thin-filament phosphorylation is altered in CHF and is directly responsible for changes in thin-filament function (19). Protein kinase A (PKA) and C (PKC) signaling are potential mediators of thin-filament phosphorylation, and an increase in myocardial tissue expression of several PKC isoforms has been demonstrated (2, 19). Although PKC phosphorylation sites are present on skeletal muscle troponin I and troponin T, skeletal muscle troponin I lacks the PKA-specific, NH2-terminal phosphorylation sites present on cardiac troponin I (8). Thus it could be argued that our inability to observe alterations in skeletal muscle thin-filament function relates to the absence of PKA regulatory sites. We do not believe this is the case, however, since phorbol ester activation of PKC in rat myocardium results in a similar reduction in maximal cardiac thin-filament force production as failing human myocardial thin filaments, thus implicating PKC-mediated phosphorylation of troponin in human heart failure (19).
In direct contrast with the pathophysiology of human myocardium (19), our data indicate that the functional deficit in skeletal muscle of patients with cardiomyopathy is not the result of altered thin-filament function. Differences between the two tissues could relate to their patterns of use. In patients with CHF, the myocardium is subjected to increased load, which causes myocyte hypertrophy and remodeling. In contrast, skeletal muscle use is decreased in CHF (30), with consequent atrophy (29). Alterations in cellular regulatory proteins related to these different muscle use patterns could affect thin-filament function differently in cardiac and skeletal muscle. In addition, the response of the cardiac and skeletal muscle to circulating neurohumoral activation may differ. For example, in an animal model of heart failure, Krankel et al. (13) recently observed a threefold increase in cytokine-induced expression of protein phosphatase 2A in skeletal muscle. This phosphatase demonstrates a high specificity to thin-filament regulatory proteins and may counteract any effect of increased PKC activity in skeletal muscle.
Another factor to consider in the interpretation of our findings is that thin filaments were isolated from biopsies that contain multiple fiber types (i.e., fast- and slow-twitch fibers). There are pronounced functional differences between fiber types with greater shortening velocity and increased maximal power output in fast-twitch relative to slow-twitch fibers (1, 15). Although many functional differences between fiber types have been ascribed to myosin composition (1, 15), variation in the composition of thin-filament proteins, such as troponin C, I, and T isoforms (20, 22, 23), could also contribute. This point is particularly relevant to our results since our laboratory (31) and others (36) have shown alterations in fiber-type distribution in CHF, with a shift toward a more fast-twitch phenotype. To directly test whether variation in fiber type affects thin-filament function, we isolated native thin filaments from rat EDL and soleus muscles [composed primarily of fast-twitch and slow-twitch fibers, respectively (32)] and characterized their function using the in vitro motility assay. No difference in thin-filament unloaded shortening, calcium sensitivity, or isometric force was demonstrated between these muscles of vastly different fiber-type composition. Although some studies have suggested modest increases in calcium sensitivity in chemically skinned slow and fast single muscle fibers (4), these differences could be caused by factors unrelated to the thin filament, such as thick-filament proteins and/or other myofilament lattice proteins. In this respect, our data are the first to assess functional differences of thin filaments isolated from different muscle fiber types. Thus any differences in fiber-type distribution between patients and controls would not impact our ability to detect an effect of heart failure on thin-filament function. Perhaps most importantly, we found nearly identical MHC isoform distributions between heart failure patients and controls in this study (see RESULTS), suggesting that any differences in muscle fiber-type distribution between groups would be minor and would not impact group differences in thin-filament function.
Our findings also suggest that heart failure does not affect myosin function with respect to unloaded shortening velocity or isometric force production. In our studies, myosin was isolated from tissue samples that contained multiple muscle fiber types (i.e., MHC I, IIa, and IIx isoforms). Although this will not impact myosin force production, since prior studies have found little to no differences in force production among fiber types (1, 15), there are well-described differences in actin sliding velocity between MHC I and II species (12). In particular, previous in vitro motility experiments using mixtures of slow (MHC I) and fast myosins (MHC IIa and IIx) have demonstrated that slow myosin will induce drag on the fast myosin, greatly impeding the sliding speed of the actin filaments (11). Because of this, our sliding velocity data represent an average value for all MHC isoforms (I, IIa, and IIx). Our values for sliding velocity from isolated MHC, however, agree well with prior studies using MHC isolated from individual muscle fibers (12). Importantly, since there were no differences in MHC isoform composition between the heart failure and control groups (see RESULTS), variation in MHC isoform content should not impact group differences in sliding velocity.
We should note that our procedures for isolation of both myosin and thin filaments require the use of reducing agents. Thus we cannot dismiss the possibility that our experimental preparation eliminated oxidative modifications to myosin and/or thin filaments that could impair contractile function. In animal models, there is evidence for increased generation of reactive oxygen species in skeletal muscle (33). Although increased reactive oxygen species can impair muscle performance (24), the extent to which oxidative modifications alter skeletal muscle function in humans in vivo has not been clearly defined.
Our findings suggest that heart failure does not alter isolated thin filament or myosin function. Therefore, the questions remains as to what causes deficits in whole muscle function in heart failure patients. Recent studies from our laboratory have shown that heart failure promotes a reduction in skeletal muscle MHC protein content (31), presumably via downregulation of MHC transcription (27). As we clearly demonstrate in Fig. 4A, a decrease in MHC protein content would reduce muscle force production. A similar relationship of MHC protein content to muscle force production has been demonstrated in human single muscle fibers (6, 14). In this context, and in light of the current results, we put forth the hypothesis that skeletal muscle contractile dysfunction in heart failure (26) patients is caused by reduced MHC protein content rather than by alterations in the function of individual contractile proteins.
In conclusion, although marked deficits in skeletal muscle exist in patients with heart failure, these cannot be ascribed to alterations in thin filament or myosin function. Thus other factors, such as alterations in myofibrillar protein content, likely contribute to the skeletal myopathy of heart failure.
Acknowledgments
We thank Kelly J. Begin for technical assistance.
GRANTS
This study was funded by National Institutes of Health Grants HL-077418, AG-17494 (M. J. Toth), and HL-65586 (P. Van Buren).
References
- 1.Bottinelli R, Canepari M, Pellegrino MA, Reggiani C. Force-velocity properties of human skeletal muscle fibres: myosin heavy chain isoform and temperature dependence. J Physiol. 1996;495:573–586. doi: 10.1113/jphysiol.1996.sp021617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Bowling N, Walsh RA, Song G, Estridge T, Sandusky GE, Fouts RL, Mintze K, Pickard T, Roden R, Bristow MR, Sabbah HN, Mizrahi JL, Gromo G, King GL, Vlahos CJ. Increased protein kinase C activity and expression of Ca2+-sensitive isoforms in the failing human heart. Circulation. 1999;99:384–391. doi: 10.1161/01.cir.99.3.384. [DOI] [PubMed] [Google Scholar]
- 3.Brooks SP, Storey KB. Bound and determined: a computer program for making buffers of defined ion concentrations. Anal Biochem. 1992;201:119–126. doi: 10.1016/0003-2697(92)90183-8. [DOI] [PubMed] [Google Scholar]
- 4.Brotto MA, Biesiadecki BJ, Brotto LS, Nosek TM, Jin JP. Coupled expression of troponin T and troponin I isoforms in single skeletal muscle fibers correlates with contractility. Am J Physiol Cell Physiol. 2006;290:C567–C576. doi: 10.1152/ajpcell.00422.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Canepari M, Rossi R, Pellegrino MA, Orrell RW, Cobbold M, Harridge S, Bottinelli R. Effects of resistance training on myosin function studied by the in vitro motility assay in young and older men. J Appl Physiol. 2005;98:2390 –2395. doi: 10.1152/japplphysiol.01103.2004. [DOI] [PubMed] [Google Scholar]
- 6.D’Antona G, Pellegrino MA, Adami R, Rossi R, Carlizzi CN, Canepari M, Saltin B, Bottinelli R. The effect of ageing and immobilization on structure and function of human skeletal muscle fibres. J Physiol. 2003;552:499–511. doi: 10.1113/jphysiol.2003.046276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Dorn GW, Force T. Protein kinase cascades in the regulation of cardiac hypertrophy. J Clin Invest. 2005;115:527–537. doi: 10.1172/JCI24178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Gordon AM, Homsher E, Regnier M. Regulation of contraction in striated muscle. Physiol Rev. 2000;80:853–924. doi: 10.1152/physrev.2000.80.2.853. [DOI] [PubMed] [Google Scholar]
- 9.Harrington D, Anker SD, Chua TP, Webb-Peploe KM, Ponikowski PP, Poole-Wilson PA, Coats AJ. Skeletal muscle function and its relation to exercise tolerance in chronic heart failure. J Am Coll Cardiol. 1997;30:1758 –1764. doi: 10.1016/s0735-1097(97)00381-1. [DOI] [PubMed] [Google Scholar]
- 10.Harrington D, Coats AJ. Skeletal muscle abnormalities and evidence for their role in symptom generation in chronic heart failure. Eur Heart J. 1997;18:1865–1872. doi: 10.1093/oxfordjournals.eurheartj.a015194. [DOI] [PubMed] [Google Scholar]
- 11.Harris DE, Work SS, Wright RK, Alpert NR, Warshaw DM. Smooth, cardiac and skeletal muscle myosin force and motion generation assessed by cross-bridge mechanical interactions in vitro. J Muscle Res Cell Motil. 1994;15:11–19. doi: 10.1007/BF00123828. [DOI] [PubMed] [Google Scholar]
- 12.Hook P, Larsson L. Actomyosin interactions in a novel single muscle fiber in vitro motility assay. J Muscle Res Cell Motil. 2000;21:357–365. doi: 10.1023/a:1005614212575. [DOI] [PubMed] [Google Scholar]
- 13.Krankel N, Adams V, Gielen S, Linke A, Erbs S, Schuler G, Hambrecht R. Differential gene expression in skeletal muscle after induction of heart failure: impact of cytokines on protein phosphatase 2A expression. Mol Genet Metab. 2003;80:262–271. doi: 10.1016/s1096-7192(03)00132-x. [DOI] [PubMed] [Google Scholar]
- 14.Larsson L, Li X, Edstrom L, Eriksson LI, Zackrisson H, Argentini C, Schiaffino S. Acute quadriplegia and loss of muscle myosin in patients treated with nondepolarizing neuromuscular blocking agents and corticosteroids: mechanisms at the cellular and molecular levels. Crit Care Med. 2000;28:34 – 45. doi: 10.1097/00003246-200001000-00006. [DOI] [PubMed] [Google Scholar]
- 15.Larsson L, Moss RL. Maximum velocity of shortening in relation to myosin isoform composition in single fibres from human skeletal muscles. J Physiol. 1993;472:595– 614. doi: 10.1113/jphysiol.1993.sp019964. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Lunde PK, Dahlstedt AJ, Bruton JD, Lannergren J, Thoren P, Sejersted OM, Westerblad H. Contraction and intracellular Ca2+ handling in isolated skeletal muscle of rats with congestive heart failure. Circ Res. 2001;88:1299 –1305. doi: 10.1161/hh1201.092041. [DOI] [PubMed] [Google Scholar]
- 17.Mann DL, Bristow MR. Mechanisms and models in heart failure: the biomechanical model and beyond. Circulation. 2005;111:2837–2849. doi: 10.1161/CIRCULATIONAHA.104.500546. [DOI] [PubMed] [Google Scholar]
- 18.Noguchi T, Camp P, Alix SL, Gorga JA, Begin KJ, Leavitt BJ, Ittleman FP, Alpert NR, LeWinter MM, VanBuren P. Myosin from failing and non-failing human ventricles exhibit similar contractile properties. J Mol Cell Cardiol. 2003;35:91–97. doi: 10.1016/s0022-2828(02)00282-1. [DOI] [PubMed] [Google Scholar]
- 19.Noguchi T, Hunlich M, Camp PC, Begin KJ, El Zaru M, Patten R, Leavitt BJ, Ittleman FP, Alpert NR, LeWinter MM, VanBuren P. Thin filament-based modulation of contractile performance in human heart failure. Circulation. 2004;110:982–987. doi: 10.1161/01.CIR.0000139334.43109.F9. [DOI] [PubMed] [Google Scholar]
- 20.Parmacek MS, Leiden JM. Structure and expression of the murine slow/cardiac troponin C gene. J Biol Chem. 1989;264:13217–13225. [PubMed] [Google Scholar]
- 21.Perreault CL, Gonzalez-Serratos H, Litwin SE, Sun X, Franzini-Armstrong C, Morgan JP. Alterations in contractility and intracellular Ca2+ transients in isolated bundles of skeletal muscle fibers from rats with chronic heart failure. Circ Res. 1993;73:405– 412. doi: 10.1161/01.res.73.2.405. [DOI] [PubMed] [Google Scholar]
- 22.Perry SV. Troponin T: genetics, properties and function. J Muscle Res Cell Motil. 1998;19:575– 602. doi: 10.1023/a:1005397501968. [DOI] [PubMed] [Google Scholar]
- 23.Perry SV. Troponin I: inhibitor or facilitator. Mol Cell Biochem. 1999;190:9 –32. [PubMed] [Google Scholar]
- 24.Reid MB. Invited Review: Redox modulation of skeletal muscle contraction: what we know and what we don’t. J Appl Physiol. 2001;90:724 –731. doi: 10.1152/jappl.2001.90.2.724. [DOI] [PubMed] [Google Scholar]
- 25.Reiser PJ, Portman MA, Ning XH, Schomisch MC. Human cardiac myosin heavy chain isoforms in fetal and failing adult atria and ventricles. Am J Physiol Heart Circ Physiol. 2001;280:H1814 –H1820. doi: 10.1152/ajpheart.2001.280.4.H1814. [DOI] [PubMed] [Google Scholar]
- 26.Szentesi P, Bekedam MA, van Beek-Harmsen BJ, van der Laarse WJ, Zaremba R, Boonstra A, Visser FC, Stienen GJ. Depression of force production and ATPase activity in different types of human skeletal muscle fibers from patients with chronic heart failure. J Appl Physiol. 2005;99:2189 –2195. doi: 10.1152/japplphysiol.00542.2005. [DOI] [PubMed] [Google Scholar]
- 27.Toth MJ, Ades PA, LeWinter MM, Tracy RP, Tchernof A. Skeletal muscle myofibrillar mRNA expression in heart failure: relationship to local and circulating hormones. J Appl Physiol. 2006;100:35– 41. doi: 10.1152/japplphysiol.00570.2005. [DOI] [PubMed] [Google Scholar]
- 28.Toth MJ, Ades PA, Tischler MD, Tracy RP, LeWinter MM. Immune activation is associated with reduced skeletal muscle mass and physical function in chronic heart failure. Int J Cardiol. 2006;109:179 –187. doi: 10.1016/j.ijcard.2005.06.006. [DOI] [PubMed] [Google Scholar]
- 29.Toth MJ, Gottlieb SS, Fisher ML, Poehlman ET. Skeletal muscle atrophy and peak oxygen consumption in heart failure. Am J Cardiol. 1997;79:1267–1269. doi: 10.1016/s0002-9149(97)00098-2. [DOI] [PubMed] [Google Scholar]
- 30.Toth MJ, Gottlieb SS, Goran MI, Fisher ML, Poehlman ET. Daily energy expenditure in free-living heart failure patients. Am J Physiol Endocrinol Metab. 1997;272:E469 –E475. doi: 10.1152/ajpendo.1997.272.3.E469. [DOI] [PubMed] [Google Scholar]
- 31.Toth MJ, Matthews DE, Ades PA, Tischler MD, Van Buren P, Previs M, LeWinter MM. Skeletal muscle myofibrillar protein metabolism in heart failure: relationship to immune activation and functional capacity. Am J Physiol Endocrinol Metab. 2005;288:E685–E692. doi: 10.1152/ajpendo.00444.2004. [DOI] [PubMed] [Google Scholar]
- 32.Toth MJ, Palmer BM, LeWinter MM. Effect of heart failure on skeletal muscle myofibrillar protein content, isoform expression and calcium sensitivity. Int J Cardiol. 2006;107:211–219. doi: 10.1016/j.ijcard.2005.03.024. [DOI] [PubMed] [Google Scholar]
- 33.Tsutsui H, Ide T, Hayashidani S, Suematsu N, Shiomi T, Wen J, Nakamura K, Ichikawa K, Utsumi H, Takeshita A. Enhanced generation of reactive oxygen species in the limb skeletal muscles from a murine infarct model of heart failure. Circulation. 2001;104:134 –136. doi: 10.1161/01.cir.104.2.134. [DOI] [PubMed] [Google Scholar]
- 34.van Der Velden J, Klein LJ, Zaremba R, Boontje NM, Huybregts MA, Stooker W, Eijsman L, de Jong JW, Visser CA, Visser FC, Stienen GJ. Effects of calcium, inorganic phosphate, and pH on isometric force in single skinned cardiomyocytes from donor and failing human hearts. Circulation. 2001;104:1140 –1146. doi: 10.1161/hc3501.095485. [DOI] [PubMed] [Google Scholar]
- 35.VanBuren P, Alix SL, Gorga JA, Begin KJ, LeWinter MM, Alpert NR. Cardiac troponin T isoforms demonstrate similar effects on mechanical performance in a regulated contractile system. Am J Physiol Heart Circ Physiol. 2002;282:H1665–H1671. doi: 10.1152/ajpheart.00938.2001. [DOI] [PubMed] [Google Scholar]
- 36.Vescovo G, Serafini F, Facchin L, Tenderini P, Carraro U, Dalla LL, Catani C, Ambrosio GB. Specific changes in skeletal muscle myosin heavy chain composition in cardiac failure: differences compared with disuse atrophy as assessed on microbiopsies by high resolution electrophoresis. Heart. 1996;76:337–343. doi: 10.1136/hrt.76.4.337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Wilson JR, Mancini DM. Factors contributing to the exercise limitation of heart failure. J Am Coll Cardiol. 1993;22:93–98. doi: 10.1016/0735-1097(93)90469-h. [DOI] [PubMed] [Google Scholar]
- 38.Wolff MR, Buck SH, Stoker SW, Greaser ML, Mentzer RM. Myofibrillar calcium sensitivity of isometric tension is increased in human dilated cardiomyopathies: role of altered beta-adrenergically mediated protein phosphorylation. J Clin Invest. 1996;98:167–176. doi: 10.1172/JCI118762. [DOI] [PMC free article] [PubMed] [Google Scholar]