

Abstract
There is growing recognition that psychological stress influences pain. Hormones that comprise the physiological response to stress (e.g. corticosterone; CORT) may interact with effectors of neuropathic pain. To test this hypothesis, mice received a spared nerve injury (SNI) after exposure to 60 min restraint stress. In stressed mice, allodynia was consistently increased. The mechanism(s) underlying the exacerbated pain response involves CORT acting via glucocorticoid receptors (GRs); RU486, a GR antagonist, prevented the stress-induced increase in allodynia whereas exogenous administration of CORT to non-stressed mice reproduced the allodynic response caused by stress. Since nerve injury-induced microglial activation has been implicated in the onset and propagation of neuropathic pain, we evaluated cellular and molecular indices of microglial activation in the context of stress. Activation of dorsal horn microglia was accelerated by stress; however, this effect was transient and was not associated with the onset or maintenance of a pro-inflammatory phenotype. Stress-enhanced allodynia was associated with increased dorsal horn extracellular signal-regulated kinase phosphorylation (pERK). ERK activation could indicate a stress-mediated increase in glutamatergic signaling, therefore mice were treated prior to SNI and stress with memantine, an N-methyl-D-aspartate receptor (NMDAR) antagonist. Memantine prevented stress-induced enhancement of allodynia after SNI. These data suggest that the hormonal responses elicited by stress exacerbate neuropathic pain through enhanced central sensitization. Moreover, drugs that inhibit glucocorticoids (GCs) and/or NMDAR signaling could ameliorate pain syndromes caused by stress.
Keywords: pain, glucocorticoids, inflammation, microglia, glutamate, ERK
Introduction
Individuals suffering from cancer, stroke, spinal cord injury and multiple sclerosis often become debilitated by neuropathic pain. Pain likely develops as a consequence of enhanced neuro-immune signaling and central sensitization in the spinal cord (Campbell and Meyer, 2006; Ji and Strichartz, 2004; Tsuda et al., 2005). Since psychosocial stress is often endured alongside these conditions (Gold et al., 2005; Scadding and Koltzenburg, 2006; Strang, 1998) and clinical observations suggest that stress increases susceptibility to develop pain and exacerbates existing pain (Ashkinazi and Vershinina, 1999; DeLeo, 2006; Greco et al., 2004; Nicholson and Martelli, 2004; Turner et al., 2002), it is important to understand how stress affects the development and severity of neuropathic pain.
Stress hormones, e.g., glucocorticoids (GCs), could enhance pain-like behaviors (Blackburn-Munro and Blackburn-Munro, 2003). Conceptually, this may seem paradoxical since synthetic GCs are used to treat inflammatory conditions that cause pain including peripheral nerve injury. However, GCs also play a central role in neuroplasticity (Joels and Krugers, 2007; Revest et al., 2005; Wang et al., 2005). Glucocorticoid receptor (GR) expression is increased in spinal cord dorsal horn neurons after nerve injury in rats (Wang et al., 2004; Wang et al., 2005) and mice (Takasaki et al., 2005). Adrenalectomy (Wang et al., 2004) or intrathecal delivery of a GR antagonist (RU486) or GR anti-sense oligonucleotides reduces neuropathic pain caused by nerve injury (Takasaki et al., 2005; Wang et al., 2004). Also, intrathecal delivery of dexamethasone, a synthetic GC, exacerbates pain-like behaviors (Wang et al., 2004). Collectively, these data suggest that exogenous GCs can influence the onset or maintenance of neuropathic pain; however, the impact of stress has not been evaluated in this context.
The purpose of the current study was to test the novel hypothesis that stress increases neuropathic pain. The results show that stress potentiates nerve injury-induced tactile allodynia via a mechanism involving GCs acting at GRs and glutamate receptor-mediated ERK activation in dorsal horn neurons (Daulhac et al., 2006; Ji et al., 1999; Karim et al., 2001; Kohno et al., 2008; Lever et al., 2003). Indeed, pERK was increased in the spinal cord dorsal horn in stressed/nerve-injured mice and pre-stress treatment with memantine, an NMDAR antagonist, prevented stress-enhanced allodynia. These novel data reveal that psychosocial stress, a natural consequence of most debilitating human diseases, can markedly exacerbate neuropathic pain and perhaps other pain-like syndromes (e.g., inflammatory pain) (Chover-Gonzalez et al., 2000; Khasar et al., 2008; Khasar et al., 2005; Rivat et al., 2007).
Methods
Animals
Adult female C57BL/6 mice (Taconic, Germantown, NY, USA) were group-housed in standard cages with ad libitum access to food and water. Mice were maintained in a vivarium with controlled temperature (∼20° C) on a 12 hr light/dark cycle and were randomly assigned to experimental groups after a two week habituation period. Behavioral testing was performed during the light cycle. Mice were 8-10 weeks of age at the time of surgery. All procedures were conducted in accordance with protocols approved by the Ohio State University Institutional Laboratory Animal Care and Use Committee and with the guidelines of the Committee for Research and Ethical Issues of IASP.
Restraint stress protocol
Mice assigned to experimental groups incorporating stress were placed individually into well-ventilated polypropylene tubes (2.8 cm internal diameter, 9.7 cm length) for 60 min. The restraint tube permits minimal, confined movement including postural adjustments. Mice not subjected to stress (No Stress groups) remained undisturbed in their home cages.
Drugs
Mifepristone (RU486; 50 mg/kg) and corticosterone (CORT; 1.5 mg/kg) were prepared in a sterile peanut oil vehicle (Veh). Memantine (MEM; 20 mg/kg) was prepared in sterile water vehicle. Drugs were administered in a 0.1 ml volume via intraperitoneal (i.p.) injection. All drugs were obtained from Sigma-Aldrich (St. Louis, MO, USA) and were prepared fresh daily. The concentration of CORT used in Experiment #2 was empirically defined to reproduce stress-induced plasma CORT concentrations in our lab and others (DeVries et al., 1995; Sugo et al., 2002). For in vitro experiments using CORT, two preparations were tested: CORT (0.1 μM) dissolved in 0.0005% DMSO and 0.005% EtOH and water-soluble CORT-2-hydroxymethyl-β-cyclodextrin (HBC) dissolved in media (both from Sigma). Lipopolysacchande (LPS; E. coli 055:B5; lot no. 067K4056) was obtained from Sigma. Mouse recombinant tumor necrosis factor-α (rTNF-α) was obtained from eBioscience (San Diego, CA, USA).
Peripheral nerve injury procedure
The spared nerve injury (SNI) procedure was performed as described previously by Shields et al. (Shields et al., 2003). Under isoflurane anesthesia (4% induction, 1.5% maintenance) in oxygen-enriched air and following shaving and aseptic preparation, an incision of the skin and biceps femoris muscle was introduced to expose the sciatic nerve and its three terminal branches at the upper-thigh level. Two of the branches, the sural and common peroneal nerves, were tightly ligated with 7-0 silk suture (Genzyme Biosurgety, Fall River, MA) and transected distal to the ligature. Subsequently, 1-2 mm of each nerve was resected, approximately 1 mm distal to the ligature. The tibial nerve was not disturbed. The muscle/fascia layer and skin layer were closed separately with 5-0 nylon suture (Syneture, Norwalk, CT).
Behavioral Testing
Punctate mechanical sensitivity was analyzed by measuring threshold response to von Frey filaments (Stoelting Co., Wood Dale, IL). Monofilament stimulation was applied to the midline of the plantar surface of each hind paw using the up-down method for threshold sensitivity (Chaplan et al., 1994). The stimulus intensity threshold represents the smallest force that elicited repeated withdrawal of the hind paw during 10 trials (≥ 50% response sensitivity to smallest force). Threshold values represent von Frey hair handle markings that correspond to log10 of (10× force in mg), as displayed in Figs 1&6 (Chacur et al., 2001).
Figure 1.
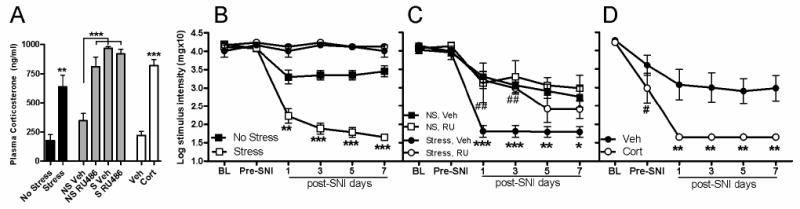
Stress and corticosterone exacerbate nerve-injury induced allodynia. A) Acute stress (60 min restraint) increases plasma CORT (vs. No Stress (NS)). By binding GR, RU486 treatment increases plasma CORT (vs. Vehicle (Veh)). CORT treatment also increases plasma CORT (vs. Veh). B) Acute stress prior to SNI decreases the pain threshold (von Frey hair measurements) in the ipsilateral hind paw on post-SNI days 1, 3, 5, & 7 compared to the NS group. Stress has no effect on sham-operated mice (vs. NS; black circle = Sham NS; white circle = Sham Stress). C) When RU486 (RU) was used to block GRs, stress-induced potentiation of mechanical allodynia is reduced on post-SNI days 1 & 3 compared to Veh, as indicated by “##.” Asterisks (*) indicate the difference between Veh + NS and Veh + Stress allodynia on post-SNI days 1, 3, 5, & 7. D) Treatment with CORT prior to SNI decreases the pain threshold in the ipsilateral hind paw on post-SNI days 1, 3, 5, & 7 compared to Veh. Compared to Baseline (BL), CORT reduces pre-SNI thresholds (#p<0.0001). Results are expressed as the mean ± SEM; B,C,D; n=5-6/group SNI, n=2/group Sham; y-axis values represent von Frey hair handle markings. *p<0.05; ** or ##p<0.01;***p<0.0001.
Figure 6.
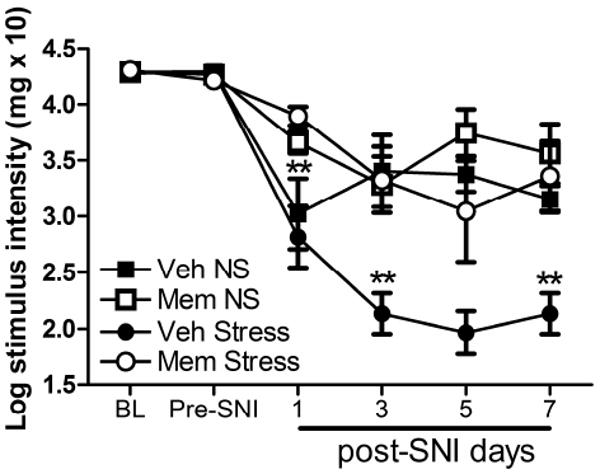
Stress-induced potentiation of allodynia is NMDA receptor-dependent. Pre-stress treatment with memantine (MEM) blocks the effects of stress on allodynia (post-SNI days 1, 3, 5, and 7) after nerve injury (**p<0.01 vs. Veh + Stress). Results are expressed as mean ± SEM; n=6-7/group; y-axis values represent von Frey hair handle markings.
Behavioral assessments were made without stress prior to SNI (Baseline; BL), 1 d after stress but prior to SNI (Pre-SNI) and on days 1, 3, 5, and 7 d post- SNI (Post-SNI 1, 3, 5, 7; Table 1). Table 1 summarizes the pharmacological and behavioral paradigm utilized in these studies. The paradigm incorporates two stress sessions; however, a single session immediately prior to SNI is sufficient to enhance allodynia (data not shown). Mice were acclimated to the testing procedure for 20 min each day for two days prior to the start of the experiment and for 10 min prior to the onset of each session. All behavioral measurements were conducted by an investigator that was unaware of the treatment groups.
Table 1.
| Baseline (BL; day -2) | Pre-SNI (day -1) | SNI (day 0) | Post-SNI (days 1, 3, 5, 7) |
|---|---|---|---|
| 1. von Frey test 2. Treatment (≥ 30 min after BL von Frey test) |
von Frey test (to verify no effect of Treatment w/o injury) |
1. Treatment 2. SNI |
von Frey test |
| Experiment | Treatment | ||
| 1 & 4 | No Stress or Stress (60 min, immediately prior to SNI) | ||
| 2 | Veh or RU486 (60 min prior to No Stress or Stress; Stress as above) | ||
| 3 | Veh or CORT (60 min prior to SNI) | ||
| 5 | Veh or MEM (60 min prior to No Stress or Stress; Stress as above) | ||
Determination of plasma corticosterone concentration
At baseline (day -2 of the experiment), a blood sample was obtained from the periorbital sinus following restraint or injection of CORT. A sample also was collected from No Stress mice at baseline. On day 8 post-SNI, prior to transcardial perfusion, a blood sample was obtained from the periorbital sinus. All blood samples were collected within a 2 h time period beginning 3-5 h after the onset of the light cycle. After centrifugation, plasma was collected and stored at -70°C. Levels of plasma CORT were measured in duplicate samples using an 125I radioimmunoassay kit (ICN Pharmaceuticals, Costa Mesa, CA).
Histological analyses
Mice were anesthetized with a ketamine/xylazine (80 mg kg−1/10 mg kg−1; i.p.) cocktail prior to transcardial perfusion with 30 ml 0.1 M phosphate-buffered saline (PBS, pH 7.4) followed by 100 ml of 4% paraformaldehyde (sample size for post-SNI tissue collection: n=4 for 1 d; n=5-6 mice/group for 8 d). After perfusion, spinal cords were rapidly removed then post-fixed for 2 h, followed by a rinse and overnight immersion in 0.2 phosphate buffer (PB) solution. Tissues were cryoprotected in 30% sucrose in 0.2 M PBS for 48 h. Spinal cords were blocked in 8 mm segments centered on the L-5 level, embedded in optimal cutting temperature compound (OCT; Tissue-Tek, VWR International, West Chester, PA) and frozen at -80°C. Transverse serial sections (10 μm) were cut using a cryostat and thaw-mounted on SuperFrost Plus slides (Fisher Scientific, Houston, TX), then stored at -20°C until use. After drying at room temperature, slides were rinsed with 0.1 M phosphate buffer solution (PBS) and overlaid with blocking solution for 1 h. To identify the central projections of injured axons, slides were processed for isolectin-B4 staining (IB4, Griffonia simplicifolia; Sigma; (Beggs and Salter, 2007)). After a brief 6% H2O2–MeOH incubation, slides were rinsed in 0.1 M PBS and incubated in Dulbecco's PBS plus 0.1% Triton X-100 (Sigma) for 30 mm, followed by biotinylated-IB4 (5 μg/ml) for 2 h at room temperature. After rinsing, slides were incubated in streptavidin-conjugated AlexaFluor 488. To label microglia, slides were incubated with anti-rabbit Iba-1 (Ito et al., 1998) (1:750; Wako, Richmond, VA) overnight at 4°C. Slides were then rinsed in 0.1 M PBS and incubated with either biotinylated goat anti-rabbit antibody (Vector, Burlingame, CA) or AlexaFluor 546-conjugated antibody in blocking solution. For immunoperoxidase labeling, slides were rinsed in 0.1 M PBS and incubated in 3% H2O2 to quench endogenous tissue peroxidase. Bound antibody was visualized using the Elite-ABC reagent (Vector) followed by DAB substrate (Vector). To label activated ERK, slides were processed for antigen retrieval according to the manufacturer's instructions (Dako Target Retrieval Solution, Carpinteria, CA). Slides were incubated with anti-rabbit pERK (1:500; Cell Signaling Technology, Beverly, MA) overnight at 4° C. Sections were subsequently washed and incubated for 1-2 h with goat anti-rabbit AlexaFluor 546-conjugated antibody. While only 3-4 mm of spinal cord is necessary for analyses of L4-L6 segments, an expanse of 8 mm was collected initially to visualize any potential spreading of microglial reactivity beyond the affected segments. Loss of IB4 staining (Beggs & Salter, 2007; Shields et al., 2003) with a concurrent increase in morphological indices of microglial activation were used to identify spinal cord sections affected by injury. Analyses were conducted within this range of tissue sections. For analysis of labeling, the area containing the spinal cord dorsal horn was digitized under high-power using a Zeiss Axioplan 2 imaging microscope equipped with a digital camera (Zeiss Axiocam). Using an MCID Elite 6.0 image analysis system (Imaging Research, St. Catherines, Ontario, Canada), digitized images were quantified by outlining the superficial dorsal horn then quantifying areas of positive immunoreactivity (IR) relative to the total sample area, (expressed as proportional area (PA))(Kigerl et al., 2006). Iba-1 and p-ERK labeling were evaluated across the rostro-caudal extent of the 8 mm segment (as described above); however, quantitative analyses were restricted to a series of five sequential sections (∼200 μm apart) centered on the area clearly affected by injury as defined above. All data were obtained by an investigator that was unaware of the treatment groups.
Laser capture microdissection
Mice were anesthetized as above and intracardially perfused at 3 days post-SNI with DEPC-treated 0.1 M PBS (n=5/group; No Stress and Stress). The lumbar enlargement of the spinal cord containing L4-L6 was blocked, embedded in OCT then flash frozen in N-methylbutane cooled by dry ice. Transverse serial sections (10 μm) were cut using a cryostat, then mounted on RNase free P.A.L.M. membrane slides (Zeiss, Thornwood, NY, USA) and stored at -80° C. The laser capture microdissection (LCM) procedure used here has been described previously by our lab (Kigerl et al., 2007; Longbrake et al., 2007). Briefly, sections were quick-stained with hematoxylin (Vector) and the dorsal horn region containing activated microglia (see Fig. 2) was circumscribed using the P.A.L.M. Microbeam laser capture system fit with a 5× cutting objective. Ipsilateral and contralateral dorsal horn tissue (∼6 mm2) was collected separately in RNase-free tubes containing 20 μL of lysis buffer (RNaqueous Micro kit, Ambion, Austin, TX, USA). RNA was isolated using the RNaqueous Micro kit (Ambion) using the manufacturer's protocol optimized for LCM samples. Samples were DNase treated (RNaqueous kit, Ambion) and cDNA was prepared by reverse transcription with SuperScript II and random primers (Invitrogen, Carlsbad, CA, USA).
Figure 2.
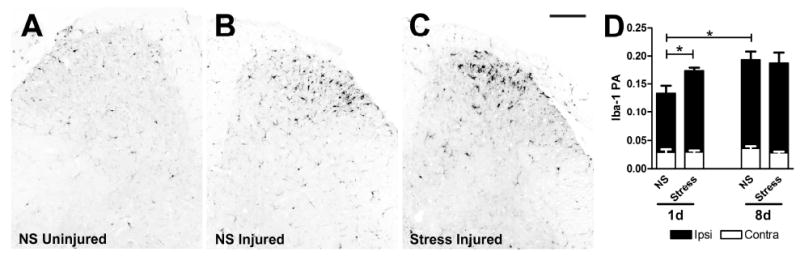
Nerve-injury induced microglial reactivity is transiently increased by stress. Dorsal horn Iba-1 immunoreactivity (IR) is increased ipsilateral to the nerve injury (B) compared to contralateral (uninjured) side (A) in all mice (representative No Stress (NS) group shown in A&B; *p<0.0001). C) Acute stress increases ipsilateral Iba-1 IR on post-SNI day 1 compared to NS. By 8 days, Iba-1 IR is not different between Stress and NS groups. D) Quantitative analysis of Iba-1 IR as illustrated in A-C (RA=proportional area; see Methods for details of quantitative analysis; n=4-6/group). Black bars = ipsilateral dorsal horn; White bars = contralateral dorsal horn. Results are expressed as mean ± SEM. Scale bar in C = 100μm.
Quantitative RT-PCR (qRT-PCR)
To determine stress-induced effects on microglial activation and subsequent indices of neuroinflammation, select cytokine and microglial markers were analyzed from mRNA isolated from LCM samples. Gene-specific primer pairs for IL-1β, IL-6, TNF-α, and TLR4 were used as described previously (Kigerl et al., 2007). The primer pair for amplification of CDllb was forward 5′-GGATCATAGGCGCCCACTT-3′ and reverse 5′-TCCTTACCCCCACTCAGAGACT-3′; for IL-1α, forward 5′-CAGGATGTGGACAAACAC-3′ and reverse 5′-GCTCACGAACAGTTGTGAATCTG-3′. Gene expression was determined using qRT-PCR and compared between No Stress and Stress mice (n=4-5/group). Primer sequence specificity was confirmed by performing BLAST analysis for highly similar sequences against known sequence databases. PCR reactions were carried out in triplicate using 1 μL cDNA/reaction and SYBR Green master mix (Applied Biosystems, Foster City, CA, USA) in 20 μL reactions. PCR product was measured using SYBR Green fluorescence collected on an Applied Biosystems 7300 system (Ririe et al., 1997). Standard curves were generated for each gene using a control cDNA dilution series. Melting point analyses were performed for each reaction to confirm single amplified products. ΔΔCt analysis was used to normalize gene data to 18s ribosomal RNA expression, then each ipsilateral value was expressed as fold change from its own contralateral value. This analysis was used for between-group comparisons (No Stress vs. Stress ipsilateral gene expression). Within-group comparisons reflect ipsilateral vs. contralateral (injury-induced) gene expression.
In vitro corticosterone experiments
The immortalized BV-2 microglia cell line was used to examine the priming effect of CORT on LPS or TNF-α –induced IL-1β gene expression. Cells were treated with CORT (0.1 μM or Veh) for 60 mm or 24 h, then replaced with LPS (10 or 100 ng/ml, or Veh) or TNF- α (10 ng/ml, or Veh) for 60 mm, 6 h or 24 h at 37° C in a 5% C02 humidified incubator. The Veh control for LPS and TNF-α was cell culture media (10% fetal bovine serum (Hyclone), 5% penicillin/streptamycin and 5% glutamax in DMEM (Gibco/Invitrogen). Independent studies have shown that GCs are not purely immunosuppressive; they can enhance certain immune functions, especially when present in low concentrations (Drew and Chavis, 2000; Lim et al., 2007; MacPherson et al., 2005; Viswanathan and Dhabhar, 2005; Zhu et al., 2007). Therefore, we compared varying CORT concentrations over a range from 0.1 μM to 1 mM (at 10× intervals) in an effort to identify an optimal priming dose. Since our goal was to prime rather than suppress microglial function, we used the lowest dose that did not suppress LPS-induced IL-1β gene expression. RNA was isolated using the Trizol method (Invitrogen) and IL-lβ transcript was measured using qRT-PCR as described above. A minimum of three independent experiments was conducted for each in vitro manipulation.
To determine the capacity for CORT to prime intracellular signaling, thereby providing an explanation for stress-mediated increases in allodynia, we evaluated NF-κB signaling using 3T3 fibroblasts either transiently transfected or stably expressing an NF-κB responsive luciferase reporter (generously provided by Dr. Denis Guttridge). 3T3 stable cell lines were generated by seeding at a density of 2×105 cells in 6 cm dishes 24 h prior to transfection. Cotransfection of seeded fibroblasts with 4 μg of reporter plasmid 3×κB-Luc and 1 μg of pcDNA3 (Invitrogen) was performed using Superfect reagent as recommended by the manufacturer (Qiagen). At 48 h post-transfection, the cells were trypsinized and cultured at 1/30 their density followed by selection in 1 mg/ml of Geneticin (G418; Life Technologies). Cells were allowed to expand under G418 selection and individual clones were selected as described by Guttridge et al. (Guttridge et al., 1999). Cells were treated with CORT (0.1 μM or Veh) for 60 mm, then replaced with TNF-α (10 ng/ml, or Veh) for 60 min at 37°C in a 5% CO2 humidified incubator. Transcriptional activity was measured in three independent experiments according to previously described methods (Guttridge et al., 1999).
Data analysis
Behavioral data for Experiments 1, 3, & 4 were analyzed using one-way repeated measures analysis of variance (ANOVA). Behavioral data for Experiments 2 & 5 were analyzed using two-way (drug × stress) repeated measures ANOVA. Post-hoc analyses were conducted using Bonferroni's or Tukey-Kramer tests (for unequal sample sizes). Factorial ANOVA was used for histological analyses. Student's t test was used to analyze behavioral data for individual time points and between-group gene expression analyses. Within-group gene expression analyses were conducted using paired t-test. For statistical analyses and graphing, we used Statview for Windows 5.0.1 (SAS Institute, Cary, NC) and GraphPad Prism 5.00 for Windows, respectively (GraphPad Software, San Diego California USA). An α-level of p < 0.05 was used as an indication of statistical significance.
Results
Acute stress increases allodynia induced by nerve injury
To confirm that restraint stress elicited a physiologically appropriate response, we measured plasma CORT immediately after restraint. Circulating CORT was increased by ∼400% compared to the No Stress group (F(l, 10) = 16.97, p < 0.01; 637.5 ± 99.8 ng/ml vs. 176.6 ± 50.6 ng/ml; n=6/group; Fig. 1A). Consistent with previous observations (Bomholt et al., 2005; Ulrich-Lai et al., 2006), CORT measurements 8 days post-SNI revealed no stress- or injury-induced effects (p > 0.05; data not shown). In the absence of nerve injury, stress had no effect on paw withdrawal threshold (p > 0.05 vs. No Stress at Pre-SNI; Fig. 1B). Likewise, after sham surgery, stress had no effect on paw withdrawal threshold (p > 0.05 vs. No Stress; n=2/group; Fig. 1B). As expected, allodynia was observed following SNI as described previously (F(l, 10) = 103.51, p < 0.0001 vs. Baseline (or vs. Sham: (F(l, 14) = 12.68, p < 0.01) (Shields et al., 2003). This is a decrease in gram force threshold of ∼80% (0.3g vs. 1.5g) from Baseline. This pain-like response was consistently enhanced by stress (F(1, 10)=87.20, p < 0.0001 vs. No Stress; n=6/group; Fig. 1B).
RU486, a glucocorticoid receptor antagonist, mitigates stress-induced enhancement of allodynia after SNI
As in Experiment 1, allodynia was observed in all mice after SNI (F (3, 16)=94.31, p < 0.0001 vs. Baseline) and stress increased allodynia (F(3, 16) = 14.41, p < 0.0001 vs. No Stress + Veh). To determine if stress-mediated increases in circulating CORT contribute to enhanced allodynia, RU486 was injected prior to subjecting mice to stress and SNI. RU486 markedly suppressed allodynia in stressed mice (p < 0.001 vs. Veh; n=5/group; Fig. 1C) but had no effect on allodynia caused by nerve injury alone (p > 0.05 vs. Veh). At Baseline and Pre-SNI time points, there were no differences between groups (p > 0.05).
Corticosterone increases nerve injury-induced allodynia
We next tested whether exogenous CORT mimicked the effects of restraint stress. At Baseline or Pre-SNI time points, paw withdrawal thresholds were not different between CORT or Veh-treated mice (p > 0.05). However, 24 hours after administering exogenous CORT, paw withdrawal thresholds were reduced relative to Baseline values (p < 0.0001; baseline vs. pre-SNI, Figure 1D). Vehicle treatment had no significant effect on withdrawal threshold during pre-SNI testing (p > 0.05 vs. Baseline). After SNI, allodynia developed in all mice (F (1, 8)=30.75, p < 0.0001 vs. Baseline) but was exacerbated in mice treated with CORT prior to SNI (F (1, 8) = 16.58, p < 0.01 vs. Veh; n=5/group; Fig. 1D).
Activation of microglia in spinal cord dorsal horn is transiently increased by stress
In response to most types of peripheral nerve injury, spinal cord microglia change shape and increase expression of different cell surface antigens. These changes often predict the induction of pro-inflammatory functions in microglia that contribute to the onset and maintenance of pain-like behaviors (Narita et al., 2006; Raghavendra et al., 2003; Tsuda et al., 2005). To determine whether this model of SNI consistently activates microglia and whether this response is exacerbated by stress, we first measured dorsal horn immunoreactivity for the microglial marker, Iba-1. In each experiment, SNI induced morphological indices of microglial reactivity ipsilateral to the nerve injury (t (8) = 10.81, p < 0.0001 vs. contralateral dorsal horn; Fig. 2A-D). Microglial activation was evident by 1 day post-lesion and increased further by 8 days (Fig. 2D). In stressed mice, microglial reactivity was increased at one day post-SNI (t (6) =2.89, p < 0.05, vs. No Stress; n=4/group; Fig. 2D); however, by 8 days, the effect of stress had dissipated (n=5-6/group; Fig. 2D).
Next, to better understand the potential functional implications of the enhanced morphological activation of microglia, we used laser capture microdissection to isolate injured and uninjured (contralateral) spinal cord dorsal horn regions populated by activated and resting microglia, respectively, at 3 days post-SNI. From these samples we measured changes in expression of CD11b, TLR4, IL-1α, IL-1β, TNF-α, and IL-6 mRNA. Consistent with the data from Fig. 2, we found a small but non-significant induction of CD11b (a microglia-specific marker) mRNA after SNI (Fig. 3A) that was enhanced by stress (t (4)=3.63, p < 0.05 vs. contralateral; n=5/group; data not shown). Expression of CD11b and TLR4 transcript in injured dorsal horn tissue was increased by stress (t (7)=2.67, p < 0.05 and t(6)=4.03, p < 0.01, respectively, vs. No Stress ipsilateral; n=4-5/group; Fig. 3A, B). However, expression of downstream cytokine transcripts, IL-1β, IL-6, TNF-α, and IL-1α was unaffected by SNI or stress (p > 0.05 for all comparisons; Fig. 3C-F).
Figure 3.
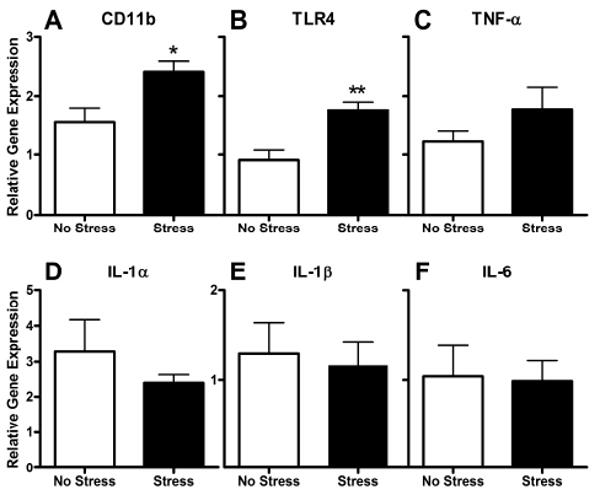
Dorsal horn pro-inflammatory gene expression is not altered by stress 3 days post-SNI. A, B) Stress increases CD11b and TLR4 gene expression, indicative of enhanced microglial activation (also see Fig. 2). C-F) In contrast, the expression of pro-inflammatory genes known to be associated with neuropathic pain is not increased by SNI or Stress. Data are normalized to the internal control gene (18s), and expressed relative to their contralateral value. Results are expressed as mean ± SEM; n=4-5/group. *p<0.05; **p<0.01.
Corticosterone neither enhances nor suppresses inflammatory responses in vitro
Elevated transcript for CD11b and TLR4 (Fig. 3) suggests that microglia are primed by stress. The absence of stress-induced cytokines, however, may indicate that any priming effect of stress-derived glucocorticoids (GCs) on microglial function preceded the timing of our analyses (3 days post-SNI). Thus, two in vitro assays were used to test the hypothesis that stress-induced GCs could prime intracellular signaling in microglia. Indeed, acute stress prior to surgery has been shown to enhance inflammation, and stress-induced CORT may alert immune cells to impending injury (Dhabhar, 2003; Viswanathan and Dhabhar, 2005).
First, to determine if GCs can enhance the function of microglia (e.g., in response to a stimulus released in dorsal horn after SNI), we examined the effect of CORT pre-treatment on BV-2 cells stimulated with LPS or TNF-α. LPS is the prototypical ligand for toll-like receptor 4 (TLR4), the activation of which contributes to cytokine production and pain after nerve injury (Tanga et al., 2005). TNF-α is produced after injury and also contributes to neuropathic pain (DeLeo et al., 1997; Hao et al., 2007; Schafers et al., 2003a; Schafers et al., 2003b; Zanella et al., 2008). Further rationale for the use of LPS and TNF-α derives from the observed stress-induced increase in TLR4 (LCM data; Fig. 3), that, when activated, drives NF-κB activation and subsequent TNF-α production. When CORT was applied to cells for 1 or 24 hours (based on the in vivo stress and CORT pre-treatment paradigm and time course of development of allodynia, respectively; see Fig. 1) then replaced with LPS or TNF-α for 1, 6 or 24 hours, IL-1β gene expression was generally unaffected (p > 0.05 vs. Veh pre-treatment). In one out of three experiments, CORT pre-treatment increased IL-1β mRNA; however, the reason for the inconsistency could not be determined and no contamination was evident. Because ethanol, a component of the vehicle in which CORT was prepared, suppresses TLR4 function (Dai and Pruett, 2006), we next tested water-soluble CORT. Again, CORT pre-treatment had no effect on LPS- or TNF-α-induced IL-1β gene expression (p > 0.05 vs. Veh+LPS/TNF-α; Fig. 4A). LPS-induced nitric oxide production was also unaffected by CORT pre-treatment (data not shown).
Figure 4.
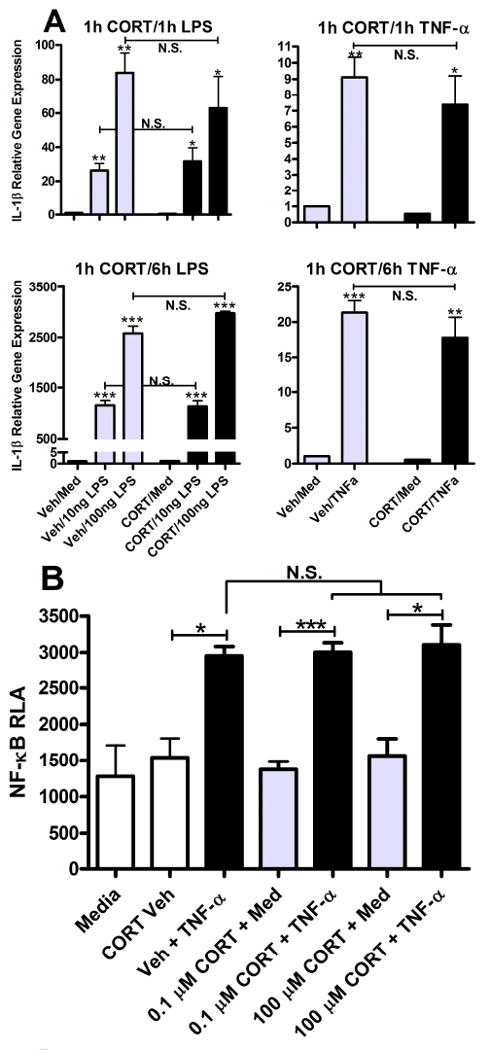
Glucocorticoids do not prime pro-inflammatory gene expression or NF-κB activity. A) LPS (10 and 100 ng/ml) and TNF-α (10 ng/ml) for 1 or 6 h increase IL-lβ transcript (vs. media) in BV-2 cells. Pre-treatment with CORT (0.1 μM) for 1 h has no effect on LPS- or TNF-α-induced IL-1β gene expression. B) TNF-α (10 ng/ml) for 1 h increases NF-κB activity (vs. media & CORT vehicle). Pre-treatment with CORT (0.1 μM) for 1 h has no effect on TNF-induced NF-κB activity. Relative luciferase activity (RLA) was determined by normalizing to β-gal. Results are expressed as mean ± SEM. *p<0.05; ***p<0.0001.
To broaden the analysis further, we analyzed CORT effects on priming of NF-κB transcription. NF-κB operates upstream of numerous pro-inflammatory mediators that are known to cause or be associated with pain (Meunier et al., 2007; Tegeder et al., 2004). NIH3T3 cells were either stably or transiently transfected with an NF-κB responsive luciferase reporter, 3×κB. Cells were primed with CORT for 1 hour then were stimulated with TNF-α (10 ng/ml) for 1 hour. Transcriptional activity was measured by luciferase assay as previously described (Guttridge et al., 1999). Although TNF-α markedly enhanced NF-κB activity (p < 0.05 vs. Med/CORT Veh), CORT pre-treatment did not increase (or decrease) this response (p > 0.05 vs. Veh + TNF-α; Fig. 4B).
Stress increases phosphorylation of ERK in the dorsal horn after SNI
Because stress-enhanced allodynia was not accompanied by a persistent pro-inflammatory microglial response, we next questioned whether the effects of stress were a result of enhanced central sensitization. Indeed, acute stress and GCs can sensitize neurons to injury-induced activation in part by increasing glutamatergic signaling via NMDA receptors (Ahmed et al., 2006; Kim et al., 1996; Norris and Strickland, 2007; Wang et al., 2005; Yang et al., 2008). Activation of glutamate receptors has been implicated in stimulation-evoked ERK activation in spinal cord dorsal horn neurons (Daulhac et al., 2006; Ji et al., 1999; Karim et al., 2001; Kohno et al., 2008; Lever et al., 2003), therefore we determined the effect of stress on dorsal horn pERK immunoreactivity. As shown in Fig. 5, stress increased pERK labeling in the superficial laminae one day after SNI (t (38) =4.58, p < 0.05 vs. No Stress; n=4/group). Consistent with previous work (Ji et al., 1999; Song et al., 2005; Zhuang et al., 2005), pERK-positive cells exhibited neuronal morphology. We did not test the effects of stress alone (with sham injury) on pERK induction.
Figure 5.
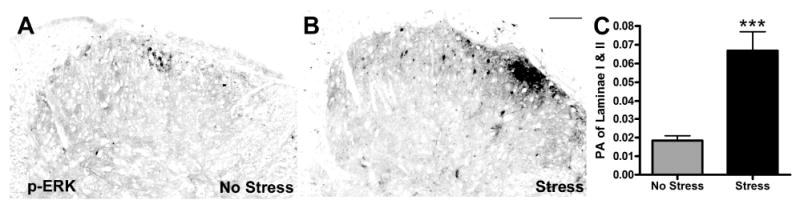
Stress increases nerve injury-induced pERK. A&B) pERK labeling in superficial dorsal horn at 1 day post-SNI is increased by stress. C) Quantitative analysis of pERK labeling confirms stress-induced increases in pERK (p < 0.0001 vs. No Stress (NS); PA=proportional area). Results are expressed as mean ± SEM; n=4/group. Scale bar in B = 100 μm.
Memantine blocks stress-enhanced allodynia after SNI
To determine whether the stress-mediated increase in pERK predicts or reflects enhanced signaling via NMDA receptors and thus “central sensitization”, we pre-treated mice with memantine (MEM), a moderate affinity NMDA receptor antagonist. MEM pre-treatment attenuated the development of allodynia in both Stess and No Stress mice; however, this effect was only evident at 1 day post-SNI (p < 0.01 vs. Veh). In contrast, MEM prevented the enhanced pain-like behavior caused by stress for the duration of the testing (F (3, I5)=9.6l, p < 0.001 vs. Veh+Stress; n=6-7/group; Fig. 6).
Discussion
The present data show that stress exacerbates pain-like behavior caused by peripheral nerve injury. Two lines of evidence reveal a causal role for glucocorticoids (GCs) in this response. First, exogenous CORT reproduced the effects of stress. Second, pharmacological inhibition of glucocorticoid receptors (GRs) with RU486 blunted the potentiating effects of stress on nerve injury-induced allodynia. Recently, it was shown that nerve injury increases spinal GRs (Takasaki et al., 2005; 2006; Wang et al., 2004; Wang et al., 2005). Thus, the potential for stress-induced GCs to exert effects on spinal cord neurons or glia is enhanced by nerve injury. Despite the novelty of these data, the precise cellular and molecular intermediates affected by circulating GCs that results in exacerbated neuropathic pain are unknown. We tested two distinct mechanisms of neuropathic pain as substrates for CORT effects: microglial reactivity and neuronal sensitivity.
Acute stress activates inflammatory processes (Burns et al., 2008; Steptoe et al., 2007; Viswanathan and Dhabhar, 2005) and neuroinflammation has been implicated in the induction of neuropathic pain in models of central and peripheral nervous system injury. Therefore we predicted that resident CNS “immune cells”, i.e., microglia (Gehrmann et al., 1995; Streit et al., 1988), would exist in a hyperactivated state in stressed mice. Although we observed that stress accelerated morphological (Iba-1 labeling) and phenotypic (CD11b and TLR4 mRNA) indices of microglial activation elicited by SNI, and that these changes temporally corresponded with the onset of an exaggerated allodynia (∼24-72 hours post-SNI), these changes were not maintained. Moreover, we found no evidence that stress or GCs could enhance pro-inflammatory signaling in vivo or in vitro. It is possible that had we used primary microglia instead of BV-2 cells, GC-mediated priming would have become evident. Indeed, Horvath et al. (2008) have shown that compared to BV-2 cells, primary microglia are more responsive to pro-inflammatory signaling. Still, we found little evidence of enhanced microglial activation or cytokine signaling in vivo (see above and Fig. 3). Collectively, these data were surprising given that there is a large body of evidence linking microglia and pro-inflammatory cytokines (mRNA and protein) to the induction of neuropathic pain (DeLeo and Yezierski, 2001; Detloff et al., 2008; Milligan and Watkins, 2009; Narita et al., 2006; Tsuda et al., 2005). However, independent studies have shown that acute stress, akin to that used in this report, can induce rapid (within 6 hours) and transient morphological changes in microglia without evoking changes in pro-inflammatory cytokines (Sugama et al., 2007).
Although stress accelerated the microglial response to SNI without an apparent increase in expression of pro-inflammatory cytokine mRNA or NFκB transcription, we still cannot entirely dismiss a role for activated microglia in contributing to the enhanced pain-like state experienced by stressed mice. Indeed, changes in mRNA do not always faithfully predict changes in synthesis or release of that protein. Also, microglia release other mediators, including brain derived neurotrophic factor (BDNF), that we did not measure but that can enhance neuronal excitability. In response to nerve injury, activation of purinergic receptors (e.g., P2X4) on microglia elicits BDNF release, which in turn increases neuronal excitability (Coull et al., 2005; Tsuda et al., 2003; Ulmann et al., 2008). Interestingly, enhancement of BDNF synthesis is observed following acute stress (Smith et al., 1995; Yang et al., 2008). To our knowledge, the regulation of BDNF production after stress or in response to exogenous GCs has not been established in the spinal cord after nerve injury. Thus, it is feasible that GCs facilitate microglia-neuron communication via BDNF. This could facilitate or act in parallel with stress and GC-mediated activation of mitogen activated protein kinases (MAPKs) (Meller et al., 2003; Norris and Strickland, 2007; Revest et al., 2005; Sananbenesi et al., 2003) and NMDA receptor activation in neurons (Cho and Little, 1999; Jing et al., 2008; Qi et al., 2005; Wang et al., 2005; Wong et al., 2007; Yang et al., 2008). Other glial cells, particularly astrocytes, have well-documented roles in neuropathic pain (Kawasaki et al., 2008; Tanga et al., 2004; Zhuang et al., 2006) and are responsive to corticosterone exposure (Melcangi et al., 1997; Ovadia et al., 1984; Vielkind et al., 1990), thus could play a role in stress-induced pain enhancement (LaCroix-Fralish and DeLeo, 2007). Given the potential for these different stress-inducible pathways to “sensitize” or activate glia and/or neurons, it is intriguing that stress alone failed to elicit allodynia (Fig. 1B). In contrast, exogenous Cort was able to cause modest tactile allodynia in the absence of nerve injury; however, this “pain-like” behavior was variable and modest compared to that caused by nerve injury alone. Only when GCs were combined with nerve injury did we observe a consistent and sustained allodynia.
Most of the data in this report suggest that the combination of stress and nerve injury triggers a complex interplay between GCs, GRs and enhanced glutamate transmission. Specifically, the stress-induced increase in activation of extracellular signal-regulated protein kinase (pERK) in spinal cord dorsal horn and the ability of memantine, an open-channel NMDA receptor antagonist, to block stress-enhanced allodynia (see Figs. 5&6), suggest that these pathways converge to cause central sensitization. Published data support this notion. Neuronal and glial glutamate transporters in the spinal cord are downregulated and glutamate uptake is reduced after nerve injury (Binns et al., 2005; Hughes et al., 2004; Sung et al., 2003). Importantly, EAAC1 downregulation is regulated by GRs; RU486 or GR antisense oligonucleotide treatment restores EAAC1 expression after nerve injury (Wang et al., 2006). These data support the possibility that stress-induced activation of GR results in decreased glutamate uptake and subsequent NMDA receptor activation. Indeed, stress or corticosterone has been shown to decrease glutamate transporter expression (Jacobsson et al., 2006; Madrigal et al., 2003) and increase extracellular glutamate (César Venero, 1999; Lowy et al., 1993; Moghaddam et al., 1994; Reznikov et al., 2007). GCs also augment nerve injury-induced expression of neuronal NMDA receptors, a phenomenon reversible by RU486 (Wang et al., 2005). Activation of NMDA and metabotropic glutamate receptors are implicated in ERK activation in dorsal horn spinal cord neurons and the subsequent induction of pain (Daulhac et al., 2006; Ji et al., 1999; Karim et al., 2001; Kawasaki et al., 2004; Kohno et al., 2008; Lever et al., 2003; Xu et al., 2008). Thus, suppression of neuronal and glial glutamate transporters by stress and GCs could impair removal of glutamate from the extracellular space and prime glutamatergic transmission through MAPK activation and upregulation of ionotropic and/or metabotropic glutamate receptors.
Clearly, more work is needed before we fully understand the mechanisms underlying the exaggerated pain-like behavior that we observed in stressed mice. However, this study has important clinical implications as the data show that stress may predict the induction and pathogenesis of chronic pain (Diatchenko et al., 2006). The significance of psychosocial effects on pain is emphasized by the delayed onset of neuropathic pain in various diseases and neurological disorders including cancer, post-herpetic neuralgia, amputation, HIV, ischemia and spinal cord injury. This temporal gap between insult and pain onset provides an opportunity for clinical intervention and underscores the need for greater understanding of the mechanisms that control communication between stress hormones, neurons and CNS glia.
Acknowledgments
The authors thank Dr. Ming Wang, Viy McGaughy, and Wenmin Lai for their expert technical assistance, and Dr. Denis Guttridge for supplying NF-κB reporter 3T3 cells. We are grateful to Dr. Fiona Holmes for helping us develop the SNI model in our lab, and to Dr. John Gensel for assistance in statistical analyses. Funding was provided by NIH NS37846 (PGP) and 1R01NR010806 (ACD).
Footnotes
The authors declare no conflict of interest pertaining to the data described herein.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Ahmed T, Frey JU, Korz V. Long-term effects of brief acute stress on cellular signaling and hippocampal LTP. J Neurosci. 2006;26:3951–8. doi: 10.1523/JNEUROSCI.4901-05.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ashkinazi I, Vershinina EA. Pain sensitivity in chronic psychoemotional stress in humans. Neurosci Behav Physiol. 1999;29:333–7. doi: 10.1007/BF02465346. [DOI] [PubMed] [Google Scholar]
- Beggs S, Salter MW. Stereological and somatotopic analysis of the spinal microglial response to peripheral nerve injury. Brain Behav Immun. 2007;21:624–33. doi: 10.1016/j.bbi.2006.10.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Binns BC, Huang Y, Goettl VM, Hackshaw KV, Stephens RL., Jr Glutamate uptake is attenuated in spinal deep dorsal and ventral horn in the rat spinal nerve ligation model. Brain Res. 2005;1041:38–47. doi: 10.1016/j.brainres.2005.01.088. [DOI] [PubMed] [Google Scholar]
- Blackburn-Munro G, Blackburn-Munro R. Pain in the brain: are hormones to blame? Trends Endocrinol Metab. 2003;14:20–7. doi: 10.1016/s1043-2760(02)00004-8. [DOI] [PubMed] [Google Scholar]
- Bomholt SF, Mikkelsen JD, Blackburn-Munro G. Normal hypothalamo-pituitary-adrenal axis function in a rat model of peripheral neuropathic pain. Brain Res. 2005;1044:216–26. doi: 10.1016/j.brainres.2005.03.005. [DOI] [PubMed] [Google Scholar]
- Burns VE, Edwards KM, Ring C, Drayson M, Carroll D. Complement cascade activation after an acute psychological stress task. Psychosom Med. 2008;70:387–96. doi: 10.1097/PSY.0b013e31816ded22. [DOI] [PubMed] [Google Scholar]
- Campbell JN, Meyer RA. Mechanisms of neuropathic pain. Neuron. 2006;52:77–92. doi: 10.1016/j.neuron.2006.09.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- César Venero JB. Rapid glucocorticoid effects on excitatory amino acid levels in the hippocampus: a microdialysis study in freely moving rats. European Journal of Neuroscience. 1999;11:2465–2473. doi: 10.1046/j.1460-9568.1999.00668.x. [DOI] [PubMed] [Google Scholar]
- Chacur M, Milligan ED, Gazda LS, Armstrong C, Wang H, Tracey KJ, Maier SF, Watkins LR. A new model of sciatic inflammatory neuritis (SIN): induction of unilateral and bilateral mechanical allodynia following acute unilateral peri-sciatic immune activation in rats. Pain. 2001;94:231–44. doi: 10.1016/S0304-3959(01)00354-2. [DOI] [PubMed] [Google Scholar]
- Chaplan SR, Bach FW, Pogrel JW, Chung JM, Yaksh TL. Quantitative assessment of tactile allodynia in the rat paw. J Neurosci Methods. 1994;53:55–63. doi: 10.1016/0165-0270(94)90144-9. [DOI] [PubMed] [Google Scholar]
- Cho K, Little HJ. Effects of corticosterone on excitatory amino acid responses in dopamine-sensitive neurons in the ventral tegmental area. Neuroscience. 1999;88:837–45. doi: 10.1016/s0306-4522(98)00264-4. [DOI] [PubMed] [Google Scholar]
- Chover-Gonzalez AJ, Jessop DS, Tejedor-Real P, Gibert-Rahola J, Harbuz MS. Onset and severity of inflammation in rats exposed to the learned helplessness paradigm. Rheumatology (Oxford) 2000;39:764–71. doi: 10.1093/rheumatology/39.7.764. [DOI] [PubMed] [Google Scholar]
- Colburn RW, DeLeo JA, Rickman AJ, Yeager MP, Kwon P, Hickey WF. Dissociation of microglial activation and neuropathic pain behaviors following peripheral nerve injury in the rat. J Neuroimmunol. 1997;79:163–75. doi: 10.1016/s0165-5728(97)00119-7. [DOI] [PubMed] [Google Scholar]
- Coull JA, Beggs S, Boudreau D, Boivin D, Tsuda M, Inoue K, Gravel C, Salter MW, De Koninck Y. BDNF from microglia causes the shift in neuronal anion gradient underlying neuropathic pain. Nature. 2005;438:1017–1021. doi: 10.1038/nature04223. [DOI] [PubMed] [Google Scholar]
- Dai Q, Pruett SB. Ethanol suppresses LPS-induced Toll-like receptor 4 clustering, reorganization of the actin cytoskeleton, and associated TNF-alpha production. Alcohol Clin Exp Res. 2006;30:1436–44. doi: 10.1111/j.1530-0277.2006.00172.x. [DOI] [PubMed] [Google Scholar]
- Daulhac L, Mallet C, Courteix C, Etienne M, Duroux E, Privat AM, Eschalier A, Fialip J. Diabetes-Induced Mechanical Hyperalgesia Involves Spinal Mitogen-Activated Protein Kinase Activation in Neurons and Microglia via N-Methyl-D-aspartate-Dependent Mechanisms. Mol Pharmacol. 2006;70:1246–1254. doi: 10.1124/mol.106.025478. [DOI] [PubMed] [Google Scholar]
- DeLeo JA. Basic science of pain. J Bone Joint Surg Am Suppl. 2006;88 2:58–62. doi: 10.2106/JBJS.E.01286. [DOI] [PubMed] [Google Scholar]
- DeLeo JA, Colburn RW, Rickman AJ. Cytokine and growth factor immunohistochemical spinal profiles in two animal models of mononeuropathy. Brain Res. 1997;759:50–7. doi: 10.1016/s0006-8993(97)00209-6. [DOI] [PubMed] [Google Scholar]
- DeLeo JA, Yezierski RP. The role of neuroinflammation and neuroimmune activation in persistent pain. Pain. 2001;90:1–6. doi: 10.1016/s0304-3959(00)00490-5. [DOI] [PubMed] [Google Scholar]
- Detloff MR, Fisher LC, McGaughy V, Longbrake EE, Popovich PG, Basso DM. Remote activation of microglia and pro-inflammatory cytokines predict the onset and severity of below-level neuropathic pain after spinal cord injury in rats. Exp Neurol. 2008;212:337–47. doi: 10.1016/j.expneurol.2008.04.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DeVries AC, DeVries MB, Taymans S, Carter CS. Modulation of pair bonding in female prairie voles (Microtus ochrogaster) by corticosterone. Proc Natl Acad Sci U S A. 1995;92:7744–8. doi: 10.1073/pnas.92.17.7744. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dhabhar FS. Stress, leukocyte trafficking, and the augmentation of skin immune function. Ann N Y Acad Sci. 2003;992:205–17. doi: 10.1111/j.1749-6632.2003.tb03151.x. [DOI] [PubMed] [Google Scholar]
- Diatchenko L, Nackley AG, Slade GD, Fillingim RB, Maixner W. Idiopathic pain disorders--pathways of vulnerability. Pain. 2006;123:226–30. doi: 10.1016/j.pain.2006.04.015. [DOI] [PubMed] [Google Scholar]
- Drew PD, Chavis JA. Inhibition of microglial cell activation by Cortisol. Brain Res Bull. 2000;52:391–6. doi: 10.1016/s0361-9230(00)00275-6. [DOI] [PubMed] [Google Scholar]
- Gehrmann J, Matsumoto Y, Kreutzberg GW. Microglia: intrinsic immuneffector cell of the brain. Brain Res Brain Res Rev. 1995;20:269–87. doi: 10.1016/0165-0173(94)00015-h. [DOI] [PubMed] [Google Scholar]
- Gold SM, Mohr DC, Huitinga I, Flachenecker P, Sternberg EM, Heesen C. The role of stress-response systems for the pathogenesis and progression of MS. Trends Immunol. 2005;26:644–52. doi: 10.1016/j.it.2005.09.010. [DOI] [PubMed] [Google Scholar]
- Greco CM, Rudy TE, Manzi S. Effects of a stress-reduction program on psychological function, pain, and physical function of systemic lupus erythematosus patients: a randomized controlled trial. Arthritis Rheum. 2004;51:625–34. doi: 10.1002/art.20533. [DOI] [PubMed] [Google Scholar]
- Guttridge DC, Albanese C, Reuther JY, Pestell RG, Baldwin AS., Jr NF-kappaB controls cell growth and differentiation through transcriptional regulation of cyclin D1. Mol Cell Biol. 1999;19:5785–99. doi: 10.1128/mcb.19.8.5785. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hao S, Mata M, Glonoso JC, Fink DJ. Gene transfer to interfere with TNF[alpha] signaling in neuropathic pain. Gene Ther. 2007;14:1010–1016. doi: 10.1038/sj.gt.3302950. [DOI] [PubMed] [Google Scholar]
- Horvath RJ, Nutile-McMenemy N, Alkaitis MS, Deleo JA. Differential migration, LPS-induced cytokine, chemokine, and NO expression in immortalized BV-2 and HAPI cell lines and primary microglial cultures. J Neurochem. 2008;107:557–69. doi: 10.1111/j.1471-4159.2008.05633.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hughes DI, Polgar E, Shehab SA, Todd AJ. Peripheral axotomy induces depletion of the vesicular glutamate transporter VGLUT1 in central terminals of myelinated afferent fibres in the rat spinal cord. Brain Res. 2004;1017:69–76. doi: 10.1016/j.brainres.2004.05.054. [DOI] [PubMed] [Google Scholar]
- Ito D, Imai Y, Ohsawa K, Nakajima K, Fukuuchi Y, Kohsaka S. Microglia-specific localisation of a novel calcium binding protein, Iba1. Brain Res Mol Brain Res. 1998;57:1–9. doi: 10.1016/s0169-328x(98)00040-0. [DOI] [PubMed] [Google Scholar]
- Jacobsson J, Persson M, Hansson E, Ronnback L. Corticosterone inhibits expression of the microglial glutamate transporter GLT-1 in vitro. Neuroscience. 2006;139:475–83. doi: 10.1016/j.neuroscience.2005.12.046. [DOI] [PubMed] [Google Scholar]
- Ji RR, Baba H, Brenner GJ, Woolf CJ. Nociceptive-specific activation of ERK in spinal neurons contributes to pain hypersensitivity. Nat Neurosci. 1999;2:1114–1119. doi: 10.1038/16040. [DOI] [PubMed] [Google Scholar]
- Ji RR, Strichartz G. Cell signaling and the genesis of neuropathic pain. Sci STKE. 2004;2004:reE14. doi: 10.1126/stke.2522004re14. [DOI] [PubMed] [Google Scholar]
- Jing H, Iwasaki Y, Nishiyama M, Taguchi T, Tsugita M, Taniguchi Y, Kambayashi M, Hashimoto K. Multisignal regulation of the rat NMDA1 receptor subunit gene--a pivotal role of glucocorticoid-dependent transcription. Life Sci. 2008;82:1137–41. doi: 10.1016/j.lfs.2008.03.022. [DOI] [PubMed] [Google Scholar]
- Joels M, Krugers HJ. LTP after stress: up or down? Neural Plast. 2007;2007:93202. doi: 10.1155/2007/93202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karim F, Wang CC, Gereau RWt. Metabotropic glutamate receptor subtypes 1 and 5 are activators of extracellular signal-regulated kinase signaling required for inflammatory pain in mice. J Neurosci. 2001;21:3771–9. doi: 10.1523/JNEUROSCI.21-11-03771.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kawasaki Y, Kohno T, Zhuang ZY, Brenner GJ, Wang H, Van Der Meer C, Befort K, Woolf CJ, Ji RR. Ionotropic and metabotropic receptors, protein kinase A, protein kinase C, and Src contribute to C-fiber-induced ERK activation and cAMP response element-binding protein phosphorylation in dorsal horn neurons, leading to central sensitization. J Neurosci. 2004;24:8310–8321. doi: 10.1523/JNEUROSCI.2396-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kawasaki Y, Xu ZZ, Wang X, Park JY, Zhuang ZY, Tan PH, Gao YJ, Roy K, Corfas G, Lo EH, et al. Distinct roles of matrix metalloproteases in the early- and late-phase development of neuropathic pain. Nat Med. 2008;14:331–6. doi: 10.1038/nm1723. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khasar SG, Burkham J, Dina OA, Brown AS, Bogen O, Alessandri-Haber N, Green PG, Reichling DB, Levine JD. Stress induces a switch of intracellular signaling in sensory neurons in a model of generalized pain. J Neurosci. 2008;28:5721–30. doi: 10.1523/JNEUROSCI.0256-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khasar SG, Green PG, Levine JD. Repeated sound stress enhances inflammatory pain in the rat. Pain. 2005;116:79–86. doi: 10.1016/j.pain.2005.03.040. [DOI] [PubMed] [Google Scholar]
- Kigerl KA, Lai W, Rivest S, Hart RP, Satoskar AR, Popovich PG. Toll-like receptor (TLR)-2 and TLR-4 regulate inflammation, gliosis, and myelin sparing after spinal cord injury. J Neurochem. 2007;102:37–50. doi: 10.1111/j.1471-4159.2007.04524.x. [DOI] [PubMed] [Google Scholar]
- Kigerl KA, McGaughy VM, Popovich PG. Comparative analysis of lesion development and intraspinal inflammation in four strains of mice following spinal contusion injury. J Comp Neurol. 2006;494:578–94. doi: 10.1002/cne.20827. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim JJ, Foy MR, Thompson RF. Behavioral stress modifies hippocampal plasticity through N-methyl-D-aspartate receptor activation. Proc Natl Acad Sci U S A. 1996;93:4750–3. doi: 10.1073/pnas.93.10.4750. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kohno T, Wang H, Amaya F, Brenner GJ, Cheng JK, Ji RR, Woolf CJ. Bradykinin Enhances AMPA and NMDA Receptor Activity in Spinal Cord Dorsal Horn Neurons by Activating Multiple Kinases to Produce Pain Hypersensitivity. J Neurosci. 2008;28:4533–4540. doi: 10.1523/JNEUROSCI.5349-07.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- LaCroix-Fralish ML, DeLeo JA. Steroid Hormone Regulation of Astrocyte Function: Implications for Central Nervous System Sensitization. In: DeLeo JA, Sorkin LS, Watkins LR, editors. Immune and Glial Regulation of Pain. Seattle: IASP Press; 2007. [Google Scholar]
- Lever IJ, Pezet S, McMahon SB, Malcangio M. The signaling components of sensory fiber transmission involved in the activation of ERK MAP kinase in the mouse dorsal horn. Mol Cell Neurosci. 2003;24:259–70. doi: 10.1016/s1044-7431(03)00200-8. [DOI] [PubMed] [Google Scholar]
- Lim HY, Muller N, Herold MJ, van den Brandt J, Reichardt HM. Glucocorticoids exert opposing effects on macrophage function dependent on their concentration. Immunology. 2007;122:47–53. doi: 10.1111/j.1365-2567.2007.02611.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Longbrake EE, Lai W, Ankeny DP, Popovich PG. Characterization and modeling of monocyte-derived macrophages after spinal cord injury. J Neurochem. 2007;102:1083–94. doi: 10.1111/j.1471-4159.2007.04617.x. [DOI] [PubMed] [Google Scholar]
- Lowy MT, Gault L, Yamamoto BK. Adrenalectomy attenuates stress-induced elevations in extracellular glutamate concentrations in the hippocampus. J Neurochem. 1993;61:1957–60. doi: 10.1111/j.1471-4159.1993.tb09839.x. [DOI] [PubMed] [Google Scholar]
- MacPherson A, Dinkel K, Sapolsky R. Glucocorticoids worsen excitotoxin-induced expression of pro-inflammatory cytokines in hippocampal cultures. Exp Neurol. 2005;194:376–83. doi: 10.1016/j.expneurol.2005.02.021. [DOI] [PubMed] [Google Scholar]
- Madrigal JL, Caso JR, de Cristobal J, Cardenas A, Leza JC, Lizasoain I, Lorenzo P, Moro MA. Effect of subacute and chronic immobilisation stress on the outcome of permanent focal cerebral ischaemia in rats. Brain Res. 2003;979:137–45. doi: 10.1016/s0006-8993(03)02892-0. [DOI] [PubMed] [Google Scholar]
- Melcangi RC, Magnaghi V, Cavarretta I, Riva MA, Martini L. Corticosteroid effects on gene expression of myelin basic protein in oligodendrocytes and of glial fibrillary acidic protein in type 1 astrocytes. J Neuroendocrinol. 1997;9:729–33. doi: 10.1046/j.1365-2826.1997.00621.x. [DOI] [PubMed] [Google Scholar]
- Meller E, Shen C, Nikolao TA, Jensen C, Tsimberg Y, Chen J, Gruen RJ. Region-specific effects of acute and repeated restraint stress on the phosphorylation of mitogen-activated protein kinases. Brain Res. 2003;979:57–64. doi: 10.1016/s0006-8993(03)02866-x. [DOI] [PubMed] [Google Scholar]
- Meunier A, Latremoliere A, Dominguez E, Mauborgne A, Philippe S, Hamon M, Mallet J, Benoliel JJ, Pohl M. Lentiviral-mediated targeted NF-kappaB blockade in dorsal spinal cord glia attenuates sciatic nerve injury-induced neuropathic pain in the rat. Mol Ther. 2007;15:687–97. doi: 10.1038/sj.mt.6300107. [DOI] [PubMed] [Google Scholar]
- Milligan ED, Watkins LR. Pathological and protective roles of glia in chronic pain. Nat Rev Neurosci. 2009;10:23–36. doi: 10.1038/nrn2533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moghaddam B, Bolnao ML, Stein-Behrens B, Sapolsky R. Glucocorticoids mediate the stress-induced extracellular accumulation of glutamate. Brain Res. 1994;655:251–4. doi: 10.1016/0006-8993(94)91622-5. [DOI] [PubMed] [Google Scholar]
- Narita M, Yoshida T, Nakajima M, Narita M, Miyatake M, Takagi T, Yajima Y, Suzuki T. Direct evidence for spinal cord microglia in the development of a neuropathic pain-like state in mice. J Neurochem. 2006;97:1337–48. doi: 10.1111/j.1471-4159.2006.03808.x. [DOI] [PubMed] [Google Scholar]
- Nicholson K, Martelli MF. The problem of pain. J Head Trauma Rehabil. 2004;19:2–9. doi: 10.1097/00001199-200401000-00002. [DOI] [PubMed] [Google Scholar]
- Norris EH, Strickland S. Modulation of NR2B-regulated contextual fear in the hippocampus by the tissue plasminogen activator system. Proc Natl Acad Sci U S A. 2007;104:13473–8. doi: 10.1073/pnas.0705848104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ovadia H, Vlodavsky I, Abramsky O, Weidenfeld J. Binding of hormonal steroids to isolated oligodendroglia and astroglia grown in vitro on a naturally produced extracellular matrix. Clin Neuropharmacol. 1984;7:307–11. doi: 10.1097/00002826-198412000-00006. [DOI] [PubMed] [Google Scholar]
- Qi AQ, Qiu J, Xiao L, Chen YZ. Rapid activation of JNK and p38 by glucocorticoids in primary cultured hippocampal cells. J Neurosci Res. 2005;80:510–7. doi: 10.1002/jnr.20491. [DOI] [PubMed] [Google Scholar]
- Raghavendra V, Tanga F, DeLeo JA. Inhibition of microglial activation attenuates the development but not existing hypersensitivity in a rat model of neuropathy. J Pharmacol Exp Ther. 2003;306:624–30. doi: 10.1124/jpet.103.052407. [DOI] [PubMed] [Google Scholar]
- Revest JM, Di Blasi F, Kitchener P, Rouge-Pont F, Desmedt A, Turiault M, Tranche F, Piazza PV. The MAPK pathway and Egr-1 mediate stress-related behavioral effects of glucocorticoids. Nat Neurosci. 2005;8:664–72. doi: 10.1038/nn1441. [DOI] [PubMed] [Google Scholar]
- Reznikov LR, Grillo CA, Piroli GG, Pasumarthi RK, Reagan LP, Fadel J. Acute stress-mediated increases in extracellular glutamate levels in the rat amygdala: differential effects of antidepressant treatment. Eur J Neurosci. 2007;25:3109–14. doi: 10.1111/j.1460-9568.2007.05560.x. [DOI] [PubMed] [Google Scholar]
- Ririe KM, Rasmussen RP, Wittwer CT. Product differentiation by analysis of DNA melting curves during the polymerase chain reaction. Anal Biochem. 1997;245:154–60. doi: 10.1006/abio.1996.9916. [DOI] [PubMed] [Google Scholar]
- Rival C, Laboureyras E, Laulin JP, Le Roy C, Richebe P, Simonnet G. Non-nociceptive environmental stress induces hyperalgesia, not analgesia, in pain and opioid-experienced rats. Neuropsychopharmacology. 2007;32:2217–28. doi: 10.1038/sj.npp.1301340. [DOI] [PubMed] [Google Scholar]
- Sananbenesi F, Fischer A, Schrick C, Spiess J, Radulovic J. Mitogen-activated protein kinase signaling in the hippocampus and its modulation by corticotropin-releasing factor receptor 2: a possible link between stress and fear memory. J Neurosci. 2003;23:11436–43. doi: 10.1523/JNEUROSCI.23-36-11436.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scadding JW, Koltzenburg M. Painful peripheral neuropathies. In: McMahon SB, Koltzenburg M, editors. Wall and Melzack's Textbook of Pain. Churchill: Elsevier; 2006. pp. 973–999. [Google Scholar]
- Schafers M, Lee DH, Brors D, Yaksh TL, Sorkin LS. Increased Sensitivity of Injured and Adjacent Uninjured Rat Primary Sensory Neurons to Exogenous Tumor Necrosis Factor-alpha after Spinal Nerve Ligation. J Neurosci. 2003a;23:3028–3038. doi: 10.1523/JNEUROSCI.23-07-03028.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schafers M, Svensson CI, Sommer C, Sorkin LS. Tumor Necrosis Factor-alpha Induces Mechanical Allodynia after Spinal Nerve Ligation by Activation of p38 MAPK in Primary Sensory Neurons. J Neurosci. 2003b;23:2517–2521. doi: 10.1523/JNEUROSCI.23-07-02517.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shields SD, Eckert WA, 3rd, Basbaum AI. Spared nerve injury model of neuropathic pain in the mouse: a behavioral and anatomic analysis. J Pain. 2003;4:465–70. doi: 10.1067/s1526-5900(03)00781-8. [DOI] [PubMed] [Google Scholar]
- Smith MA, Makino S, Kim SY, Kvetnansky R. Stress increases brain-derived neurotropic factor messenger ribonucleic acid in the hypothalamus and pituitary. Endocrinology. 1995;136:3743–50. doi: 10.1210/endo.136.9.7649080. [DOI] [PubMed] [Google Scholar]
- Song XS, Cao JL, Xu YB, He JH, Zhang LC, Zeng YM. Activation of ERK/CREB pathway in spinal cord contributes to chronic constrictive injury-induced neuropathic pain in rats. Acta Pharmacol Sin. 2005;26:789–98. doi: 10.1111/j.1745-7254.2005.00123.x. [DOI] [PubMed] [Google Scholar]
- Steptoe A, Hamer M, Chida Y. The effects of acute psychological stress on circulating inflammatory factors in humans: a review and meta-analysis. Brain Behav lmmun. 2007;21:901–12. doi: 10.1016/j.bbi.2007.03.011. [DOI] [PubMed] [Google Scholar]
- Strang P. Cancer pain--a provoker of emotional, social and existential distress. Acta Oncol. 1998;37:641–4. doi: 10.1080/028418698429973. [DOI] [PubMed] [Google Scholar]
- Streit WJ, Graeber MB, Kreutzberg GW. Functional plasticity of microglia: a review. Glia. 1988;1:301–7. doi: 10.1002/glia.440010502. [DOI] [PubMed] [Google Scholar]
- Sugama S, Fujita M, Hashimoto M, Conti B. Stress induced morphological microglial activation in the rodent brain: involvement of interleukin-18. Neuroscience. 2007;146:1388–99. doi: 10.1016/j.neuroscience.2007.02.043. [DOI] [PubMed] [Google Scholar]
- Sugo N, Hurn PD, Morahan MB, Hattori K, Traystman RJ, DeVries AC. Social stress exacerbates focal cerebral ischemia in mice. Stroke. 2002;33:1660–4. doi: 10.1161/01.str.0000016967.76805.bf. [DOI] [PubMed] [Google Scholar]
- Sung B, Lim G, Mao J. Altered expression and uptake activity of spinal glutamate transporters after nerve injury contribute to the pathogenesis of neuropathic pain in rats. J Neurosci. 2003;23:2899–910. doi: 10.1523/JNEUROSCI.23-07-02899.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takasaki I, Kurihara T, Saegusa H, Zong S, Tanabe T. Effects of glucocorticoid receptor antagonists on allodynia and hyperalgesia in mouse model of neuropathic pain. Eur J Pharmacol. 2005;524:80–3. doi: 10.1016/j.ejphar.2005.09.045. [DOI] [PubMed] [Google Scholar]
- Tanga FY, Nutile-McMenemy N, DeLeo JA. The CNS role of Toll-like receptor 4 in innate neuroimmunity and painful neuropathy. Proc Natl Acad Sci U S A. 2005;102:5856–61. doi: 10.1073/pnas.0501634102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tanga FY, Raghavendra V, DeLeo JA. Quantitative real-time RT-PCR assessment of spinal microglial and astrocytic activation markers in a rat model of neuropathic pain. Neurochem Int. 2004;45:397–407. doi: 10.1016/j.neuint.2003.06.002. [DOI] [PubMed] [Google Scholar]
- Tegeder I, Niederberger E, Schmidt R, Kunz S, Guhnng H, Ritzeler O, Michaelis M, Geisslinger G. Specific Inhibition of IkappaB kinase reduces hyperalgesia in inflammatory and neuropathic pain models in rats. J Neurosci. 2004;24:1637–45. doi: 10.1523/JNEUROSCI.3118-03.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsuda M, Inoue K, Salter MW. Neuropathic pain and spinal microglia: a big problem from molecules in “small” glia. Trends Neurosci. 2005;28:101–7. doi: 10.1016/j.tins.2004.12.002. [DOI] [PubMed] [Google Scholar]
- Tsuda M, Shigemoto-Mogami Y, Koizumi S, Mizokoshi A, Kohsaka S, Salter MW, Inoue K. P2X4 receptors induced in spinal microglia gate tactile allodynia after nerve injury. Nature. 2003;424:778–83. doi: 10.1038/nature01786. [DOI] [PubMed] [Google Scholar]
- Turner JA, Jensen MP, Warms CA, Cardenas DD. Catastrophizing is associated with pain intensity, psychological distress, and pain-related disability among individuals with chronic pain after spinal cord injury. Pain. 2002;98:127–34. doi: 10.1016/s0304-3959(02)00045-3. [DOI] [PubMed] [Google Scholar]
- Ulmann L, Hatcher JP, Hughes JP, Chaumont S, Green PJ, Conquet F, Buell GN, Reeve AJ, Chessell IP, Rassendren F. Up-regulation of P2X4 receptors in spinal microglia after peripheral nerve injury mediates BDNF release and neuropathic pain. J Neurosci. 2008;28:11263–8. doi: 10.1523/JNEUROSCI.2308-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ulrich-Lai YM, Xie W, Meij JT, Dolgas CM, Yu L, Herman JP. Limbic and HPA axis function in an animal model of chronic neuropathic pain. Physiol Behav. 2006;88:67–76. doi: 10.1016/j.physbeh.2006.03.012. [DOI] [PubMed] [Google Scholar]
- Vielkind U, Walencewicz A, Levine JM, Bohn MC. Type II glucocorticoid receptors are expressed in oligodendrocytes and astrocytes. J Neurosci Res. 1990;27:360–73. doi: 10.1002/jnr.490270315. [DOI] [PubMed] [Google Scholar]
- Viswanathan K, Dhabhar FS. Stress-induced enhancement of leukocyte trafficking into sites of surgery or immune activation. Proc Natl Acad Sci U S A. 2005;102:5808–13. doi: 10.1073/pnas.0501650102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang S, Lim G, Yang L, Sung B, Mao J. Downregulation of spinal glutamate transporter EAAC1 following nerve injury is regulated by central glucocorticoid receptors in rats. Pain. 2006;120:78–85. doi: 10.1016/j.pain.2005.10.015. [DOI] [PubMed] [Google Scholar]
- Wang S, Lim G, Zeng Q, Sung B, Ai Y, Guo G, Yang L, Mao J. Expression of central glucocorticoid receptors after peripheral nerve injury contributes to neuropathic pain behaviors in rats. J Neurosci. 2004;24:8595–605. doi: 10.1523/JNEUROSCI.3058-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang S, Lim G, Zeng Q, Sung B, Yang L, Mao J. Central glucocorticoid receptors modulate the expression and function of spinal NMDA receptors after peripheral nerve injury. J Neurosci. 2005;25:488–95. doi: 10.1523/JNEUROSCI.4127-04.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wong TP, Howland JG, Robillard JM, Ge Y, Yu W, Titterness AK, Brebner K, Liu L, Weinberg J, Christie BR, et al. Hippocampal long-term depression mediates acute stress-induced spatial memory retrieval impairment. Proc Natl Acad Sci U S A. 2007;104:11471–6. doi: 10.1073/pnas.0702308104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu Q, Garraway SM, Weyerbacher AR, Shin SJ, Inturrisi CE. Activation of the neuronal extracellular signal-regulated kinase 2 in the spinal cord dorsal horn is required for complete Freund's adjuvant-induced pain hypersensitivity. J Neurosci. 2008;28:14087–96. doi: 10.1523/JNEUROSCI.2406-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang PC, Yang CH, Huang CC, Hsu KS. Phosphatidylinositol 3-Kinase Activation Is Required for Stress Protocol-induced Modification of Hippocampal Synaptic Plasticity. J Biol Chem. 2008;283:2631–2643. doi: 10.1074/jbc.M706954200. [DOI] [PubMed] [Google Scholar]
- Zanella JM, Burnght EN, Hildebrand K, Hobot C, Cox M, Christoferson L, McKay WF. Effect of etanercept, a tumor necrosis factor-alpha inhibitor, on neuropathic pain in the rat chronic constriction injury model. Spine. 2008;33:227–34. doi: 10.1097/BRS.0b013e318162340a. [DOI] [PubMed] [Google Scholar]
- Zhu XY, Liu YJ, Diao F, Fan J, Lu J, Xu RB. Role of glucocorticoids and glucocorticoid receptor in priming of macrophages caused by glucocorticoid receptor blockade. Endocrine. 2007;31:130–7. doi: 10.1007/s12020-007-0019-6. [DOI] [PubMed] [Google Scholar]
- Zhuang ZY, Gerner P, Woolf CJ, Ji RR. ERK is sequentially activated in neurons, microglia, and astrocytes by spinal nerve ligation and contributes to mechanical allodynia in this neuropathic pain model. Pain. 2005;114:149–59. doi: 10.1016/j.pain.2004.12.022. [DOI] [PubMed] [Google Scholar]
- Zhuang ZY, Wen YR, Zhang DR, Borsello T, Bonny C, Strichartz GR, Decosterd I, Ji RR. A peptide c-Jun N-terminal kinase (JNK) inhibitor blocks mechanical allodynia after spinal nerve ligation: respective roles of JNK activation in primary sensory neurons and spinal astrocytes for neuropathic pain development and maintenance. J Neurosci. 2006;26:3551–60. doi: 10.1523/JNEUROSCI.5290-05.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]