

Abstract
We investigate the role of Rab4, a Ras-like small GTPase coordinating protein transport from the endosome to the plasma membrane, on the recycling and activation of endogenous β-adrenergic receptor (β-AR) in HL-1 cardiac myocytes in vitro and transgenic mouse hearts in vivo. β1-AR, the predominant subtype of β-AR in HL-1 cardiac myocytes, was internalized after stimulation with isoproterenol (ISO) and fully recycled at 4 h upon ISO removal. Transient expression of Rab4 markedly facilitated recycling of internalized β-AR to the cell surface and enhanced β-AR signaling as measured by ISO-stimulated cAMP production. Transgenic overexpression of Rab4 in the mouse myocardium significantly increased the number of β-AR in the plasma membrane and augmented cAMP production at the basal level and in response to ISO stimulation. Rab4 overexpression induced concentric cardiac hypertrophy with a moderate increase in ventricle/body weight ratio and posterior wall thickness and a selective up-regulation of the β-myosin heavy chain gene. These data provide the first evidence indicating that Rab4 is a rate-limiting factor for the recycling of endogenous β-AR and augmentation of Rab4-mediated traffic enhances β-AR function in cardiac myocytes.
β-Adrenergic receptors (ARs)2 are members of the seven transmembrane spanning G protein-coupled receptor (GPCR) family and play a critical role in the regulation of cardiac function in response to cate-cholamine stimulation (1–3). Three subtypes, β1-AR, β2-AR, and β3-AR, have been identified in the mammalian hearts. β1-AR and β2-AR are major mediators of cardiac contractility through coupling to heterotrimeric G proteins to regulate the activation of adenylyl cyclases, which in turn modulates production of intracellular cAMP and activation of protein kinase A. β1-AR couples to the stimulatory G protein Gs, whereas β2-AR couples to both Gs and the inhibitory G protein Gi (1, 4, 5).
The number of β-AR at the cell surface at a certain time determines the amplitude of functional response to β-AR stimulation by extracellular hormones. This number of cell surface receptors is regulated by precise receptor intracellular trafficking and targeting. Intracellular trafficking of the receptors is a dynamic process and highly coordinated by many regulatory factors at distinct organelles (6, 7). After being synthesized, folded, and assembled in the endoplasmic reticulum (ER), the β-ARs are transported from the ER to the plasma membrane through the Golgi apparatus, where posttranslational modifications (e.g. glycosylation) occur. Upon stimulation with agonists, the β-ARs at the plasma membrane may undergo internalization to the endosome (8, 9). The internalization involves phosphorylation of β-ARs by at least two kinases, protein kinase A and G protein receptor kinase (GRK), and subsequent binding of the phosporylated receptors to arrestins, which serve as adaptor proteins recruiting components of the transport machinery to the clathrin-coated pits, initiating formation of the early endosome (10, 11). The internalized receptors in the early endosome may be sorted to the lysosome for degradation or to the recycling endosome for return to the plasma membrane (12–14).
Rab proteins are Ras-like small GTPases that regulate vesicular protein transport in both endocytosis and exocytosis (6, 7, 15–17). Although most of the Rab GTPases identified are ubiquitous and highly conserved in their structure and function, each Rab GTPase has a distinct intracellular localization and regulates discrete protein transport steps in secretory and endocytic pathways (15–17). For example, Rab5 modulates protein transport from the plasma membrane to the endosome, whereas Rab4 is specifically involved in the transport from the early endosome to the plasma membrane (7, 17). For GPCRs, Rab4 has been demonstrated to regulate the recycling of internalized β2-AR, neurokinin 1 receptor, and CB1 cannabinoid receptor (9, 18–20). However, these studies were carried out in systems overexpressing individual receptors and the function of Rab4 was manipulated through expressing dominant-negative Rab4 mutants to attenuate endogenous Rab4 function. These studies have demonstrated that Rab4 is essential for the recycling of internalized GPCRs back to the plasma membrane. Importantly, endogenous Rab4 expression is augmented in transgenic mouse hearts overexpressing β2-AR, which develop heart failure (21), suggesting that endogenous Rab4 level may be altered in certain pathological conditions. However, the effects of increase wild-type Rab4 expression on the GPCR recycling and signaling and on the cardiac function have not been studied.
In this report, we investigated the effect of augmentation of Rab4 function on the recycling and activation of endogenous β-AR in both cardiac myocytes in vitro and mouse hearts in vivo. Our data demonstrated that increased wild-type Rab4 expression facilitated recycling to the plasma membrane and signaling of β-AR in cultured HL-1 cardiac myocytes. Our results also showed that cardiac specific overexpression of wild-type Rab4 augmented the membrane targeting and function of β-AR. Furthermore, overexpression of wild-type Rab4 induced cardiac hypertrophy with preserved contractile function. These data provide the first evidence indicating that endogenous Rab4 expression level is a rate-limiting factor for the recycling of endogenous β-AR and that augmentation of Rab4-mediated traffic enhances β-AR function in cardiac myocytes.
EXPERIMENTAL PROCEDURES
Materials
Antibodies against Rab1, Rab4, Rab5, Gs, Gi, Gβ, GRK2, and calregulin were purchased from Santa Cruz Biotechnology, Inc. Anti-GM130 antibody was from BD Transduction Laboratories. Antibody against Na+-K+-ATPase was from Affinity Bio-Reagents (Golden, CO). Anti-FLAG M2 monoclonal antibody, isoproterenol (ISO), alprenolol, atenolol, ICI118,551, and forskolin were from Sigma. [125I]Iodocyanopindolol (specific activity = 2000 Ci/mmol) and [3H]CGP12177 (specific activity = 51 Ci/mmol) were from Amersham Biosciences. All other materials were obtained as described elsewhere (22, 23).
Culture and Transfection of HL-1 Cardiomyocytes
HL-1 myocytes were plated onto 12-well plates at a density of 4 × 105 cells/well and cultured in Claycomb medium supplemented with 10% fetal bovine serum, 100 units/ml penicillin, 100 μg/ml streptomycin, 0.1 mM norepinephrine, and 2 mM L-glutamine as described previously (23). Rab4 was tagged with FLAG epitope at the amino terminus of Rab4 (FLAG-Rab4) by PCR using a primer GACTACAAGGACGACGATGACAAG coding a peptide DYKDDDDK. The FLAG epitope has been used to label a number of proteins resulting in tagged-proteins with similar characteristics to their respective wild-types (24). After 24-h culture without norepinephrine, HL-1 myocytes were transiently transfected with 2 μg of Rab4 or the pcDNA3 vector using Lipofectamine 2000 reagent (Invitrogen) as described previously (23).
Ligand Binding in Intact HL-1 Cardiomyocytes
Intact cell ligand binding was used to measure cell surface expression of β-AR. HL-1 myocytes were cultured on 12-well plates and incubated with [3H]CGP12177 at a concentration of 20 nM for 2 h at room temperature. To measure the expression of β1-AR and β2-AR subtypes, the HL-1 cells were preincubated with the β1-AR-selective antagonist atenolol (20 μM) or the β2-AR-selective antagonist ICI118,551 (20 μM) for 30 min. The nonspecific binding was determined in the presence of alprenolol (20 μM). After washing twice with ice-cold phosphate-buffered saline (1 ml each time), the cells were digested with 1 ml of 1 M NaOH. The radioactivity was counted by liquid scintillation spectrometry in 5 ml of Ecoscint A scintillation solution (National Diagnostics, Inc., Atlanta, GA).
Measurement of β-AR Internalization and Recycling in HL-1 Myocytes
β-AR internalization in response to stimulation with ISO and recycling of internalized receptors were determined as essentially described (9, 12) with modifications. Briefly, HL-1 myocytes were cultured on 12-well plates and transfected as described above. At 48 h after transfection, the cells were incubated with ISO at a concentration of 10 μM for different times at 37 °C to initiate receptor internalization. The cells were washed twice with 1 ml of ice-cold Dulbecco’s modified Eagle’s medium to remove ISO and allowed to recover for different time periods (from 15 to 240 min). β-AR expression at the cell surface was then determined by ligand binding as described above.
Generation of Rab4 Transgenic Mice
Transgenic mice overexpressing Rab4 in the myocardium were generated essentially as described (21). The cDNA encoding FLAG-Rab4 was cloned into exon three of the full-length mouse α-myosin heavy chain (MHC) promoter (21). The entire 7.7-kb transgene fragment containing the entire α-MHC promoter, the complete FLAG-Rab4 cDNA, and a human growth hormone polyadenylation signal sequence was released from the plasmid backbone by digestion with BamHI and was used for microinjection into pronuclei of fertilized mouse oocytes (FVB/N background) using standard techniques (Pennington Biomedical Research Institute, Louisiana State University, Baton Rouge, LA). Transgenic mice were identified by Southern blot or PCR analysis using genomic DNA extracted from mouse tails. All studies were performed in Rab4 transgenic mice and nontransgenic (NTG) siblings at 22 weeks old in accordance with protocols approved by the Louisiana State University Health Sciences Center Institutional Animal Care and Use Committee.
Measurement of Cardiac β-AR Expression
β-AR density was measured as described (25), with modifications. Briefly, myocardial membranes were prepared by homogenization of both ventricles with a Polytron in buffer containing 20 mM Tris-HCl, pH 7.4, 1 mM EDTA, and 150 mM NaCl supplemented with Complete Mini protease inhibitor mixture (Roche Applied Science, Mannheim, Germany) and centrifuged at 500 × g for 10 min at 4 °C. The supernatant was then centrifuged at 10,000 × g for 40 min. Final membrane preparations were re-suspended in binding buffer containing 75 mM Tris-HCl, pH 7.4, 12.5 mM MgCl2, 2 mM EDTA at a concentration of 2.0 mg/ml. Receptor binding of myocardial membranes was performed using the nonselective β-AR ligand [125I]iodocyanopindolol. Reactions were conducted in 500 μl of binding buffer containing 25 μg of membrane proteins and 400 pM [125I]iodocyanopindolol for 1 h. Nonspecific binding was determined in the presence of 20 μM alprenolol. The reactions were terminated by vacuum filtration through glass-fiber filters. The radioactivity was counted in a γ counter. All assays were performed in duplicate, and receptor density was expressed as fmol/mg of membrane protein.
Measurement of cAMP Production
cAMP production in response to stimulation with ISO or forskolin was measured in the presence of 3-isobutyl-1-methylxanthine (0.5 mM), a phosphodiesterase inhibitor, by using cAMP enzymeimmunoassay system (Biotrak, Amersham Biosciences) as described (26). For measurement of cAMP production by membrane fractions prepared from NTG and Rab4 transgenic mouse ventricles, an aliquot of membrane fraction (about 0.8 μg of protein) was transferred into microtiter plates and then incubated with anti-cAMP antiserum, followed by the incubation with cAMP-peroxidase. After washing and addition of substrate, peroxidase activity was measured by spectrometry. cAMP concentrations were calculated based on the competition of cAMP in samples with a fixed quantity of peroxidase-labeled cAMP.
For measurement of cAMP production in cultured cardiomyocytes, HL-1 cells were cultured in 12-well plates and transfected with 2 μg of Rab4 or pcDNA3 as described above. After 48 h, the cells were stimulated with increasing concentrations of ISO (from 10−9 to 10−5 M) or forskolin (100 μM) for 10 min at room temperature. The reactions were stopped by aspirating the medium and then the cells were lysed using 200 μl of dodecyltrimethylammonium (2.5%). One-hundred μl of cell lysate was used to determine cAMP concentration as described above.
Measurement of Cardiac Hypertrophy
Morphometric analysis and histological examination of Masson’s trichrome- and hematoxylin-eosin-stained ventricles used standard techniques as described previously (21). Cardiac gene expression was assayed by RNA dot blot analysis using total RNA (3 μg/dot) extracted from ventricles of NTG and transgenic mice and 32P-labeled oligonucleotides as probes (21, 27). Radiolabeled RNA dots were quantitated with a PhosphorImager (Amersham Biosciences), and expression of each cardiac gene was normalized to glyceraldehyde-3-phosphate dehydrogenase expression.
Echocardiography
Mice were anesthetized with avertin (250 mg/kg, intraperitoneal). Cardiac ultrasound studies were performed on Rab4 transgenic mice and NTG sibling controls at 22 weeks old using a SSA770 Aplio Ultrasound system (Toshiba America Medical Systems, Tustin, CA) with a 1204AX linear array transducer scanning at 14 MHz center frequency. Depth setting was 2 cm with a 0.75-cm electronic focus and two-dimensional imaging frame rate of 238 Hz. Two-dimensional guided M-mode studies of the left ventricle at the level of the papillary muscles were performed. M-mode measurements were made using the leading-edge to leading-edge method, as per American Society of Echocardiography guidelines (28). The left ventricular end-systolic dimension (ESD) was determined as the point in time when the posterior left ventricular wall was most anterior and left ventricular end-diastolic dimension (EDD) when the posterior LV wall was most posterior. Left ventricular fractional shortening (the proportion of blood ejected during systole compared with maximal ventricular capacity in diastole) was measured as: (EDD − ESD)/EDD. Both sonographer and interpreter were blinded to the identity of experimental groups.
Immunoblot Analysis
Western blot analysis of protein expression was carried out as described previously (22, 23). Proteins were separated by SDS-PAGE and transferred onto polyvinylidene difluoride membranes. The signal was detected using ECL Reagent Plus (PerkinElmer Life Sciences, Boston, MA) with a Fujifilm Luminescent Image Analyzer (LAS-1000plus) and quantitated using Image Gauge Program (Version 3.4). Protein loading and transfer efficiency were evaluated by Amido Black staining of the membrane after immunoblotting.
Statistical Analysis
Data are expressed as the mean ± S.E. Differences were evaluated using Student’s t test. p < 0.05 was considered as statistically significant.
RESULTS
Effect of Transient Expression of Rab4 on the Recycling of Internalized β-AR in HL-1 Cardiomyocytes
To determine whether Rab4 is involved in the regulation of endogenous β-AR recycling in cardiac myocytes, we choose HL-1 cardiomyocytes, an immortal cardiac muscle cell line that proliferates and retains phenotypic characteristics of cardiomyocytes (29). We first determined the relative expression of β1-AR and β2-AR in HL-1 myocytes by ligand binding in intact cells in the presence or absence of the β1-AR-selective antagonist atenolol or the β2-AR-selective antagonist ICI118,551. β1-AR is the predominant subtype of β-AR in HL-1 cardiac myocyte, whereas β2-AR expression is markedly lower than that of β1-AR (Fig. 1A).
FIGURE 1. Cell surface expression and internalization of β-AR in HL-1 cardiac myocytes.

A, cell surface expression of total β-AR, β1-AR, and β2-AR measured by intact cell ligand binding. HL-1 myocytes were cultured in 12-well dishes and incubated with [3H]CGP12177 as described under “Experimental Procedures.” Cell surface expression of β1-AR and β2-AR was determined in the presence of atenolol and ICI118,551, respectively. Nonspecific binding obtained in the presence of alprenolol was substracted from the value presented. The data shown are the percentage of the total β-AR binding (2242 ± 151 cpm) and are presented as the means ± S.E. of six separate experiments. B, time-dependent internalization of β-AR. HL-1 cells were exposed to 10 μM ISO at 37 °C for different time periods and the β-AR binding sites left at the cell surface were determined as described in A. C, dose-dependent internalization of β-AR. HL-1 cells were stimulated with different concentrations of ISO for 30 min and the cell surface expression of β-AR was measured. In B and C, the data are presented as percentage of β-AR obtained in absence of ISO (2382 ± 212 cpm) and presented as means ± S.E. of six independent determinations. *, p < 0.05 versus the data obtained in the absence of ISO.
Internalization of β-AR in response to stimulation with ISO in HL-1 cardiac myocytes was then characterized. ISO stimulation induced internalization of plasma membrane β-AR in a time- and dose-dependent manner (Fig. 1, B and C). Cell surface expression of β-AR was reduced by 23% after ISO stimulation for 15 min and the internalization reached the maximum after simulation for 30–60 min (Fig. 1B). The cell surface expression of β-AR was significantly attenuated by 57% in HL-1 myocytes after stimulation with ISO at a concentration of 10 μM for 30 min (Fig. 1C).
In the next series of experiments we determined whether increased Rab4 function could modulate the recycling of internalized β-AR in HL-1 myocytes. HL-1 myocytes were transiently transfected with FLAG-tagged Rab4. Rab4 expression was then determined by Western blotting using FLAG high affinity monoclonal and Rab4 antibodies. The FLAG antibody detected only exogenously transfected Rab4, whereas the Rab4 antibody detected both transfected FLAG-Rab4 and endogenous Rab4. Rab4 expression was about four times higher in the FLAG-Rab4 transfected cells than endogenous Rab4 in cells transfected with the pcDNA3 vector (Fig. 2A). β-AR expression at the cell surface as measured by intact cell ligand binding was about the same in cells transfected with the pcDNA vector and Rab4 (specific binding in pcDNA-transfected cells: 2242 ± 151 cpm and in Rab4-transfected cells: 2171 ± 237 cpm, n = 3 each in duplicate).
FIGURE 2. Effect of transient expression of Rab4 on the recycling of internalized β-AR in HL-1 myocytes.
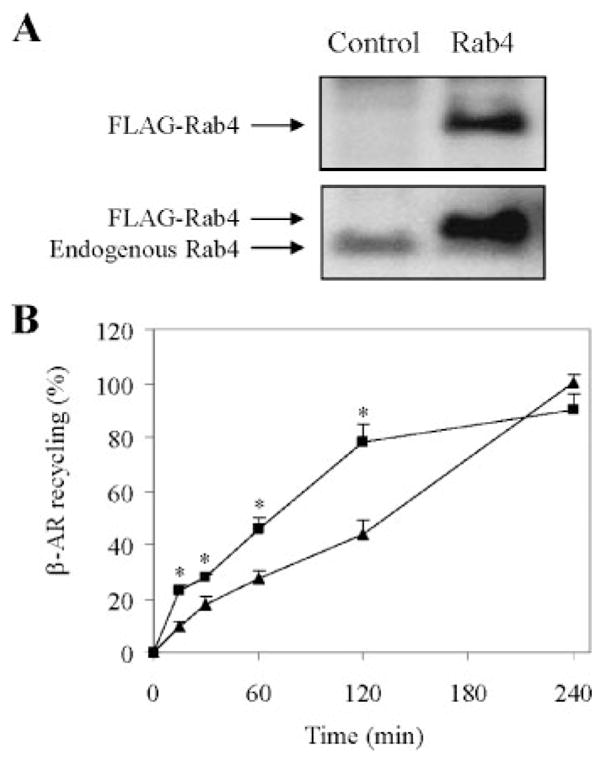
A, immunoblot analysis of Rab4 expression. HL-1 myocytes were transiently transfected with FLAG-tagged Rab4 or the pcDNA3 vector (Control). Rab4 expression was determined by Western blotting using anti-FLAG (upper panel) and Rab4 antibodies (lower panel). Rab4 antibodies detected both exogenous and endogenous Rab4. B, effect of Rab4 on the recycling of ISO-mediated internalized β-AR. HL-1 cells were transiently transfected for 48 h with Rab4 (squares) or the pcDNA3 vector (triangles), stimulated with ISO (10 μM) for 30 min, and allowed to recover for 15, 30, 60, 120, and 240 min at 37 °C. Cell surface expression of β-AR was measured by ligand binding as described in the legend of Fig. 1. β-AR expression at the cell surface are 2382 ± 212 cpm in the absence of ISO and 952 ± 317 cpm after 30 min exposure to ISO. The data shown are the percentage of β-AR recycling in the cell transfected with pcDNA3 and recovered for 240 min after ISO stimulation and presented as the means ± S.E. of six independent experiments. *, p < 0.05 versus control at the same time points.
The HL-1 myocytes were treated by ISO for 30 min to initiate internalization and then allowed to recover for various period of time (15, 30, 60, 120, and 240 min). After 4 h, β-ARs were fully recycled back to the plasma membrane in myocytes transfected with the pcDNA3 vector(Fig. 2B). However, the recycling of internalized β-AR was much faster in HL-1 myocytes transfected with Rab4 and reached the maximal recovery after 2 h (Fig. 2B). At 4 h after recycling, the expression of β-AR at the cell surface is similar in HL-1 myocytes transfected with control plasmid and Rab4. These data indicate that increased Rab4 function by transient expression of wild-type Rab4 facilitates the recycling of ISO-internalized endogenous β-AR, mostly β1-AR, in HL-1 cardiomyocytes.
Effect of Transient Expression of Rab4 on β-AR Signaling in HL-1 Cardiomyocytes
To determine whether Rab4-faciliated recycling of internalized β-AR could modulate β-AR signaling, we measured the effect of transient expression of Rab4 on cAMP production in HL-1 cardiomyocytes. HL-1 myocytes were stimulated with increasing concentration of ISO (from 10−9 to 10−5 M) and intracellular cAMP concentrations were then measured. cAMP production in response to ISO stimulation at concentrations from 10−8 to 10−5 M was significantly higher in myocytes transfected with Rab4 than myocytes transfected with the pcDNA3 vector Fig. 3. To determine whether increased cAMP production in Rab4-transfected HL-1 myocytes is due to the alteration of adenylyl cyclase activity, we measured cAMP production in response to stimulation with forskolin, which directly activates adenylyl cyclases, bypassing the plasma membrane receptors. Forskolin-stimulated cAMP production was almost the same in Rab4 and pcDNA-transfected HL-1 myocytes (7109 ± 780 pmol/well versus 7235 ± 861 pmol/well, n = 3, p > 0.05). These data indicate that Rab4 is able to regulate β-AR signaling through modulating β-AR recycling.
FIGURE 3. Effect of transient expression of Rab4 on ISO-stimulated cAMP production in HL-1 myocytes.

HL-1 myocytes cultured on 12-well plates were transiently transfected with FLAG-tagged Rab4 (squares) or the pcDNA3 vector (triangles) and stimulated with increasing concentrations of ISO (from 10−9 to 10−5 M) for 10 min. cAMP concentrations were determined as described under “Experimental Procedures.” Rab4 expression did not significantly influence cAMP production at basal level (Rab4-transfected cells: 381 ± 136 and pcDNA3-transfected cells: 452 ± 67 pmol/well, n = 4, p > 0.05). The data shown are fold increased over the basal and presented as the means ± S.E. of four independent determinations. *, p < 0.05 versus control at the same ISO concentration.
Effect of Transgenic Overexpression of Rab4 on β-AR Expression in the Plasma Membrane
Preceding data indicate that increased Rab4 function facilitates recycling of internalized endogenous β-AR in cultured HL-1 myocytes. To determine whether increased Rab4 function could influence β-AR recycling in cardiac myocytes in vivo, we generated transgenic mice cardiac-specifically expressing FLAG-tagged Rab4. Transgenic mice were identified by Southern blot and PCR analyses of genomic DNA extracted from mouse tails. Rab4 expression in the ventricles of Rab4 transgenic and NTG mice was determined by Western blot analysis using anti-Rab4 and FLAG antibodies. Rab4 expression in transgenic mouse ventricles was increased by about 12-fold compared with NTG siblings (Fig. 4A). Expression of Rab1 and Rab5 was the same in Rab4 transgenic and NTG mouse ventricles (Fig. 4A), indicating that cardiac overexpression of Rab4 had no compensatory effects on the expression of other closely related Rab GTPases.
FIGURE 4. Effect of transgenic expression of Rab4 in the myocardium on the density of β-AR in the plasma membrane.

A, immunoblot analysis of Rab expression levels in hearts from transgenic mice overexpressing Rab4 and NTG mice. Fifty μg of total homogenate prepared from ventricles of four Rab4 and four NTG mice was separated by 12% SDS-PAGE and transferred onto a polyvinylidene difluoride membrane. Rab4 expression levels were detected by Western blotting using anti-Rab4 and anti-FLAG antibodies and Rab1 and Rab5 expression by Rab-isoform specific antibodies. B, immunoblot analysis of the ER marker calregulin, the Golgi marker GM130, and the plasma membrane marker Na+-K+-ATPase in cytosolic and membrane fractions. Myocardial cytosolic and plasma membrane fractions were prepared by homogenization and centrifugation at 10,000 × g as described under “Experimental Procedures.” Twenty-five μg of protein from the cytosolic and membrane fractions was analyzed. The blots shown are representatives of three separate experiments. C, specific [125I]iodocyanopindolol binding to membrane fractions prepared from Rab4 and NTG mouse ventricles. Twenty-five μg of membrane protein was incubated with the nonselective β-AR ligand [125I]iodocyanopindolol (400 pM) in a total volume of 500 μl of binding buffer for 1 h. Specific binding was performed in duplicate and nonspecific binding determined in the presence of 20 μM alprenolol. Receptor density was expressed as fmol/mg membrane protein and presented as the means ± S.E. (n = 6). *, p < 0.05 versus NTG. D, Expression of Gs, Gi, Gβ, and GRK2 in four Rab4 and four NTG mouse hearts. Fifty μg of total homogenate prepared from mouse ventricles was analyzed.
We next determined whether Rab4 overexpression could alter the density of β-AR in the plasma membrane by radioligand binding. As β-AR are synthesized in the ER and transported to the plasma membrane through the Golgi apparatus, any contamination of the ER and/or the Golgi in the plasma membrane fractions would influence the actual number of the receptors in the plasma membrane. Thus, we first determined whether the plasma membrane preparations contained the ER and/or Golgi by measuring the expression of the ER marker calregulin, the Golgi marker GM130, and the plasma membrane marker Na+-K+-ATPase by immunoblotting. Both calregulin and GM130 were exclusively detected in the cytosolic fraction but not in the membrane fraction, whereas Na+-K+-ATPase was detected in the membrane fraction but not in the cytosolic fraction (Fig. 4B). These data indicate that the membrane preparations did not contain the ER and the Golgi.
β-AR density was significantly increased in the membrane fraction from Rab4 transgenic mouse ventricles by 22% (Fig. 4C) compared with that from NTG. In contrast to the increased β-AR density, expression of Gs, Gi, Gβ, and GRK2, molecules involved in the β-AR signaling, was not altered in Rab4 transgenic mice (Fig. 4D). These data indicate that increased Rab4 function by overexpressing wild-type Rab4 selectively augments β-AR expression in the plasma membrane.
Effect of Transgenic Overexpression of Rab4 on β-AR Signaling
We then determined whether enhanced Rab4 expression in the plasma membrane in vivo could activate β-AR signaling. cAMP production in response to stimulation with ISO was measured using membrane preparations from Rab4 transgenic and NTG mouse ventricles. Consistent with the increased β-AR density in the plasma membrane, ISO-stimulated cAMP production was significantly augmented by 3.3-fold in ventricles from Rab4 transgenic mice as compared with NTG controls (Fig. 5A). cAMP production at basal level was also significantly increased by 1.6-fold in Rab4 transgenic mouse hearts (Fig. 5A). ISO-promoted cAMP production is doubled in Rab4 transgenic mice compared with NTG controls (Fig. 5B). In contrast to ISO stimulation, cAMP production in response to forskolin stimulation was the same in Rab4 transgenic and NTG controls (Fig. 5A). Together with no change observed at the expression level of Gs, Gi, Gβ, and GRK2, these data suggest that increased β-AR signaling in the Rab4 transgenic mouse heart is solely due to the increased plasma membrane expression of β-AR.
FIGURE 5. cAMP production in Rab4 transgenic and NTG mice.
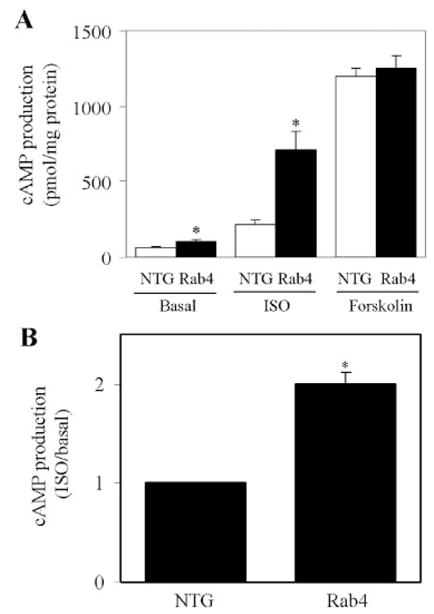
A, ventricular membrane fractions from Rab4 transgenic and NTG mice were prepared as described in the legend of Fig. 4. cAMP production in response to stimulation with ISO (10 μM) and forskolin (100 μM) in the presence of the phosphodiesterase inhibitor 3-isobutyl-1-methylxanthine (0.5 mM) was measured as described under “Experimental Procedures.” The data shown are the means ± S.E. of four Rab4 transgenic and four NTG mice. *, p < 0.05 versus respective NTG. B, ISO-induced cAMP production in NTG and Rab4 transgenic mice. The data are presented as fold increase over the basal values in ventricle membrane fractions (n = 4; *, p < 0.05 versus NTG).
Effect of Transgenic Overexpression of Rab4 on Cardiac Hypertrophy and Function
The absolute heart and ventricle weights were significantly increased in Rab4 transgenic mice at 22 weeks old as compared with age-matched NTG controls (heart weight: NTG, 0.15 ± 0.01 and Rab4, 0.18 ± 0.01 g, n = 12, p < 0.05; ventricle weight: NTG, 0.13 ± 0.01 and Rab4, 0.16 ± 0.01 g, n = 12, p < 0.05). There was no difference in body weight between Rab4 transgenic and NTG mice (NTG, 32.4 ± 1.1 and Rab4, 31.2 ± 0.7 g), resulting in an increase in heart and ventricle weight-to-body weight ratio in the Rab4 mice (Fig. 6A). In contrast, the lung and liver weights and their index to body weight were the same between the Rab4 transgenic and NTG mice (lung weight: NTG, 0.21 ± 0.02 and Rab4, 0.22 ± 0.02 g; liver weight: NTG, 1.50 ± 0.29 and Rab4, 1.57 ± 0.29 g). Pathological and histological examination of Rab4 transgenic mouse hearts showed an enlargement in left ventricular wall thickness without cardiomyocyte necrosis, myocardial fibrosis, and myofibrillar disarray (Fig. 6, B and C). Left ventricular myocyte sizes in Rab4 transgenic mice were enlarged (Fig. 6C). These data indicate that increased expression of Rab4 GTPase in myocardium induces cardiac hypertrophy.
FIGURE 6. Effect of Rab4 overexpression on cardiac hypertrophy and function.
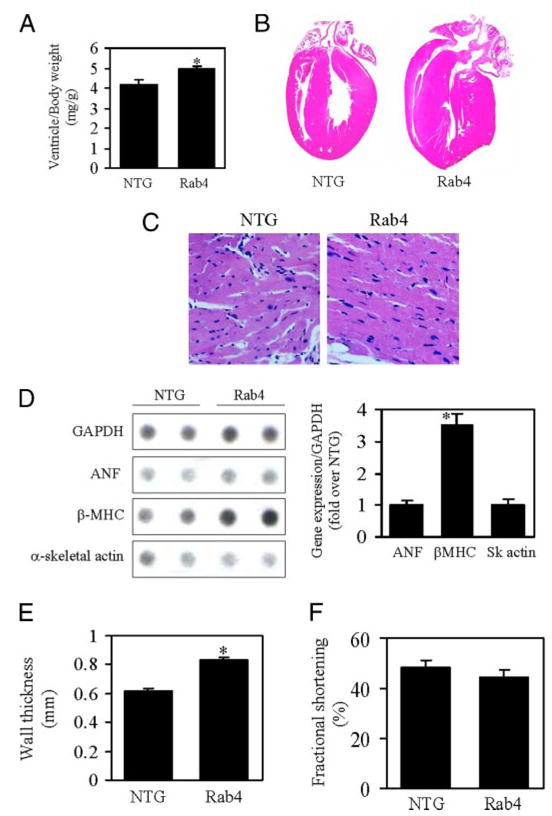
A, ventricle/body weight in Rab4 transgenic and NTG mice. B, representative cross-sections of hearts stained with hematoxylin and eosin from Rab4 transgenic and NTG mice at 22 weeks of ages. The heart from Rab4 transgenic mouse shows concentric remodeling with thicker left ventricular wall and smaller left ventricular chamber. C, histological analysis of left ventricles stained with Masson’s trichrome from transgenic and NTG mice, showing decreased myocyte number per microscopic field and enlarged myocyte size in Rab4 transgenic mice. D, effect of Rab4 overexpression on the expression of cardiac hypertrophy-associated genes. Representative RNA dot blots from two NTG and two Rab4 transgenic mice are shown (left panel). Total RNA was extracted from ventricles of NTG and transgenic mice. RNA dot blotting was carried out as described under “Experimental Procedures” using 3 μg of RNA per dot. Quantitative data of mRNA expression normalized to glyceraldehyde-3-phosphate dehydrogenase (GAPDH) (right panel) are shown. E, echocardiographic analysis of posterior wall thickness of NTG and Rab4 transgenic mouse hearts. F, left ventricular fractional shortening in NTG and Rab4 transgenic mice. Values are the means ± S.E. (n = 7–12; *, p < 0.05 versus NTG).
Increased expression of cardiac fetal genes is associated with cardiac hypertrophy. To determine whether Rab4 overexpression-induced cardiac hypertrophy, as reflected by increased cardiac mass, is accompanied by an increased expression of hypertrophy-associated genes, we quantified the expression of atrial netriuretic peptide, β-MHC and α-skeletal actin by RNA dot blot. β-MHC expression normalized to the mRNA expression of glyceraldehyde-3-phosphate dehydrogenase was increased by 3.5-fold in Rab4 transgenic mouse hearts as compared with those from NTG mice (Fig. 6D). In contrast, expression of atrial netriuretic peptide and α-skeletal actin genes was not altered. These data indicate that Rab4 overexpression selectively up-regulates expression of β-MHC.
In vivo M-mode echocardiography was used to determine the effect of Rab4 overexpression on left ventricular dimension at end-diastole (EDD) and end-systole (ESD) and posterior left ventricular wall thickness at end-diastole. Consistent with morphological and gravimetric data, posterior wall thickness in Rab4 transgenic mice was significantly increased by ~30% (Fig. 6E), but both ESD and EDD were similar to NTG sibling controls (data not shown). Thus, the ratio of wall thickness to ventricular radius (h/r) was increased (NTG, 0.41 ± 0.02 and Rab4, 0.52 ± 0.03, n = 7–12, p < 0.05), which suggests concentric ventricular remodeling. Left ventricular fractional shortening, a measure of systolic contractile function, was the same in Rab4 transgenic and NTG mice (Fig. 6F). These data suggest that Rab4 overexpression induces concentric hypertrophy with preserved cardiac systolic function.
DISCUSSION
Rab4 GTPase coordinates protein transport from the endosome to the plasma membrane (15–17). Several studies have demonstrated that co-expression of the dominant-negative GDP-bound Rab4 mutant inhibits GPCR transport from the endosome to the plasma membrane after stimulation with their agonists, suggesting that normal Rab4 function is required for the recycling of internalized receptors (9, 18–20). However, the effect of augmentation of Rab4 function on the transport of endogenous GPCRs has not been examined. In this report, we investigated the influence of increased Rab4 function by overexpressing wild-type Rab4 on the transport of endogenous β-AR to the plasma membrane in both cardiac mycoytes in vitro and mouse hearts in vivo.
The most important finding in this report is that Rab4 functions as a rate-limiting factor for the transport of endogenous β-AR to the plasma membrane. We first demonstrated that the recycling of β-AR after agonist stimulation was significantly facilitated by transient expression of wild-type Rab4 in HL-1 myocytes, in which β1-AR is the predominant subtype. To define whether Rab4 could enhance β-AR recycling in the mouse heart in vivo, we generated transgenic mice overexpressing Rab4 in the myocardium and determined the effect of increased Rab4 expression on the plasma membrane expression of β-AR. Chronic expression of Rab4 in the mouse heart moderately, but significantly, increased the density of β-AR in the plasma membrane. As the same numbers of HL-1 cells were used for transfection with control and Rab4 plasmids and the size of the myocytes from Rab4 transgenic mouse hearts were enlarged, it is likely that the receptor density is increased in each myocyte expressing wild-type Rab4. Rab4 expression did not alter β-AR expression at the cell surface before ISO stimulation and after complete recycling at 4 h, suggesting that Rab4 did not alter total β-AR expression. In contrast to the β-AR, expression of Rab1, Rab5, Gs, Gi, Gβ, and GRK2 was not affected by Rab4 expression, suggesting that altered Rab4 expression did not influence total protein synthesis. As Rab4 has been well demonstrated to regulate protein transport specifically from the endosomes to the plasma memebrane (6, 7, 9, 15–20), the increase in β-AR in the membrane fraction is presumably due to the facilitated recycling of internalized β-AR from the endosome, rather then modulating other receptor trafficking steps such as export from the ER to the plasma membrane or internalization from the plasma membrane to the endosome. These data demonstrate for the first time that the augmentation of Rab4 function may enhance β-AR targeting to the plasma membrane through facilitating the recycling pathway in cardiac myocytes. These data are also the first demonstration that endogenous Rab4 expression level is a rate-limiting factor for the GPCR superfamily.
To determine whether overexpression of Rab4 could regulate β-AR signaling as a consequence of modifying β-AR recycling, we measured ISO-stimulated cAMP production in HL-1 cardiomyocytes and in membrane preparations from Rab4 transgenic and NTG mouse hearts. cAMP production in HL-1 cells in response to stimulation at the highest concentration of ISO (10−5 M) was doubled compared with the basal level. This increase is similar to the data obtained from human atrial membranes (30). As expected, the increased β-AR density in the plasma membrane led to increase in cAMP production after stimulation with ISO in both cultured myocytes and transgenic mouse hearts overexpressing Rab4. However, Rab4 expression had no effect on the cAMP production in response to forskolin stimulation, suggesting that increased cAMP production in response to ISO by Rab4 expression is not due to the alteration of adenylyl cyclase activity. In addition, Rab4 expression had no effect on the expression of other molecules involved in β-AR signal regulation, including G proteins and GRK2. Therefore, Rab4 overexpression-enhanced cAMP production in cardiomyocytes in vitro and in vivo is likely due to the increase in the β-AR expression in the plasma membrane.
Cardiac specific expression of Rab4 induced mild myocardial hypertrophy with a moderate increase in ventricular weight, enlargement of ventricular myocyte size, β-MHC expression and wall thickness, without deteriorative effect on cardiac performance based on echocardiography analysis. Our data suggest that cardiac hypertrophy observed in the Rab4 mice is possibly attributed to the enhanced β-AR recycling and signaling. However, we cannot exclude that the facilitation of the transport from the endosome to the plasma membrane of other signaling molecules may also contribute to the phenotype observed in transgenic mice overexpressing Rab4. Nevertheless, cardiac hypertrophy observed in Rab4 transgenic mouse hearts suggests that protein transport from the endosome to the plasma membrane may function as a possible regulatory site for control of cardiomyocyte growth. By virtue of the ability to regulate recycling and signaling of GPCRs in cardiac myocytes, Rab4 may play an important role in regulating cardiomyocyte growth and function.
In chronic human heart failure and in many animal models, there are several hallmark alterations in the adrenergic receptor systems that contribute to the loss of cardiac function. These alterations include a decrease in the expression and coupling of β1-AR, a decrease in the coupling of β2-AR, an increase in expression of Gi, an increase in the expression of GRK, and a decrease in the expression and/or function of adenylyl cyclases (2–5). Therefore, the β-AR signal transduction pathway has been intensively manipulated in many transgenic and knockout mouse models for developing strategies to enhance the diminished β-AR function and eventually treat chronic heart failure (3, 31, 32). The strategies to enhance β-AR signaling include using receptor agonists, increasing receptor density in the absence of agonists, blocking receptor desensitization, and expressing constitutively active mutant receptors (33, 34). Rab4 overexpression moderately increased β-AR expression in the plasma membrane, enhanced β-AR signaling, and induced cardiomyocyte growth without clear functional deterioration. These data suggest that facilitating the recycling of internalized β-AR through modifying the function of components of the vesicular transport machinery (e.g. Rab4 GTPase) has therapeutic potential as an alternative strategy to enhance receptor signaling and possibly improve myocardial function in heart failure.
Elucidation of the functional role of individual Rab GTPases in the regulation of the intracellular trafficking and signal transduction of GPCRs has just started. We have demonstrated that the transport of distinct GPCRs to the cell surface may be differentially modulated by Rab1 (22) and transgenic expression of Rab1 in the myocardium induced pathologic hypertrophic phenotypes, which is considerably different from that observed in Rab4 transgenic mice (21). Rab5 regulates the endocytic trafficking from the plasma membrane to the endosome of GPCRs including β2-AR, angiotensin II type 1, dopamine D2, μ-opioid, m4 muscarinic acetylcholine, and neurokinin 1 receptors (19, 35–37). Rab4 modulates the recycling of internalized GPCRs including β2-AR, neurokinin 1, and CB1 cannabinoid receptors (9, 18–20). We demonstrated here that recycling of β1-AR, a predominant β-AR subtype in HL-1 myocytes, is also modulated by Rab4. Similar to Rab4, Rab11 also participates in the regulation of the recycling of internalized GPCRs (7, 9, 35). Moreover, Rab7 may be involved in the targeting of GPCRs to the lysosome for degradation (7, 35). Therefore, defining the functional role of individual Rab GTPases in cardiomyocyte growth by modifying the transport of selective GPCRs at distinct steps may provide a novel foundation for the development of strategies in treating cardiac disease.
Acknowledgments
We are grateful to George Wien and Michele Smith (Toshiba America Medical Systems, Tustin, CA) for assistance in ultrasound studies.
Footnotes
This work was supported in part by National Institutes of Health Award (“Mentoring in Cardiovascular Biology”) 1P20RR018766 (program director: Stephen M. Lanier, Ph.D.) and by Louisiana Board of Regents Grant LEQSF (2002-05)-RD-A-18 (to G. W.). The costs of publication of this article were defrayed in part by the payment of page charges.
The abbreviations used are: AR, adrenergic receptor; ER, endoplasmic reticulum; GPCR, G protein-coupled receptor; NTG, non-transgenics; MHC, myosin heavy chain; EDD, end diastolic dimension; ESD, end systolic dimension; GRK, G protein receptor kinase; ISO, isoproterenol.
References
- 1.Xiang Y, Kobilka BK. Science. 2003;300:1530–1532. doi: 10.1126/science.1079206. [DOI] [PubMed] [Google Scholar]
- 2.Post SR, Hammond HK, Insel PA. Annu Rev Pharmacol Toxicol. 1999;39:343–360. doi: 10.1146/annurev.pharmtox.39.1.343. [DOI] [PubMed] [Google Scholar]
- 3.Freeman K, Lerman I, Kranias EG, Bohlmeyer T, Bristow MR, Lefkowitz RJ, Iaccarino G, Koch WJ, Leinwand LA. J Clin Invest. 2001;107:967–974. doi: 10.1172/JCI12083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Communal C, Singh K, Sawyer DB, Colucci WS. Circulation. 1999;100:2210–2212. doi: 10.1161/01.cir.100.22.2210. [DOI] [PubMed] [Google Scholar]
- 5.Lefkowitz RJ, Rockman HA, Koch WJ. Circulation. 2000;101:1634–1637. doi: 10.1161/01.cir.101.14.1634. [DOI] [PubMed] [Google Scholar]
- 6.Ulloa-Aguirre A, Janovick JA, Brothers SP, Conn PM. Traffic. 2004;5:821–837. doi: 10.1111/j.1600-0854.2004.00232.x. [DOI] [PubMed] [Google Scholar]
- 7.Duvernay MT, Filipeanu CM, Wu G. Cell Signal. 2005;17:1457–1465. doi: 10.1016/j.cellsig.2005.05.020. [DOI] [PubMed] [Google Scholar]
- 8.Krupnick JG, Benovic JL. Annu Rev Pharmacol Toxicol. 1998;38:289–319. doi: 10.1146/annurev.pharmtox.38.1.289. [DOI] [PubMed] [Google Scholar]
- 9.Seachrist JL, Anborgh PH, Ferguson SS. J Biol Chem. 2000;275:27221–27228. doi: 10.1074/jbc.M003657200. [DOI] [PubMed] [Google Scholar]
- 10.Hausdorff WP, Caron MG, Lefkowitz RJ. FASEB J. 1990;4:2881–2889. [PubMed] [Google Scholar]
- 11.Wu G, Krupnick JG, Benovic JL, Lanier SM. J Biol Chem. 1997;272:17836–17842. doi: 10.1074/jbc.272.28.17836. [DOI] [PubMed] [Google Scholar]
- 12.Morrison KJ, Moore RH, Carsrud ND, Trial J, Millman EE, Tuvim M, Clark RB, Barber R, Dickey BF, Knoll BJ. Mol Pharmacol. 1996;50:692–699. [PubMed] [Google Scholar]
- 13.Hertel C, Staehelin M. J Cell Biol. 1983;97:1538–1543. doi: 10.1083/jcb.97.5.1538. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Bohm SK, Grady EF, Bunnett NW. Biochem J. 1997;322:1–18. doi: 10.1042/bj3220001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Takai Y, Sasaki T, Matozaki T. Physiol Rev. 2001;81:153–208. doi: 10.1152/physrev.2001.81.1.153. [DOI] [PubMed] [Google Scholar]
- 16.Rosenfeld JL, Knoll BJ, Moore RH. Receptors Channels. 2002;8:87–97. [PubMed] [Google Scholar]
- 17.Seachrist JL, Ferguson SS. Life Sci. 2003;74:225–235. doi: 10.1016/j.lfs.2003.09.009. [DOI] [PubMed] [Google Scholar]
- 18.Roosterman D, Cottrell GS, Schmidlin F, Steinhoff M, Bunnett NW. J Biol Chem. 2004;279:30670–30679. doi: 10.1074/jbc.M402479200. [DOI] [PubMed] [Google Scholar]
- 19.Leterrier C, Bonnard D, Carrel D, Rossier J, Lenkei Z. J Biol Chem. 2004;279:36013–36021. doi: 10.1074/jbc.M403990200. [DOI] [PubMed] [Google Scholar]
- 20.Odley A, Hahn HS, Lynch RA, Marreez Y, Osinska H, Robbins J, Dorn GW., 2nd Proc Natl Acad Sci U S A. 2004;101:7082–7087. doi: 10.1073/pnas.0308335101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Wu G, Yussman MG, Barrett TJ, Hahn HS, Osinska H, Hilliard GM, Wang X, Toyokawa T, Yatani A, Lynch RA, Robbins J, Dorn GW., II Circ Res. 2001;89:1130–1137. doi: 10.1161/hh2401.100427. [DOI] [PubMed] [Google Scholar]
- 22.Wu G, Zhao G, He Y. J Biol Chem. 2003;278:47062–47069. doi: 10.1074/jbc.M305707200. [DOI] [PubMed] [Google Scholar]
- 23.Filipeanu CM, Zhou F, Claycomb WC, Wu G. J Biol Chem. 2004;279:41077–41084. doi: 10.1074/jbc.M405988200. [DOI] [PubMed] [Google Scholar]
- 24.Wozniak M, Saunders C, Schramm N, Keefer JR, Limbird LE. Methods Enzymol. 2002;343:530–544. doi: 10.1016/s0076-6879(02)43156-4. [DOI] [PubMed] [Google Scholar]
- 25.Schwinger RHG, Böhm M, Erdmann E. Am Heart J. 1990;119:899–904. doi: 10.1016/s0002-8703(05)80329-1. [DOI] [PubMed] [Google Scholar]
- 26.Sato M, Gettys TW, Lanier SM. J Biol Chem. 2004;279:13375–13382. doi: 10.1074/jbc.M312660200. [DOI] [PubMed] [Google Scholar]
- 27.van den Hoff MJ, Deprez RH, Monteiro M, de Boer PA, Charles R, Moorman AF. J Mol Cell Cardiol. 1997;29:629–639. doi: 10.1006/jmcc.1996.0306. [DOI] [PubMed] [Google Scholar]
- 28.Sahn DJ, DeMaria AN, Kisslo J, Weyman AE. Circulation. 1978;58:1072–1083. doi: 10.1161/01.cir.58.6.1072. [DOI] [PubMed] [Google Scholar]
- 29.Claycomb WC, Lanson NA, Jr, Stallworth BS, Egeland DB, Delcarpio JB, Bahinski A, Izzo NJ., Jr Proc Natl Acad Sci U S A. 1998;95:2979–2984. doi: 10.1073/pnas.95.6.2979. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Kilts JD, Gerhardt MA, Richardson MD, Sreeram G, Mackensen GB, Grocott HP, White WD, Davis RD, Newman MF, Reves JG, Schwinn DA, Kwatra MM. Circ Res. 2000;87:705–709. doi: 10.1161/01.res.87.8.705. [DOI] [PubMed] [Google Scholar]
- 31.Milano CA, Allen LF, Rockman HA, Dolber PC, McMinn TR, Chien KR, Johnson TD, Bond RA, Lefkowitz RJ. Science. 1994;264:582–586. doi: 10.1126/science.8160017. [DOI] [PubMed] [Google Scholar]
- 32.Ping P, Gelzer BR, Roth DA, Kiel D, Insel PA, Hammond HK. J Clin Invest. 1995;95:1271–1280. doi: 10.1172/JCI117777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Tong H, Rockman HA, Koch WJ, Steenbergen C, Murphy E. Circ Res. 2004;94:1133–1141. doi: 10.1161/01.RES.0000126048.32383.6B. [DOI] [PubMed] [Google Scholar]
- 34.Rockman HA, Chien KR, Choi DJ, Iaccarino G, Hunter JJ, Ross J, Jr, Lefkowitz RJ, Koch WJ. Proc Natl Acad Sci U S A. 1998;95:7000–7005. doi: 10.1073/pnas.95.12.7000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Dale LB, Seachrist JL, Babwah AV, Ferguson SS. J Biol Chem. 2004;279:13110–13118. doi: 10.1074/jbc.M313333200. [DOI] [PubMed] [Google Scholar]
- 36.Iwata K, Ito K, Fukuzaki A, Inaki K, Haga T. Eur J Biochem. 1999;263:596–602. doi: 10.1046/j.1432-1327.1999.00549.x. [DOI] [PubMed] [Google Scholar]
- 37.Mace G, Miaczynska M, Zerial M, Nebreda AR. EMBO J. 2005;24:3235–3246. doi: 10.1038/sj.emboj.7600799. [DOI] [PMC free article] [PubMed] [Google Scholar]