

Abstract
Type 1 diabetes (T1D) is the result of an autoimmune destruction of pancreatic β cells. The cellular and molecular defects that cause the disease remain unknown. Pluripotent cells generated from patients with T1D would be useful for disease modeling. We show here that induced pluripotent stem (iPS) cells can be generated from patients with T1D by reprogramming their adult fibroblasts with three transcription factors (OCT4, SOX2, KLF4). T1D-specific iPS cells, termed DiPS cells, have the hallmarks of pluripotency and can be differentiated into insulin-producing cells. These results are a step toward using DiPS cells in T1D disease modeling, as well as for cell replacement therapy.
Keywords: β cell, disease model, autoimmune, directed differentiation, endoderm
The study of human disease is often hindered by the lack of a good model system. The initiation of the primary disease process often occurs long before the patient shows any sign of disease. Also, relevant patient tissue can be limited and difficult to obtain. Although rodent models can give valuable insights, these rarely fully recapitulate the human disease. Although the nonobese diabetic (NOD) mouse has been enormously useful, there are justifiable concerns regarding its validity as a model for human type 1 diabetes (T1D) (1–3). Recently, human induced pluripotent stem (iPS) cells with disease genotypes have been generated as a tool for human disease modeling (4–7). Disease-relevant cell types can be generated via in vitro differentiation protocols, and in the best cases, these protocols enable an in vitro analysis of the disease pathology. Although ES cells are the gold standard for pluripotent stem (PS) cells, ES cells only model diseases that can be diagnosed or predicted by simple Mendelian genetics [e.g., cystic fibrosis (8) and Fanconi Anemia (9)]. Our focus lies in understanding T1D, a disease with complex underlying genetics and unidentified environmental triggers. For T1D, as well as other multigenic diseases, iPS cells are the best starting point, because they are derived from patient cells and, thereby, capture the disease genotype in a stem cell. To this date, it is not clear whether different forms of type 1 diabetes exist, and how genetics and environment factor influence each other in disease onset and progression.
T1D results from the destruction of insulin-producing β cells by the body's own immune system. A cure could be achieved by combining β cell replacement therapy with induction of tolerance to such cells. Cell replacement therapy for T1D requires a source of glucose-responsive, insulin-secreting cells. Promising results have been obtained by transplantation of pancreatic islets of Langerhans or pancreatic tissue, but this approach is circumscribed by the limited and irregular supply of cadaveric donor tissue, as well as the risks of treatment with immunosuppressant drugs (10, 11). An alternative source of insulin-producing cells are PS cells that can be differentiated into pancreatic β cells. For example, ES cells have been differentiated in monolayer culture along the endodermal lineage toward insulin-producing cells (12, 13). However, β cells derived from immunologically unmatched ES cells will likely be the targets of both allograft reactions and the autoimmune response that caused the initial β cell destruction.
Mouse and human fibroblasts can be used to generate iPS cells (14–16). Recently, iPS cells have been generated from fibroblasts obtained from patients with various diseases (4–7), but not for T1D. T1D-specific iPS (DiPS) cells derived from patients offer several significant advantages. First, DiPS cells would unquestionably contain the genotype responsible for the human disease. Second, DiPS cells would provide an immunologically matched autologous cell population, although dependent on improvements in differentiation protocols. Third, and the present focus of our work, patient-specific cells make possible patient-specific disease modeling wherein the initiation and progression of this poorly understood disease can be studied. Because DiPS cells can be manipulated and studied in vitro, one should be able to assess how the different cell types, including differentiated β cells, and immunocytes interact to produce a pathological phenotype. The purpose of the present study was to derive DiPS cells from patients with T1D, and to test whether these cells can be differentiated into the major target cell type, the pancreatic β cell. Extending this approach to all cell types involved in T1D could lead to an understanding of the root causes of the disease and to the development of effective prophylactic and therapeutic strategies.
Results
Generation of iPS Cells from Patients with T1D.
Skin biopsies were obtained from two Caucasian males with T1D of 11- and 27-years duration, respectively. Patient 1 was presented at age 21 with polyuria, polydypsia, and a blood glucose concentration of 680 mg/dL. Patient 2 was presented at age 3 in diabetic ketoacidosis requiring hospitalization. For both individuals, the body mass index did not exceed 22 or 23, respectively. HLA haplotypes, and other clinical data, are given in Table 1. Fibroblasts obtained from skin biopsies were cultured and infected with a combination of retroviruses encoding the transcription factors OCT4, SOX2, and KLF4 (15). Starting 4 weeks after infections, colonies were picked based on their morphological resemblance to human ES cell colonies and expanded. To test whether the cell lines expressed pluripotency markers, we verified the presence of alkaline phosphatase (AP) activity, as well as staining for antibodies to OCT4, NANOG, SOX2, TRA1-60, TRA1-81, and SSEA4. The reprogrammed cells were positive for AP activity, and were reactive to antibodies against all pluripotency markers tested (Fig. 1 A and B). Karyotype analysis and DNA fingerprinting verified that the T1D patient-specific DiPS lines were generated from the parental fibroblast line and maintained a normal karyotype (Fig. S1 and Table S1).
Table 1.
Patient information
| Patient | Race | Age/sex | Diagnosed at age of | Family history | History of ketoacidosis | Body mass index | Medication | Hemoglobin A1C, % | HLA |
|---|---|---|---|---|---|---|---|---|---|
| H1 | Caucasian | 32/male | 21 | First cousin with T1D | Yes | 22 | Insulin 45 U/day sub. cut. | 6.6 | DR1/4 |
| H2 | Caucasian | 30/male | 3 | None | Yes | 23 | Insulin 60 U/day sub. cut. | 7.2 | DR1/13 |
Fig. 1.

Generation of DiPS cells from T1D patients. DiPS lines were established from two T1D affected patient fibroblasts lines H1 (A) and H2 (B). Displayed are DiPS lines (A) H1.5 and (B) H2.4. Detection of AP activity and immunofluorescence analyses for presence of pluripotency markers OCT4, SSEA4, NANOG, TRA1-60, SOX2, and TRA1-81 are indicated. For immunofluorescence stains corresponding nuclear stains (DAPI) visualize all cells including mouse embryonic fibroblast feeder cells.
The analysis of pluripotency markers was extended to include expression of the endogenous genes encoding NANOG, OCT4, TERT, REX1, SOX2, and GDF3 by semiquantitative PCR analysis. Expression of these genes in the DiPS lines was similar to human ES cells, but was absent in the parental fibroblasts (Fig. 2A). As previously described, KLF4 is expressed in fibroblasts, as well as in human ES cells and DiPS cells (Fig. 2A) (15). Expression of β-ACTIN was used as a control for RNA recovery and to allow semiquantitative comparison of expression levels. Omission of the reverse transcription reaction was used as a control for specificity and gave no bands of expected size in the semiquantitative PCRs (Fig. 2A). A more global view of the gene expression profiles for the parental fibroblasts, DiPS, and hES lines, was obtained using DNA microarrays. Hierarchical clustering revealed that DiPS cell lines from both patients were highly similar to the human ES cell lines HUES4, HUES6, and HUES8 (17), while exhibiting low similarity to fibroblasts (Fig. 2B). The coefficient of determination (r2 = square of the correlation coefficient) was 0.72–0.74 for DiPS compared with parental fibroblasts, and 0.94–0.98 for DiPS compared with HUES cells (Table S2). We conclude that DiPS cells closely resemble human ES cells in global gene expression as we have described for iPS from primary human cells before (18).
Fig. 2.
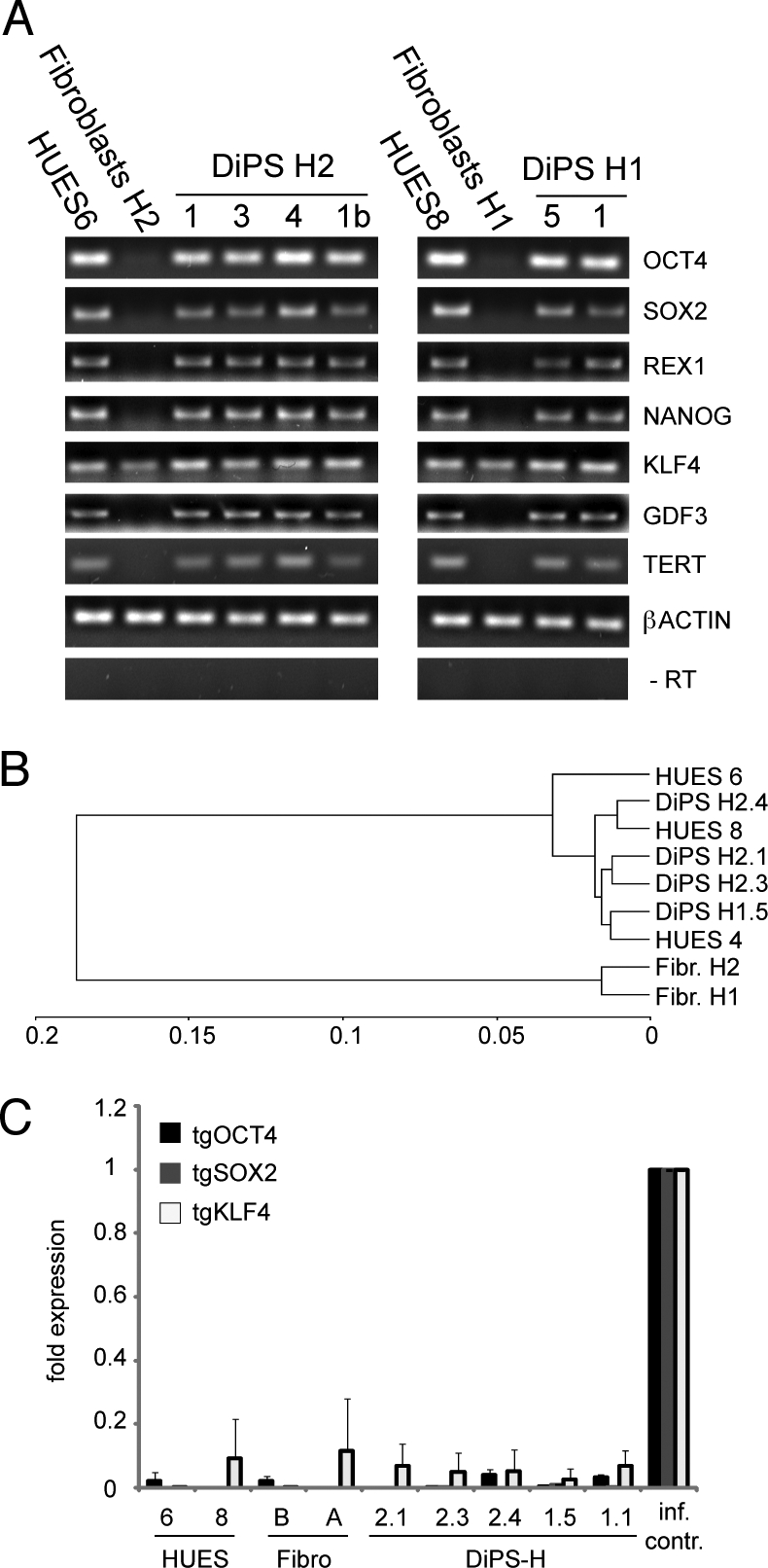
Expression analysis of patient specific DiPS cells. (A) Semiquantitative analysis of expression of OCT4, SOX2, REX1, NANOG, KLF4, GDF3, and β ACTIN. Control PCR (no RT) is included. (B) Hierarchical cluster analysis of different DiPS, HUES and fibroblast lines. (C) Quantitative assessment of viral transgene expression (tgOCT4, tgSOX2, and tgKLF4) levels. Viral transgene expression was normalized to control infected BJ fibroblasts (isolation occurred 7 days post infection). Uninfected HUES and fibroblast lines were used as controls. The experiment was performed in duplicates and the error bars represent SD.
To determine whether the viral transgenes were silenced, we performed exogenous gene-specific quantitative PCR analysis. Compared with the infected fibroblast control, the transgene expression in the DiPS cells was low or at background levels (Fig. 2C), presumably due to anticipated viral gene silencing (15). We conclude that the DiPS lines closely resemble human ES cells in their expression profiles and, like other reported iPS cells, have silenced the transgenes.
DiPS Cells Spontaneously Differentiate into Cell Types of Different Germ Layers.
DiPS cells were allowed to spontaneously differentiate in embryoid body (EB) cultures. EB formation was achieved by culturing DiPS cells in differentiation media on low-attachment plates, followed by plating onto gelatin-coated dishes for additional culture. We analyzed the morphologically differentiated cells for expression of markers for the endodermal (SOX17 and FOXA2), mesodermal (SMA), and ectodermal (TUJ1) lineages in differentiated cultures (Fig. 3A). The differentiated DiPS cells were found positive for cells of all three germ layers. Also, we verified pluripotency of DiPS cells in teratoma formation assays. After injection of DiPS cells into immunocompromised mice, DiPS cells formed teratomas containing derivatives of endoderm (glandular structures), mesoderm (cartilage), and ectoderm (nerve fibers, pigmented epithelium, and melanocytes) (Fig. 3B). We conclude that patient-specific DiPS cells can spontaneously differentiate into derivatives of all three germ layers.
Fig. 3.

Spontaneous differentiation of DiPS cells into cells of different germ layer origin. (A) In vitro differentiation of DiPS lines H1.5, H2.1, and H2.4 in EB assays was followed by monolayer culture and immunostaining for markers of ectoderm (TUJ1), mesoderm (SMA), and endoderm (FOXA2 and SOX17). An overlay with a nuclear stain (DAPI) is displayed. (B) Teratoma formation occurred after injection of DiPS into immunocompromised mice. Hematoxylin and Eosin staining of teratoma sections shows nerve fibers (N), melanocytes (M), pigmented epithelium (P), cartilage (C), and glandular structures (G).
DiPS Cells Can Be Differentiated Along the Endodermal/Pancreatic Lineage.
We applied a directed differentiation protocol to the DiPS cells to determine whether they can be differentiated toward an insulin producing/glucose responsive cell. The protocol followed a stepwise differentiation protocol that relies on the generation of intermediate precursors thought to be similar to populations present in the developing embryo. DiPS cells were subjected to a protocol that directs differentiation to definitive endoderm followed by gut tube endoderm before further differentiation into pancreatic progenitors, and finally, the β-like cell (Fig. 4A). As determined by immunofluorescence analysis for SOX17 and FOXA2 expression, DiPS lines from both patients could respond to WNT3A and Activin A treatment to differentiate into definitive endoderm, similar to human ES cells (Fig. 4B Upper). Further differentiation toward gut tube endoderm (HNF4a and HNF1b positive), and pancreatic progenitors (PDX1 and HNF6 positive) was achieved by supplying FGF10 and cyclopamine, and FGF10, cyclopamine, retinoic acid, and (−)-Indolactam V, respectively. FOXA2 expression was detected in both definitive endoderm and pancreatic progenitors (Fig. 4B) as expected from normal embryonic development (19). The expression of these transcription factors was validated by semiquantitative PCR (Fig. 4C).
Fig. 4.

Stepwise differentiation of ES/DiPS cells toward β-like cells. (A) Schematic representation of stepwise differentiation of human PS cells to β-like cells. DiPS cell lines H1.5, H2.1, and H2.4 differentiation to definitive endoderm (DE), gut tube endoderm (GTE) and pancreatic progenitors (PPs) indicated by (B) immunostaining and (C) RT-PCR. SOX, SRY (sex determining region Y)-box; FOXA2, forkhead box protein A2; HNF, hepatocyte nuclear factor; PDX1, pancreatic and duodenal homeobox 1; HB9, homeobox gene HLXB9; NKX6.1, NK6 transcription factor related, locus 1.
Further differentiation of DiPS-derived pancreatic progenitors toward the endocrine lineage yielded cells that were positive for somatostatin, glucagon, and insulin (Fig. 5A). Insulin-positive cells were C-peptide positive, excluding insulin uptake from the media. Semiquantitative PCR analysis confirmed the expression of endocrine-specific gene products of INSULIN, PDX1, NKX2.2, GLUCAGON, and SOMATOSTATIN (Fig. 5B). To address whether the insulin could be released on glucose stimulation in vitro, we exposed the DiPS-derived insulin producing cells to low or high concentrations of glucose. The DiPS-derived population released human C-peptide on glucose stimulation (Fig. 5C). The amount of released C-peptide after high (20 mM) glucose stimulation was at least 5-fold higher than after low (2.5 mM) glucose stimulation. We conclude that DiPS cells can be differentiated to insulin producing/glucose-responsive cells.
Fig. 5.

DiPS cell lines H1.5, H2.1, and H2.4 differentiate to hormone-expressing endocrine cells indicated by (A) immunostaining and (B) semiquantitative PCR. (C) The DiPS-derived C-peptide-expressing cells secreted C-peptide on glucose stimulation. The DiPS-derived populations were stimulated with 2.5 and 20 mM D-glucose, and the amount of human C-peptide released to culture supernatant was analyzed by ELISA. C-PEP, C-peptide; INS, insulin; GLU, glucagon; SS, somatostatin.
Discussion
Insight into the pathogenesis of T1D comes largely from rodent models such as the NOD mouse or the BB rat. However, the existing rodent models are not fully representative of the relevant processes in patients with T1D. Indeed, insights from rodent models have frequently not translated well to the clinic (1–3). Access to genetically-predisposed human cells whose biological status ultimately defines the disease enables previously impossible mechanistic studies in vitro, and in vivo transplant systems. We have generated human PS cells from two patients with T1D. These DiPS cells provide a starting material for patient-specific disease modeling and for testing differentiation protocols. In mice, inclusion of c-Myc as a fourth reprogramming factor is associated with lethal tumor formation in contrast to reprogramming with three factors (Oct4, Sox2, and Klf4) (20). The reprogramming process described here was achieved by using three factors (OCT4, SOX2, and KLF4), omitting the oncogene C-MYC. Consistent with previously described iPS cell lines, transgene silencing was observed in DiPS cells (4, 15, 21, 22). Induced PS cells can be generated from mouse cells without permanent or transient integration of the transforming factors in the genome (23, 24), and this approach has recently extended to human cells (25). Currently, generation of human iPS cells involves delivery of DNA in a manner that allows potential integration into the genome, but alternative approaches are likely to be available in the near future (26). In any event, insufficient characterization of the reprogramming process and its product precludes use of iPS cells in cell replacement therapy at this point. However, the generation of patient-specific PS cells paired with differentiation into cell types relevant to the disease promises to provide valuable insights into disease pathogenesis. The DiPS cells described here are pluripotent based on similarities in gene expression to human ES cells and their ability to spontaneously differentiate into cells of different germ layers. Notably, we have differentiated these patient-specific DiPS cell lines to a cell population relevant to T1D, an insulin-producing and glucose-responsive β-like cell. Previously, iPS cells generated from foreskin fibroblasts, but not from diabetes patients, using four factor reprogramming (OCT4, SOX2, KLF4, and C-MYC) were shown to differentiate to insulin-producing clusters (27), leaving open the task of generating patient-specific DiPS, and testing their potential to differentiate toward insulin-producing β-like cells. Although DiPS cells can be differentiated to insulin producing cells, the efficiency of this process is low, possibly because the differentiation protocol has not been optimized or due to variation in the differentiation propensities of human PS cells (28). Interestingly, we observed differences between DiPS lines from the same patient, potentially due to transgene reactivation or incomplete silencing. As a consequence, characterization of multiple lines and future efforts to generate DiPS cells without viral integration will help address this issue.
As observed with human ES cells, β-like cells derived from DiPS are glucose responsive, but until the differentiation protocols are improved, it is not yet possible to directly compare these cells with purified pancreatic β cells. In addition to variation in differentiation propensities, potential deviations in T1D disease onset and progression will require the generation of multiple DiPS lines to reflect the human population afflicted with T1D. The two cell lines from different patients described here represent a starting point for this larger task.
Differentiation of DiPS cells to β-like cells is relevant not only for the long term possibility of autologous cell replacement therapy, but also for disease modeling. We propose to extend this investigation to generate cell types of the immune system that may allow the generation of a cellular interaction model of T1D. This approach should provide a way to investigate T1D disease onset and progression in vitro and/or in reconstituted animal models. These in vitro and in vivo systems will also be useful for testing of preventative and therapeutic strategies.
Materials and Methods
Cell Culture.
Skin fibroblasts from T1D patients and controls were derived from explants of 3-mm dermal biopsies after informed consent under protocols approved both by Harvard University and Columbia University College of Physicians and Surgeons. Briefly, 3-mm skin biopsies were minced with scalpels into smaller pieces, and tissue fragments were placed into a 60-mm tissue culture dish under a sterile coverslip held down by sterilized silicon grease under one corner. Media was added to completely immerse the coverslip, and dishes were incubated at 37 °C in a humidified incubator (5% CO2). Media was changed every 5 days without disturbing the coverslip. Fibroblasts grew out of the tissue fragments, and when sufficiently numerous, cells were trypsinized and expanded. Subsequently, fibroblasts were maintained in fibroblast medium (DMEM supplemented with 10% FBS, glutamine, sodium pyruvate, nonessential amino acids, and penicillin/streptomycin). The resulting fibroblasts lines are referred to as fibroblast Harvard (H) lines 1 and 2.
Human ES cell and DiPS lines were cultured in human ES media (knockout DMEM supplemented with 10% knockout serum replacement, 10% human plasma fraction, 10 ng/mL bFGF, nonessential amino acids, β-mercaptoethanol, L-glutamine, and penicillin/streptomycin). Cultures were maintained on mouse embryonic fibroblast feeders and passaged enzymatically using either 0.05% Trypsin (GIBCO) or Collagenase type IV.
Reprogramming.
VSVG-coated retroviruses were generated according to standard procedures. One day before infection, 10E5 fibroblasts were seeded per well of a six well plate. Fibroblasts were infected on days 1 and 2 with a combination of OCT4, SOX2, and KLF4 containing Moloney viruses (constructs were obtained from Addgene) (15). The media was changed on day 3 to DMEM supplemented with 10% FBS, L-glutamine, penicillin/streptomycin, nonessential amino acids, and sodium pyruvate. A day later the cells were split onto gelatinized 10-cm cell culture dishes. Subsequently, the cells were fed every other day with human ES cell media. Generation of DiPS lines H2.4, H2.3, and H2.1b occurred with supplementation of the media with 1 mM valproic acid during the reprogramming process as described before (18), whereas DiPS lines H2.1, H1.1, H1.5 were generated without addition of the chemical. Colonies were picked starting ≈4 weeks after infection.
Spontaneous Differentiation.
Spontaneous differentiation through EB formation was initiated by dissociation of human DiPS cells using collagenase IV treatment, and subsequent transfer to low attachment 6-well plates in knockout DMEM supplemented with 20% knockout serum replacement, nonessential amino acid, β-mercaptoethanol, L-glutamine, and penicillin/streptomycin. After 8–10 days suspension culture, EBs were transferred to gelatin-coated plates and cultured for an additional 8–10 days as attachment culture.
For teratoma formation assays DiPS cells were collected by collagenase IV treatment, and injected s.c. into immunocompromised mice (NOD-SCID or SCID-Beige mice). Teratomas were collected 7–10 weeks after injection, and processed according to standard procedures for paraffin embedding and hematoxylin and eosin staining.
Directed Differentiation.
Directed differentiation was conducted as described (13, 29) with the following modifications: human DiPS cells were cultured on MEF feeder cells to 70–80% confluency, then treated with 25 ng/mL WNT3A (R&D systems) + 100 ng/mL Activin A (R&D systems) in advanced RPMI (A-RPMI; Invitrogen) supplemented with 1×L-Glu and 1×PS for 1 day, followed by treatment with 100 ng/mL Activin A in A-RPMI supplemented with 1×L-Glu, 1×PS and 0.2% FBS (Invitrogen). Two days later, the media was changed to 50 ng/mL FGF10 (R&D systems) + 0.25 μM KAAD-CYC (Calbiochem) in A-RPMI supplemented with 1×L-Glu, 1×PS and 2% FBS and maintained for additional 2 days. Cells were then transferred to 50 ng/mL FGF10 + 0.25 μM KAAD-CYC + 2 μM RA (Sigma) in DMEM supplemented with 1×L-Glu, 1×PS, 1× B27 (Invitrogen) and cultured for an additional 4 days. The media was then changed to 50 ng/mL FGF10 + 300 nM ILV (Axxora) in DMEM supplemented with 1×L-Glu, 1×PS, 1× B27 and cultured for an additional 4 days. Then, cells were transferred to 50 ng/mL EX-4 (Sigma) + 10 μM DAPT (Sigma) in DMEM supplemented with 1×L-Glu, 1×PS, 1× B27 and cultured for an additional 6 days. Cells were then cultured in 50 ng/mL HGF (R&D systems) + 50 ng/mL IGF1 (R&D systems) in CMRL-1066 (Invitrogen) supplemented with 1×L-Glu, 1×PS, 1× B27 for 6 days.
Immunofluorescence.
Immunofluorescence staining was performed using primary antibodies against C-peptide (4020-01; Linco), FOXA2 (07-633; Upstate), glucacon (4031; Linco), HNF6 (sc-13050; Santa Cruz Biotechnology), insulin (A0564; Dako), NANOG (ab21624; Abcam), NKX2.5 (sc-14033; Santa Cruz Biotechnology), OCT4 (sc-5279; Santa Cruz Biotechnology), PDX1 (AF2419; R&D systems), SMA (A5228; Sigma), somatostatin (A0566; Dako), SOX2 (sc-17320; Santa Cruz Biotechnology), SOX17 (AF1924; R&D systems), SSEA4 (MAB4304; Chemicon), TRA-1–60 (MAB4360; Chemicon), TRA-1–81 (MAB4381; Chemicon), and TUJ-1 (MMS-435P; Covance Research Products). Appropriate secondary antibodies were obtained from Molecular Probes.
Gene Expression Analysis.
RNA was isolated from cells using RNAeasy kit (Qiagen). For quantitative and semiquantitative PCR analysis, cDNA synthesis was performed using SuperScript III Reverse Transcriptase and Oligo (dT) primers (Invitrogen). Primers used for amplification are listed in Table S3.
For whole-genome expression analysis, Illumina Total Prep RNA amplification Kit (Ambion) was used according to manufacturer's guideline. Hybridization to Whole-Genome Expression BeadChips (HumanRef-8) was followed by analysis on an Illumina Beadstation 500. All samples were prepared in duplicates. Data analysis was conducted using manufacturer's Beadstudio software.
C-Peptide Release Assay.
C-peptide release was measured by incubating the cells in Krebs–Ringer solution containing bicarbonate and Hepes (KRBH; 129 mM NaCl/4.8 mM KCl/2.5 mM CaCl2/1.2 mM KH2PO4/1.2 mM MgSO4/5 mM NaHCO3/10 mM Hepes/0.1% BSA). The cells were incubated in KRBH buffer for 1 h to wash. The cells were incubated in KRBH buffer with 2.5 mM D-glucose for 1 h and then KRBH buffer with 20 mM D-glucose for 1 h. The C-peptide levels in culture supernatants were measured using the human C-peptide ELISA kit (Alpco Diagnostics).
Supplementary Material
Acknowledgments.
We thank Kevin Eggan for organizational help and discussions, Anastasie Kweudjeu for help with transcriptional arrays, Danwei Huangfu for helpful discussions and experimental advice, Mariko Yamaki and Adriana Tajonar for technical help, Julian McKay-Wiggan, M.D. for performing skin biopsies, and Taylor Armstron and Sunanda Babu (University of Colorado, Aurora, CO) for islet antibody and HLA typing. S.C. is supported by the postdoctoral fellowship from Juvenile Diabetes Research Foundation. D.A.M. is an Investigator of the Howard Hughes Medical Institute. This work was supported by the Harvard Stem Cell Institute (Kurtzig Fund), the Russell Berrie Foundation, the Handler Foundation, and the New York Stem Cell Foundation.
Footnotes
The authors declare no conflict of interest.
See Commentary on page 15523.
This article contains supporting information online at www.pnas.org/cgi/content/full/0906894106/DCSupplemental.
References
- 1.Roep BO. Are insights gained from NOD mice sufficient to guide clinical translation? Another inconvenient truth. Ann N Y Acad Sci. 2007;1103:1–10. doi: 10.1196/annals.1394.018. [DOI] [PubMed] [Google Scholar]
- 2.Roep BO, Atkinson M. Animal models have little to teach us about type 1 diabetes: 1. In support of this proposal. Diabetologia. 2004;47:1650–1656. doi: 10.1007/s00125-004-1517-1. [DOI] [PubMed] [Google Scholar]
- 3.von Herrath M, Nepom GT. Animal models of human type 1 diabetes. Nat Immunol. 2009;10:129–132. doi: 10.1038/ni0209-129. [DOI] [PubMed] [Google Scholar]
- 4.Dimos JT, et al. Induced Pluripotent Stem Cells Generated from Patients with ALS Can Be Differentiated into Motor Neurons. Science. 2008;321:1218–1221. doi: 10.1126/science.1158799. [DOI] [PubMed] [Google Scholar]
- 5.Ebert AD, et al. Induced pluripotent stem cells from a spinal muscular atrophy patient. Nature. 2009;457:277–280. doi: 10.1038/nature07677. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Park IH, et al. Disease-Specific Induced Pluripotent Stem Cells. Cell. 2008;134:877–886. doi: 10.1016/j.cell.2008.07.041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Soldner F, et al. Parkinson's disease patient-derived induced pluripotent stem cells free of viral reprogramming factors. Cell. 2009;136:964–977. doi: 10.1016/j.cell.2009.02.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Pickering SJ, et al. Generation of a human embryonic stem cell line encoding the cystic fibrosis mutation deltaF508, using preimplantation genetic diagnosis. Reprod Biomed Online. 2005;10:390–397. doi: 10.1016/s1472-6483(10)61801-9. [DOI] [PubMed] [Google Scholar]
- 9.Verlinsky Y, et al. Human embryonic stem cell lines with genetic disorders. Reprod Biomed Online. 2005;10:105–110. doi: 10.1016/s1472-6483(10)60810-3. [DOI] [PubMed] [Google Scholar]
- 10.Naftanel MA, Harlan DM. Pancreatic islet transplantation. PLoS Med. 2004;1:e58. doi: 10.1371/journal.pmed.0010058. quiz e75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Lakey JR, Mirbolooki M, Shapiro AM. Current status of clinical islet cell transplantation. Methods Mol Biol. 2006;333:47–104. doi: 10.1385/1-59745-049-9:47. [DOI] [PubMed] [Google Scholar]
- 12.D'Amour KA, et al. Production of pancreatic hormone-expressing endocrine cells from human embryonic stem cells. Nat Biotechnol. 2006;24:1392–1401. doi: 10.1038/nbt1259. [DOI] [PubMed] [Google Scholar]
- 13.Kroon E, et al. Pancreatic endoderm derived from human embryonic stem cells generates glucose-responsive insulin-secreting cells in vivo. Nat Biotechnol. 2008;26:443–452. doi: 10.1038/nbt1393. [DOI] [PubMed] [Google Scholar]
- 14.Takahashi K, Yamanaka S. Induction of pluripotent stem cells from mouse embryonic and adult fibroblast cultures by defined factors. Cell. 2006;126:663–676. doi: 10.1016/j.cell.2006.07.024. [DOI] [PubMed] [Google Scholar]
- 15.Takahashi K, et al. Induction of pluripotent stem cells from adult human fibroblasts by defined factors. Cell. 2007;131:861–872. doi: 10.1016/j.cell.2007.11.019. [DOI] [PubMed] [Google Scholar]
- 16.Yu J, et al. Induced pluripotent stem cell lines derived from human somatic cells. Science. 2007;318:1917–1920. doi: 10.1126/science.1151526. [DOI] [PubMed] [Google Scholar]
- 17.Cowan CA, et al. Derivation of embryonic stem-cell lines from human blastocysts. N Engl J Med. 2004;350:1353–1356. doi: 10.1056/NEJMsr040330. [DOI] [PubMed] [Google Scholar]
- 18.Huangfu D, et al. Induction of pluripotent stem cells from primary human fibroblasts with only Oct4 and Sox2. Nat Biotechnol. 2008;26:1269–1275. doi: 10.1038/nbt.1502. [DOI] [PubMed] [Google Scholar]
- 19.Monaghan AP, Kaestner KH, Grau E, Schutz G. Postimplantation expression patterns indicate a role for the mouse forkhead/HNF-3 alpha, beta and gamma genes in determination of the definitive endoderm, chordamesoderm and neuroectoderm. Development. 1993;119:567–578. doi: 10.1242/dev.119.3.567. [DOI] [PubMed] [Google Scholar]
- 20.Nakagawa M, et al. Generation of induced pluripotent stem cells without Myc from mouse and human fibroblasts. Nat Biotechnol. 2008;26:101–106. doi: 10.1038/nbt1374. [DOI] [PubMed] [Google Scholar]
- 21.Lowry WE, et al. Generation of human induced pluripotent stem cells from dermal fibroblasts. Proc Natl Acad Sci USA. 2008;105:2883–2888. doi: 10.1073/pnas.0711983105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Park IH, et al. Reprogramming of human somatic cells to pluripotency with defined factors. Nature. 2008;451:141–146. doi: 10.1038/nature06534. [DOI] [PubMed] [Google Scholar]
- 23.Stadtfeld M, Nagaya M, Utikal J, Weir G, Hochedlinger K. Induced pluripotent stem cells generated without viral integration. Science. 2008;322:945–949. doi: 10.1126/science.1162494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Okita K, Nakagawa M, Hyenjong H, Ichisaka T, Yamanaka S. Generation of mouse induced pluripotent stem cells without viral vectors. Science. 2008;322:949–953. doi: 10.1126/science.1164270. [DOI] [PubMed] [Google Scholar]
- 25.Yu J, et al. Human Induced Pluripotent Stem Cells Free of Vector and Transgene Sequences. Science. 2009;324:797–801. doi: 10.1126/science.1172482. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Zhou H, et al. Generation of Induced Pluripotent Stem Cells Using Recombinant Proteins. Cell Stem Cell. 2009;4:381–384. doi: 10.1016/j.stem.2009.04.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Tateishi K, et al. Generation of insulin-secreting islet-like clusters from human skin fibroblasts. J Biol Chem. 2008;283:31601–31607. doi: 10.1074/jbc.M806597200. [DOI] [PubMed] [Google Scholar]
- 28.Osafune K, et al. Marked differences in differentiation propensity among human embryonic stem cell lines. Nat Biotechnol. 2008;26:313–315. doi: 10.1038/nbt1383. [DOI] [PubMed] [Google Scholar]
- 29.Chen S, et al. A small molecule that directs differentiation of human ESCs into the pancreatic lineage. Nat Chem Biol. 2009;5:258–265. doi: 10.1038/nchembio.154. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.