

Abstract
Objective
Core2 1-6-N-glucosaminyltransferase-I (C2GlcNAcT-I) plays an important role in optimizing the binding functions of several selectin ligands, including P-selectin glycoprotein ligand. We used apolipoprotein E (ApoE)-deficient atherosclerotic mice to investigate the role of C2GlcNAcT-I in platelet and leukocyte interactions with injured arterial walls, in endothelial regeneration at injured sites, and in the formation of arterial neointima.
Methods and Results
Arterial neointima induced by wire injury was smaller in C2GlcNAcT-I–deficient apoE−/− mice than in control apoE−/− mice (a 79% reduction in size). Compared to controls, apoE−/− mice deficient in C2GlcNAcT-I also demonstrated less leukocyte adhesion on activated platelets in micro-flow chambers (a 75% reduction) and accumulation of leukocytes at injured areas of mouse carotid arteries was eliminated. Additionally, endothelial regeneration in injured lumenal areas was substantially faster in C2GlcNAcT-I–deficient apoE−/− mice than in control apoE−/− mice. Endothelial regeneration was associated with reduced accumulation of platelet factor 4 (PF4) at injured sites. PF4 deficiency accelerated endothelial regeneration and protected mice from neointima formation after arterial injury.
Conclusions
C2GlcNAcT-I deficiency suppresses injury-induced arterial neointima formation, and this effect is attributable to decreased leukocyte recruitment to injured vascular walls and increased endothelial regeneration. Both C2GlcNAcT-I and PF4 are promising targets for the treatment of arterial restenosis.
Percutaneous transluminal coronary intervention is a mainstay in the treatment of patients with coronary artery disease. In a great number of patients, however, this intervention results in arterial injury that causes restenosis of the vessel. Arterial restenosis even occurs in the drug-eluting stent area. Restenosis is characterized by a decrease in arterial luminal diameter of 50% or more that results from pathological intimal hyperplasia.1 Wire-induced neointima formation in the mouse carotid artery is a widely used model for mimicking the pathology of arterial neointima in patients with arterial restenosis.2
The accumulation of platelets and leukocytes on injured arterial areas is requisite for neointima formation. Immediately after arterial injury, platelets interact with the injured area via many factors, including glycoprotein Ib and glycoprotein IIb/IIIa.3, 4 Upon adherence, platelets become activated and express P-selectin, which along with integrins and other platelet-derived factors orchestrates the recruitment of leukocytes to the injured site.5-7 P-selectin glycoprotein ligand 1 (PSGL-1) is expressed on adherent leukocytes and serves as a platform to recruit more activated platelets.8 Interactions of platelet P-selectin with PSGL-1 or other P-selectin ligands presented by cells at the injured area contribute to further platelet accumulation.8 In mice, deletion or blockade of P-selectin or PSGL-1 inhibits this platelet accumulation and leukocyte adhesion, thereby suppressing the formation of arterial neointima.9-11 The roles of P-selectin and PSGL-1 in neointima formation have also been validated in other models of vascular injury 12, 13.
PSGL-1 contains sialylated and fucosylated oligosaccharides (O-glycans).8, 14 This O-glycan structure is crucial for the optimal binding of PSGL-1 to selectins.15, 16 Core2 1-6-N-glucosaminyltransferase-I (C2GlcNAcT-I), an intracellular enzyme in leukocytes, is responsible for the O-glycosylation of PSGL-1.17 C2GlcNAcT-I is important for the recruitment of Ly-6Chi mouse inflammatory monocytes to arterial vessel walls and the formation of atherosclerotic lesions.17 However, the role of C2GlcNAcT-I in the regulation of platelet accumulation, leukocyte recruitment, and neointima formation in injured arteries in vivo has yet to be clarified.
We bred C2GlcNAcT-I–deficient mice with apolipoprotein E–deficient atherosclerotic mice to generate double knockout mice (C2GlcNAcT-I−/−/apoE−/−) and their controls. Using these mice, we investigated the effect of loss of C2GlcNAcT-I on leukocyte and platelet accumulation, endothelial regeneration at injured areas of arteries, and the formation of arterial neointima. Preliminary data from these mice revealed an important role for platelet-leukocyte interactions in endothelial regeneration. To further investigate the molecular mechanisms involved in this process, we used platelet factor 4 (PF4)-deficient mice (PF4−/−) to study the role of PF4 in endothelial regeneration and neointima formation after arterial injury.
Methods
C2GlcNAcT-I−/− 18 and PF4−/− 19 mice were first crossed with C57BL/6J mice for more than 10 times, then bred with apoE−/− mice to generate double-knockout mice and their littermate controls. Carotid arteries of these mice were injured using a guide wire according to a protocol approved by the University of Minnesota Institutional Animal Care and Use Committee. Detailed methods are available in the Supplemental Material.
Results
Formation of injury-induced arterial neointima
To determine the role of C2GlcNAcT-I in the formation of arterial neointima, carotid arteries of C2GlcNAcT-I−/−/apoE−/− mice and littermate C2GlcNAcT-I+/+/apoE−/− mice were injured with a guide wire. Four weeks later, the carotid arteries were excised and processed for analysis. Injured carotid arteries from C2GlcNAcT-I-/-/apoE−/− mice exhibited neointima 4 to 6 times smaller than the neointima of C2GlcNAcT-I+/+/apoE−/− mice (Figure 1a). Additionally, the number of macrophages (Figure 1b) and smooth muscle cells (Figure 1c) in the neointima of injured arteries from C2GlcNAcT-I−/−/apoE−/− mice was reduced by 45% and 75%, respectively, compared with those from C2GlcNAcT-I+/+/apoE−/− mice. No difference was found in collagen content in neointima of both types of mice (supplementary Figure I). Notably, only one-third of arteries from these mice showed neointima and media growth, which was very minor.
Figure 1. C2GlcNAcT-I deficiency suppresses injury-induced arterial neointima formation.
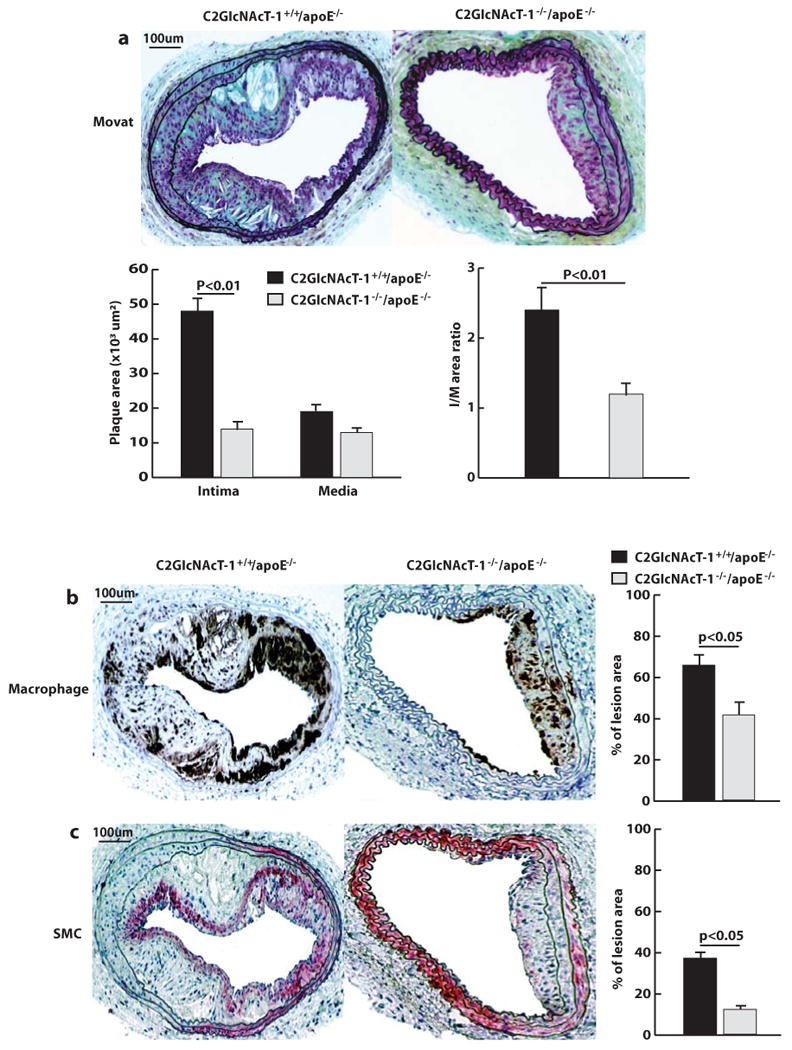
a, Cross-sections of arterial neointima stained with Movat pentachrome and quantification of the size of neointima (I), size of media (M), and ratio of intima to media (n = 12 for both groups). b, Anti-Mac-2 staining of infiltrated macrophages in arterial neointima. c, Anti-α-actin staining of vascular smooth muscle cells (SMCs) in the arterial neointima. The average numbers of cells in the neointima were obtained by analyzing 12 cross sections from 12 injured carotid arteries from mice.
Interactions of C2GlcNAcT-I-deficient leukocytes with activated platelets and injured arteries
Using an ex vivo micro-flow perfusion chamber with a shear stress of 1 dyn/cm2, we measured the tethering, rolling, and adherence of wild-type (wt) leukocytes to a surface coated with activated platelets. The number of wt leukocytes that rolled or adhered increased over time. After 5 min of perfusion, the number of rolling and adhering wt leukocytes was 800 ± 70 per mm2 and 700 ± 20 per mm2, respectively. By contrast, the number of rolling and adhering C2GlcNAcT-I-deficient leukocytes after 5 min of perfusion was only 200 ± 20 per mm2 and 180 ± 12 per mm2, respectively (Figure 2a). C2GlcNAcT-I-deficient leukocytes rolled with higher velocities than wt leukocytes did (data not shown).
Figure 2. C2GlcNAcT-I deficiency suppresses leukocyte rolling and adhesion on activated platelets and in injured arteries ex vivo and in vivo.
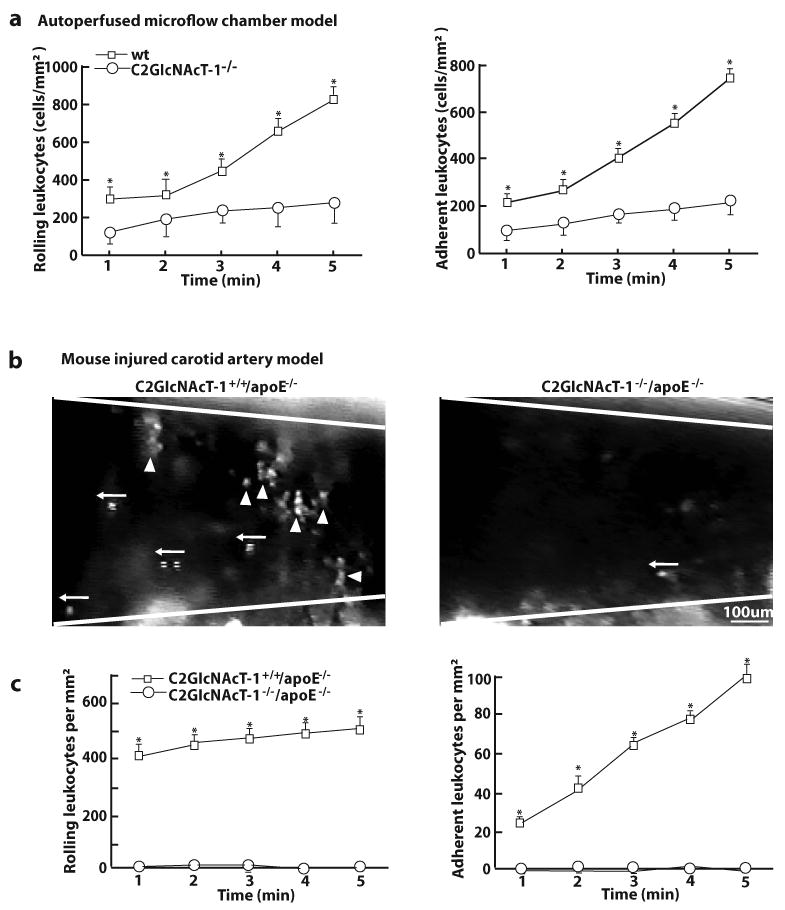
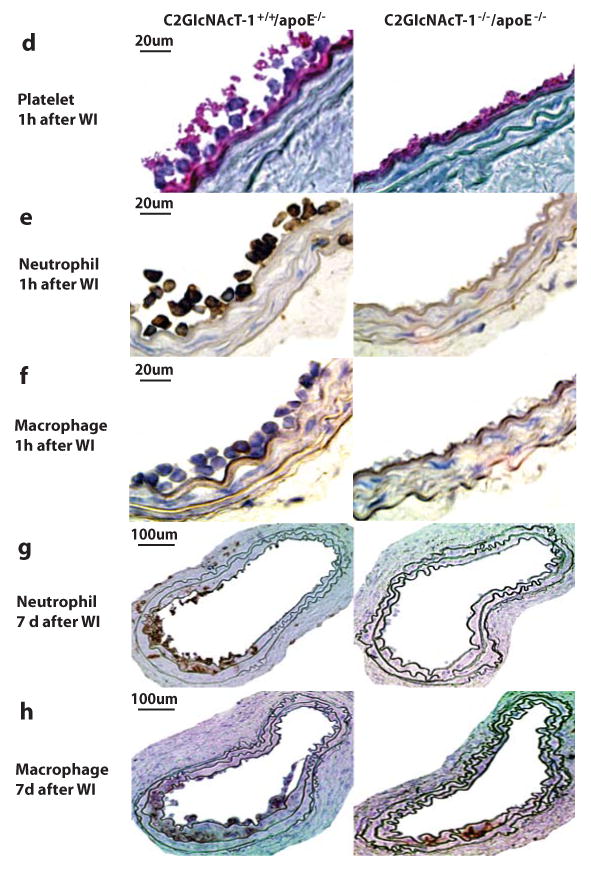
a, Leukocyte rolling and adhesion on activated platelets at 1.0 ± 0.1 dyn/cm2 within 5 min after whole blood of wt and C2GlcNAcT-I−/− mice was perfused through micro-flow chambers. b to c, Leukocyte rolling and adhesion in injured mouse carotid arteries within the first 5 min after injury. Images were obtained from videotape recordings of the epifluorescence intravital microscope with 10X objective, illustrating rolling (←) and adhering (▲) leukocytes in the injured arteries of wt (left) and C2GlcNAcT-I−/− mice (right). The data points in c represent the means of three separate experiments. *P < 0.05, C2GlcNAcT-I−/− vs. wt mice. d to f, Injured carotid arteries of C2GlcNAcT-I+/+/apoE−/− and C2GlcNAcT-I−/−/apoE−/− mice were collected at 1 hour after wire injury (WI) and immunostained with antibodies specific for platelets (d), neutrophils (e), and macrophages (f). Carotid arteries of five mice were analyzed for each group. g and h, Immunostaining for neutrophils (g) and macrophages (h) on cross-sections of carotid arteries of C2GlcNAcT-I+/+/apoE−/− and C2GlcNAcT-I−/−/apoE−/− mice. Arteries were collected at 7 days after wire injury.
We used in vivo mouse models of carotid artery and femoral artery injury and intravital epifluoresence microscopy to examine the interactions of leukocytes and platelets with injured vessel walls. Using a 10X objective, we imaged injured mouse carotid arteries and observed that leukocytes (labeled with rhodamine 6G) rolled and adhered to the injured area in C2GlcNAcT-I+/+/apoE−/− mice. However, in C2GlcNAcT-I−/−/apoE−/− mice these interactions were almost completely eliminated (Figure 2b and c). Using a 40X objective, we imaged rhodamine 6G-labeled platelets and leukocytes in injured mouse femoral arteries. In C2GlcNAcT-I+/+/apoE−/− mice, platelets appeared as small weakly fluorescent spots and leukocytes appeared as bright/large spots (supplementary Figure II, left panel). By contrast, spots for leukocytes were almost completely absent and spots for platelet were dramatically reduced in the injured femoral arterial walls of C2GlcNAcT-I−/−/apoE−/− mice (supplementary Figure II, right panel).
To further distinguish platelets and leukocytes adherent on injured arteries, we immunostained cross sections of injured arteries using specific markers. At 1 hour after arterial injury in apoE−/− mice, the denuded luminal surface was covered with platelets and leukocytes, and many platelets bound to the surface of adherent leukocytes (Figure 2d, left panel). Nearly all of the adherent leukocytes were classified as neutrophils (Figure 2e, left panel); monocytes were rare (Figure 2f, left panel). In C2GlcNAcT-I−/−/apoE−/− mice, a layer of platelets accumulated on the injured arterial wall, but no leukocytes adhered to these platelets (Figure 2d to 2f, right panels). At 7 days after wire injury, many neutrophils and macrophages in apoE−/− mice, but only a few in C2GlcNAcT-I−/−/apoE−/− mice, adhered to or infiltrated the injured arterial walls (Figure 2g and 2h).
Endothelial regeneration in the injured arteries
Re-endothelialization is an important deterrent to neointima formation.20 We used quantitative Evans blue staining to compare the regeneration of endothelial cells on the injured areas of arteries from atherosclerotic mice with and without C2GlcNAcT-I. In the arteries of C2GlcNAcT-I+/+/apoE−/− mice, re-endothelialization after wire-induced arterial injury was only 18%, 41%, and 81% at 3, 5, and 7 days, respectively; for arteries of C2GlcNAcT-I−/−/apoE−/− mice, however, re-endothelialization was 30%, 61%, and 95% (Figure 3a). We also obtained micrographs of cross-sections of injured carotid arteries that were immunostained with anti-CD31 or anti-VE-cadherin. At 3, 5, and 7 days after injury, a greater luminal circumferential area stained positively for these endothelial markers in arteries of C2GlcNAcT-I−/−/apoE−/− mice than in C2GlcNAcT-I+/+/apoE−/− arteries (Figure 3b).
Figure 3. C2GlcNAcT-I deficiency accelerates endothelial regeneration on the arterial luminal surface of the injured area.
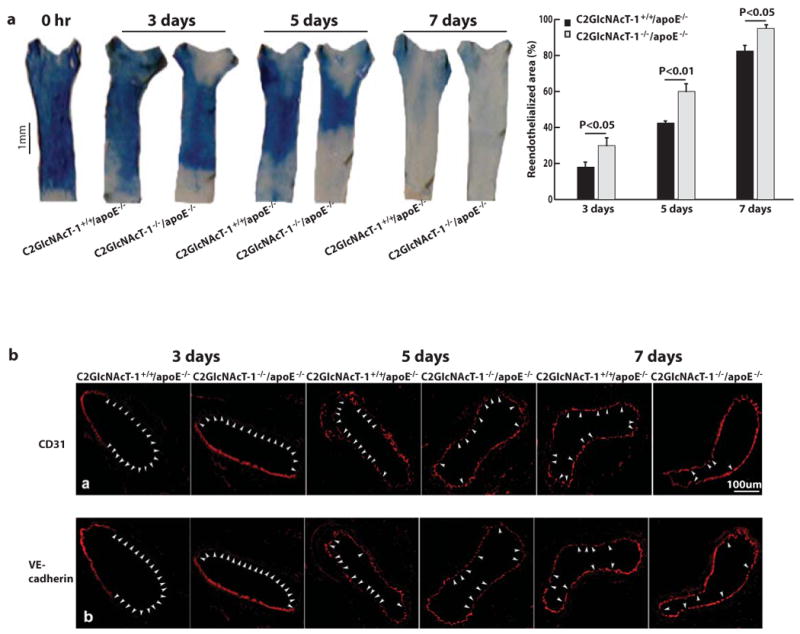
a, Evans blue staining of injured carotid arteries collected at different time points after wire injury shows the areas that were not covered with newly generated endothelial cells. Quantitative data are shown in the right panel. Results are mean values of five separate experiments for each time point. b, Representative micrographs of cross-sections of injured carotid arteries immunostained with anti-CD31 or anti-VE-cadherin. Arrowheads show the areas negative for CD31 or VE-cadherin. Carotid arteries of five mice for each time point were analyzed for each group.
PF4 and re-endothelialization of injured arteries
Recombinant platelet factor 4 (PF4) inhibits endothelial cell proliferation,21 so we immunostained the injured arteries for PF4. Nearly all areas where platelets accumulated in the injured arteries of C2GlcNAcT-I+/+/apoE−/− mice stained positively for PF4, and staining on the surface of adherent leukocytes (where platelets bound) was very robust (Figure 4a). By contrast, PF4 staining was much weaker in the injured arteries of C2GlcNAcT-I−/−/apoE−/− mice. This was further confirmed with the results of western blots. These indicated that PF4 accumulation was much greater in the injured carotid arteries of C2GlcNAcT-I+/+/apoE−/− mice than in C2GlcNAcT-I−/−/apoE−/− mice (Figure 4b).
Figure 4. PF4 in endothelial proliferation in the context of platelet-neutrophil interactions.
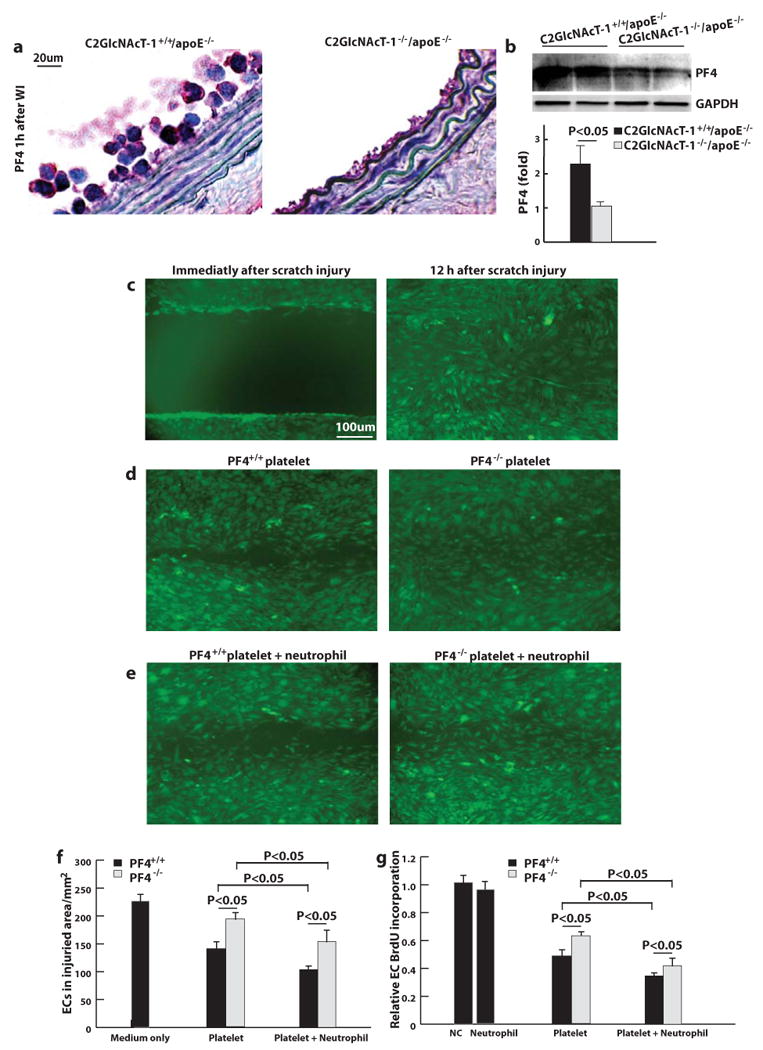
a, Anti-PF4 immunostaining with an antibody specific for PF4 (red) of cross-sections of injured carotid arteries of C2GlcNAcT-I+/+/apoE−/− and C2GlcNAcT-I−/−/apoE−/− mice. Carotid arteries were collected at 1 hour after wire injury. b, western blot for PF4 in injured carotid arteries of C2GlcNAcT-I+/+/apoE−/− and C2GlcNAcT-I−/−/apoE−/− mice. Results are mean values of three separate experiments. c to e, The covering of the wounded area with proliferating endothelial cells at 12 hour after wounding of the growing endothelial layer in the absence of additional leukocytes and platelets (right image in c), in the presence of activated platelets (d), or a mixture of neutrophils and activated platelets (e). Left images are experiments using activated platelets from PF+/+ mice whereas the right images represent experiments using activated platelets from PF−/− mice. f, Quantative analysis of endothelial proliferation under conditions as described in (c) to (e). g, Endothelial cell (EC) proliferation under the above conditions (as described in (c) to (e) was quantified by measuring BrdU incorporation. Results are mean values of five separate experiments.
To directly address the effect of platelet-released PF4 on endothelial cell proliferation, we used an in vitro model of endothelial wound repair. A sterile pipette tip dragged across confluent endothelial cell monolayers created 1-mm cell-free wounds that recovered nearly completely by ∼12 hour (Figure 4c). This process was not affected by the presence of neutrophils (data not shown). The addition of activated PF4+/+ platelets to the wounded endothelial cell monolayers significantly suppressed the recovery (Figure 4d, left panel). Furthermore, the suppression of recovery was amplified by adding a mixture of isolated neutrophils and activated PF4+/+ platelets (Figure 4e, left panel). By contrast, the addition of either activated PF4−/− platelets or a mixture of activated PF4−/− platelets with isolated neutrophils suppressed the recovery of the wounded area with endothelial cells to a much lesser extent than did PF4+/+ platelets or a mixture of both PF4+/+ platelets and neutrophils, respectively (Figure 4d, and e, right panels, and Figure 4f for quantitative data).
Analyses of 5-bromodeoxyuridine (BrdU) incorporation into endothelial cells showed that the addition of neutrophils to the wounded monolayers did not significantly affect endothelial cell proliferation. However, the addition of activated PF4+/+ platelets inhibited endothelial cell proliferation, and this inhibition was enhanced by the addition of a mixture of activated PF4+/+ platelets with isolated neutrophils. Whenever PF4−/− platelets were added, the suppression of endothelial cell proliferation was significantly reduced (Figure 4g). These experiments indicated that PF4 from activated platelets, especially in a context of platelet-neutrophil interactions, inhibited endothelial cell proliferation.
PF4 deficiency increases endothelial regeneration in injured arteries and decreases injury-induced neointima formation
We used PF4−/− mice to determine the role of platelet PF4 in endothelial regeneration and neointima formation after arterial injury in vivo. Using the 10X objective of the epifluorescence intravital microscope, we imaged injured carotid mouse arteries and observed leukocyte interactions with the injured vessels. No difference was observed in the number of rolling and adhering leukocytes in the carotid arteries of PF4−/− mice and wt mice (Figure 5a). In the mouse femoral artery injury model, using the 40X objective of the epifluorescence intravital microscope we observed similar platelet accumulations and leukocyte rolling and adhesion on the injured arteries of PF4−/− mice and wt mice (supplementary Figure III). In an atherosclerotic background, PF4 immunostaining was performed 1 hour after wire injury of arteries from PF4−/−/apoE−/− mice and PF4+/+/apoE−/− mice. Consistently, PF4 staining was positive on platelets accumulated on injured arteries of PF4+/+/apoE−/− mice and negative on those of PF4−/−/apoE−/− mice (Figure 5c). Platelet accumulation and neutrophil adhesion were observed on injured arteries from both PF4−/−/apoE−/− and PF4+/+/apoE−/− mice, and there was no significant difference between the numbers of platelets and neutrophils on the injured arteries from these mice (Figure 5b, 5d to 5e).
Figure 5. Regeneration of endothelial cells in the injured area of carotid arteries and the formation of injury-induced arterial neointima in PF4−/−/apoE−/− mice.
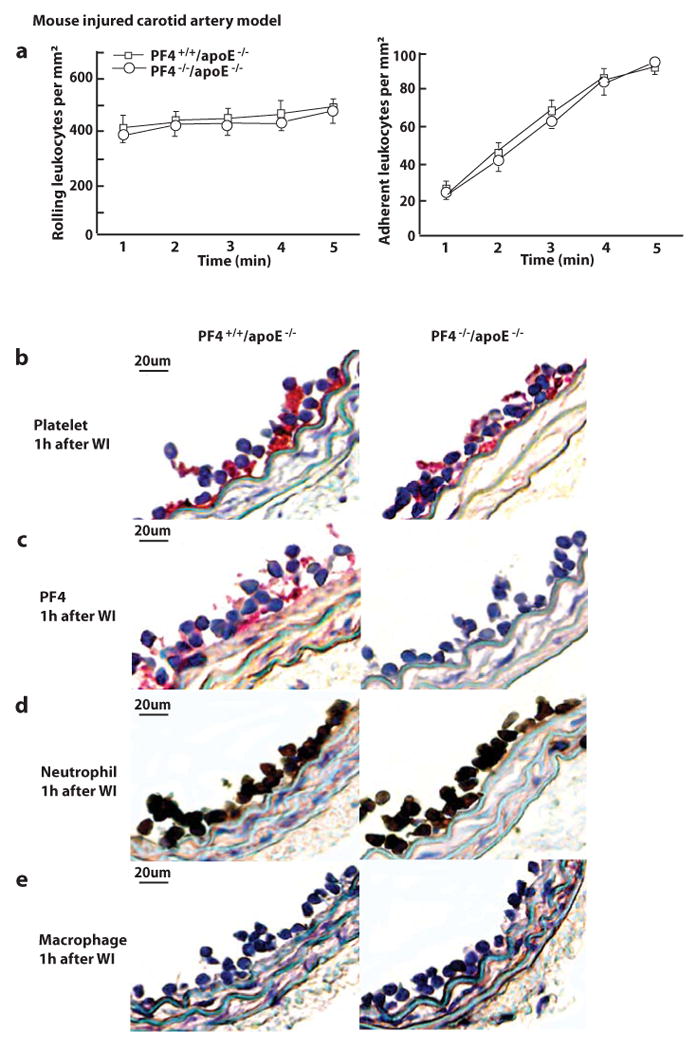
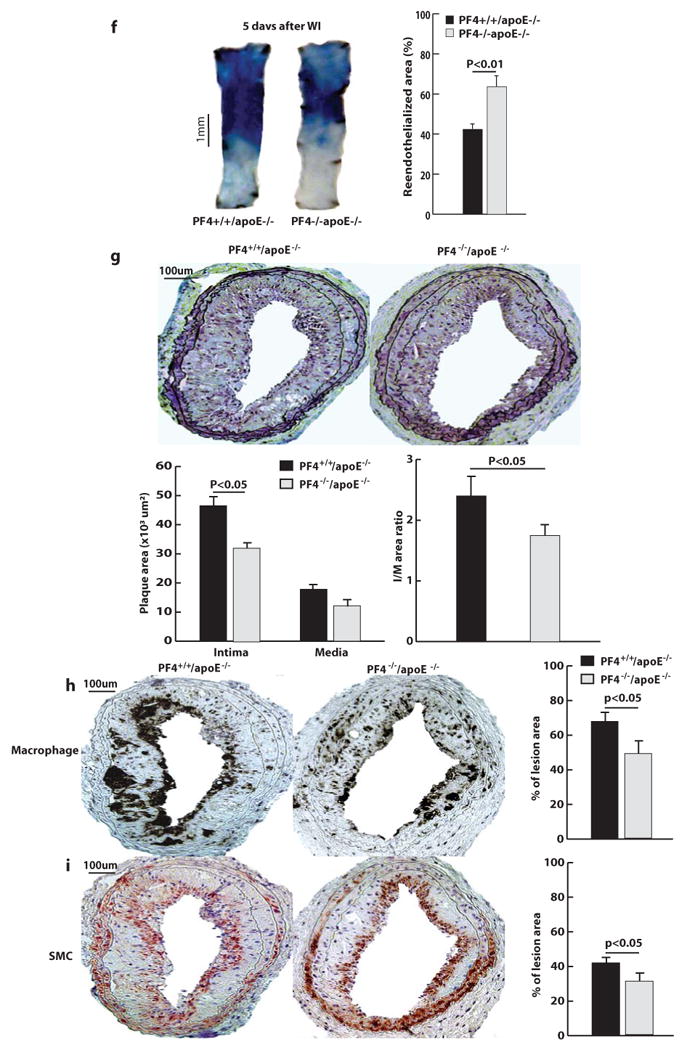
a, The number of rolling leukocytes and adherent leukocytes in injured mouse carotid arteries at 5 min after injury. Data were obtained from videotape recordings of the epifluoresence intravital microscope with 10X objective. The data points represent the means of three separate experiments. b to e, Injured carotid arteries of PF4+/+/apoE−/− and PF4−/−/apoE−/− mice were collected at 1 h after wire injury (WI) and immunostained with antibodies specific for platelets (b), PF4 (c), neutrophils (d), and macrophages (e). Carotid arteries of five mice were analyzed for each group. f, Evans blue staining of injured carotid arteries collected at 5 days after wire injury. Quantitative data on regeneration of endothelial cells in injured areas are shown in the right panels. Results are mean values of five separate experiments. g, Movat pentachrome staining of cross-sections of injured carotid arteries from PF4+/+/apoE−/− and PF4−/−/apoE−/− mice. h, Anti-Mac-2 staining of infiltrated macrophages in arterial neointima. i, Anti-α-actin staining of vascular smooth muscle cells (SMCs) in the arterial neointima. Carotid arteries were collected at 4 weeks after injury. The size of intima and media, the intima/media ratio, and the number of macrophages and SMCs in injured arteries were quantified by analyzing 12 cross-sections from 10 injured carotid arteries from mice.
Staining of wire-injured carotid arteries with Evans blue showed that at 5 days after injury, re-endothelialization was 65% in arteries of PF4−/−/apoE−/− mice but only 40% in arteries of PF4+/+/apoE−/− mice (Figure 5f). Relative to PF4+/+/apoE−/− mice, arterial neointima formation following wire injury was significantly reduced by 35% in PF4−/−/apoE−/− mice, and the size of the media in injured arteries was also reduced, although the difference was not statistically significant (Figure 5g). The number of macrophages and smooth muscle cells in neointima of PF4−/−/apoE−/− mice was reduced by 28% and 25%, respectively, compared to that of PF4+/+/apoE−/− mice (Figure 5h and i).
Discussion
This study demonstrates that C2GlcNAcT-I is critical for the formation of injury-induced arterial neointima in atherosclerotic mice. Compared to control mice, C2GlcNAcT-I−/−/apoE−/− mice were almost completely incapable of developing significant arterial neointima following wire-induced injury. The inhibition of leukocyte recruitment and rapid endothelialization of the vessel wall are important cellular mechanisms for this protection.
The compromised binding of selectin ligands in C2GlcNAcT-I−/− mice contributes to the protective effect of C2GlcNAcT-I deficiency on neointima formation. C2GlcNAcT-I deletion reduces neutrophil binding to P-, E- and L-selectin both in vitro and in vivo.18, 22, 23 Recently, we also demonstrated decreased binding of monocytes to these selectins.17 Other C2GlcNAcT-I-modified molecules such as CD34, CD43, and CD44 are speculated to be responsible for the leukocyte homing phenotype of C2GlcNAcT-I−/− mice.24 However, in CD34- or CD43-deficient mice, there is no change in the recruitment of neutrophils and monocytes to inflammatory sites.25, 26 Also, the binding of CD44 with its major ligand, hyaluronic acid, is not affected by C2GlcNAcT-I deletion.17 Therefore, C2GlcNAcT-I must affect leukocyte homing through its modification of ligands for P-, E- and L-selectins.
Our current work illustrates the dynamics of leukocyte interactions with injured arterial areas. Within the first few days of wire-induced injury, the injured arterial area is usually predominated by neutrophils and subsequently by a mixture of neutrophils and monocytes. By approximately 1 week after injury, the injured area is predominated by monocytes (data not shown), consistent with previous studies.27, 28 It has been well established that monocyte infiltration plays an important role in the formation of neointima. Ly-6Chi monocytes—important contributors to the formation of spontaneous lesions and arterial neointima—express high levels of PSGL-1 and bind selectins with high affinity.10 Without PSGL-1, Ly-6Chi monocytes cannot be effectively recruited to injured vessel walls, leading to the formation of a much smaller neointima.10 In C2GlcNAcT-I−/− mice, there is a significant defect in the binding function of selectin receptors, including PSGL-1.17 Consistent with these results, we observed that far fewer monocytes infiltrated the injured vessel wall of C2GlcNAcT-I−/−/apoE−/− mice relative to apoE−/− mice. This may be one of the underlying mechanisms for reduced arterial neointima in C2GlcNAcT-I−/−/apoE−/− mice.
Neutrophil adhesion to injured arterial areas has been long observed, but the role of neutrophils in neointima formation is uncertain.27, 29 Some studies have suggested that neutrophils directly contribute to neointima formation.29, 30 Interestingly, we found that compared to neutrophil-covered injured arterial areas of apoE−/− mice, elimination of neutrophils at the injured area in C2GlcNAcT-I−/−/apoE−/− mice promoted endothelial regeneration at the injured area. In addition, injured C2GlcNAcT-I−/−/apoE−/− arteries attracted fewer platelets and had reduced accumulation of PF4. In vitro experiments indicated that the observed inhibition of endothelial regeneration could be attributed to the increased presence of platelets and platelet-released PF4.
Endothelial recovery after percutaneous transluminal coronary intervention is crucial in the prevention of arterial restenosis.31 Slow endothelial regeneration leads to an increase in platelet accumulation, leukocyte adherence, and released inflammatory factors from leukocytes and platelets. These pathologies initiate and aggravate the inflammatory response in injured arterial wall. 31 Recombinant PF4 is a well-established potent anti-angiogenic factor.21, 32 Platelets release PF4 in large quantities,33 but they also release many other factors, including certain angiogenic factors. Consequently, the role of platelet-released PF4 within the context of the total platelet content has been uncertain with regard to endothelial cell proliferation.21 Our in vitro and in vivo studies are the first to demonstrate that platelet-released PF4 has a significant inhibitory effect on endothelial proliferation, especially in the context of neutrophil-platelet interactions. There are several possible explanations for this. First, as shown in Figure 4a, neutrophils adhering to the injured area provide a platform for more platelets to bind. Without neutrophils, very few platelets and less PF4 accumulate at the injured site. Second, PSGL-1 on neutrophils may interact with P-selectin on platelets to mediate outside-in signaling so as to activate platelets to release more PF4 from their α-granules. In P-selectin-deficient mice, platelets that accumulate on the injured arterial area are less compact,34 indicating that platelets are not fully activated in the absence of P-PSGL-1-mediated platelet-leukocyte interactions. Third, neutrophils may release enzymes to cleave PF4 to optimize its anti-angiogenic function or release other factors that synergize with PF4 in its anti-angiogenic effect.35 A variety of PF4-derived peptides inhibit endothelial cell proliferation much more potently than full-length PF4.36
Neutrophil adhesion at injured areas of PF4−/−/apoE−/− arteries was not decreased compared with PF4+/+/apoE−/− arteries. This is consistent with the early observation showing that PF4 is devoid of chemotactic activity for neutrophils.37 In PF4−/−/apoE−/− arteries, there was an increase of endothelial regeneration at injured areas. This may be the predominant underlying mechanism for decreased neointima formation in PF4−/−/apoE−/− mice, but other mechanisms may also be involved. PF4 released from platelets may bind to newly regenerated endothelium and promote monocyte recruitment. Platelet PF4 may bind to RANTES and these chemokines cooperate to recruitment monocytes to injured vessel wall.38, 39. Indeed, less accumulation of RANTES was found in the injured area of PF4−/−/apoE−/− mice than PF4+/+/apoE−/− mice (supplementary Figure IV). PF4 also can activate endothelial cells,40 and activation of newly regenerated endothelial cells on the injured area may be partially responsible for neointima formation in injured arteries of apoE−/− mice. In PF4−/−/apoE−/− mice, the level of inflammation of regenerated endothelial cells may be low as a result of PF4 absence. Additionally, protection from arterial injury in PF4−/−/apoE−/− mice may also be attributed to the high level of HDL. Consistent with the results in a previous report, 41 PF4−/−/apoE−/− mice in this study also had a higher level of HDL than PF4−/−/apoE−/− mice (data not shown).
Collectively, our data demonstrate that C2GlcNAcT-I is a promising therapeutic target to curb the formation of arterial neointima. Inhibition of C2GlcNAcT-I compromises selectin receptor function, resulting in suppression of leukocyte and platelet accumulation on the vessel wall. In addition, our results reveal an adverse effect of platelet-released PF4 on endothelial regeneration, suggesting that anti-PF4 treatment might have the beneficial effect of inhibiting arterial neointima formation.
Supplementary Material
Acknowledgments
This work was supported by AHA 0430151N, NIH HL78679 and HL080569 to Y. Huo, and by the Howard Hughes Medical Institute and NIH HL57345, HL78784 and GM62116 to J. Marth.
Reference List
- 1.Kuntz RE, Gibson CM, Nobuyoshi M, Baim DS. Generalized model of restenosis after conventional balloon angioplasty, stenting and directional atherectomy. J Am Coll Cardiol. 1993;21(1):15–25. doi: 10.1016/0735-1097(93)90712-a. [DOI] [PubMed] [Google Scholar]
- 2.Lindner V, Fingerle J, Reidy MA. Mouse model of arterial injury. Circ Res. 1993;73:792–6. doi: 10.1161/01.res.73.5.792. [DOI] [PubMed] [Google Scholar]
- 3.Andrews RK, Shen Y, Gardiner EE, Dong JF, Lopez JA, Berndt MC. The glycoprotein Ib-IX-V complex in platelet adhesion and signaling. Thromb Haemost. 1999;82(2):357–64. [PubMed] [Google Scholar]
- 4.Phillips DR, Charo IF, Scarborough RM. GPIIb-IIIa - The responsive integrin. Cell. 1991;65:359–62. doi: 10.1016/0092-8674(91)90451-4. [DOI] [PubMed] [Google Scholar]
- 5.Manka D, Forlow SB, Sanders JM, Hurwitz D, Bennett DK, Green SA, Ley K, Sarembock IJ. Critical Role of Platelet P-Selectin in the Response to Arterial Injury in Apolipoprotein-E-Deficient Mice. Arterioscler Thromb Vasc Biol. 2004;24(6):1124–9. doi: 10.1161/01.ATV.0000127619.04687.f4. [DOI] [PubMed] [Google Scholar]
- 6.Simon DI, Chen Z, Xu H, Li CQ, Dong J, McIntire LV, Ballantyne CM, Zhang L, Furman MI, Berndt MC, Lopez JA. Platelet glycoprotein ibalpha is a counterreceptor for the leukocyte integrin Mac-1 (CD11b/CD18) J Exp Med. 2000;192(2):193–204. doi: 10.1084/jem.192.2.193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Weber C. Platelets and chemokines in atherosclerosis: partners in crime. Circ Res. 2005;96(6):612–6. doi: 10.1161/01.RES.0000160077.17427.57. [DOI] [PubMed] [Google Scholar]
- 8.McEver RP, Cummings RD. Perspectives series: cell adhesion in vascular biology. role of psgl-1 binding to selectins in leukocyte recruitment. J Clin Invest. 1997;100(3):485–91. 11. doi: 10.1172/JCI119556. [review] [70 refs] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Manka D, Collins RG, Ley K, Beaudet AL, Sarembock IJ. Absence of p-selectin, but not intercellular adhesion molecule-1, attenuates neointimal growth after arterial injury in apolipoprotein e-deficient mice. Circulation. 2001;103(7):1000–5. doi: 10.1161/01.cir.103.7.1000. [DOI] [PubMed] [Google Scholar]
- 10.An G, Wang H, Tang R, Yago T, McDaniel JM, McGee S, Huo Y, Xia L. P-selectin glycoprotein ligand-1 is highly expressed on Ly-6Chi monocytes and a major determinant for Ly-6Chi monocyte recruitment to sites of atherosclerosis in mice. Circulation. 2008;117(25):3227–37. doi: 10.1161/CIRCULATIONAHA.108.771048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Phillips JW, Barringhaus KG, Sanders JM, Hesselbacher SE, Czarnik AC, Manka D, Vestweber D, Ley K, Sarembock IJ. Single injection of P-selectin or P-selectin glycoprotein ligand-1 monoclonal antibody blocks neointima formation after arterial injury in apolipoprotein E-deficient mice. Circulation. 2003;107(17):2244–9. doi: 10.1161/01.CIR.0000065604.56839.18. [DOI] [PubMed] [Google Scholar]
- 12.Bienvenu JG, Tanguay JF, Theoret JF, Kumar A, Schaub RG, Merhi Y. Recombinant soluble p-selectin glycoprotein ligand-1-Ig reduces restenosis through inhibition of platelet-neutrophil adhesion after double angioplasty in swine. Circulation. 2001;103(8):1128–34. doi: 10.1161/01.cir.103.8.1128. [DOI] [PubMed] [Google Scholar]
- 13.Wang K, Zhou Z, Zhou X, Tarakji K, Topol EJ, Lincoff AM. Prevention of intimal hyperplasia with recombinant soluble P-selectin glycoprotein ligand-immunoglobulin in the porcine coronary artery balloon injury model. J Am Coll Cardiol. 2001;38(2):577–82. doi: 10.1016/s0735-1097(01)01347-x. [DOI] [PubMed] [Google Scholar]
- 14.Norgard KE, Moore KL, Diaz S, Stults NL, Ushiyama S, McEver RP, Cummings RD, Varki A. Characterization of a specific ligand for P-selectin on myeloid cells. A minor glycoprotein with sialylated O- linked oligosaccharides. J Biol Chem. 1993;268:12764–74. [PubMed] [Google Scholar]
- 15.Kumar R, Camphausen RT, Sullivan FX, Cumming DA. Core2 beta-1,6-n-acetylglucosaminyltransferase enzyme activity is critical for P-selectin Glycoprotein Ligand-1 binding to P- selectin. Blood. 1996;88(10):3872–9. [PubMed] [Google Scholar]
- 16.Li F, Wilkins PP, Crawley S, Weinstein J, Cummings RD, McEver RP. Post-translational modifications of recombinant P-selectin glycoprotein ligand-1 required for binding to P- and E-selectin. J Biol Chem. 1996;271(6):3255–64. [PubMed] [Google Scholar]
- 17.Wang H, Tang R, Zhang W, Amirikian K, Geng Z, Geng J, Hebbel RP, Xia L, Marth JD, Fukuda M, Katoh S, Huo Y. Core2 1-6-N-Glucosaminyltransferase-I Is Crucial for the Formation of Atherosclerotic Lesions in Apolipoprotein E-Deficient Mice. Arterioscler Thromb Vasc Biol. 2009;29(2):180–7. doi: 10.1161/ATVBAHA.108.170969. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Ellies LG, Tsuboi S, Petryniak B, Lowe JB, Fukuda M, Marth JD. Core 2 oligosaccharide biosynthesis distinguishes between selectin ligands essential for leukocyte homing and inflammation. Immunity. 1998;9(6):881–90. doi: 10.1016/s1074-7613(00)80653-6. [DOI] [PubMed] [Google Scholar]
- 19.Eslin DE, Zhang C, Samuels KJ, Rauova L, Zhai L, Niewiarowski S, Cines DB, Poncz M, Kowalska MA. Transgenic mice studies demonstrate a role for platelet factor 4 in thrombosis: dissociation between anticoagulant and antithrombotic effect of heparin. Blood. 2004;104(10):3173–80. doi: 10.1182/blood-2003-11-3994. [DOI] [PubMed] [Google Scholar]
- 20.Versari D, Lerman LO, Lerman A. The importance of reendothelialization after arterial injury. Curr Pharm Des. 2007;13(17):1811–24. doi: 10.2174/138161207780831239. [DOI] [PubMed] [Google Scholar]
- 21.Bikfalvi A. Platelet factor 4: an inhibitor of angiogenesis. Semin Thromb Hemost. 2004;30(3):379–85. doi: 10.1055/s-2004-831051. [DOI] [PubMed] [Google Scholar]
- 22.Snapp KR, Heitzig CE, Ellies LG, Marth JD, Kansas GS. Differential requirements for the O-linked branching enzyme core 2 beta1-6-N-glucosaminyltransferase in biosynthesis of ligands for E-selectin and P-selectin. Blood. 2001;97(12):3806–11. doi: 10.1182/blood.v97.12.3806. [DOI] [PubMed] [Google Scholar]
- 23.Sperandio M, Thatte A, Foy D, Ellies LG, Marth JD, Ley K. Severe impairment of leukocyte rolling in venules of core 2 glucosaminyltransferase-deficient mice. Blood. 2001;97(12):3812–9. doi: 10.1182/blood.v97.12.3812. [DOI] [PubMed] [Google Scholar]
- 24.Barran P, Fellinger W, Warren CE, Dennis JW, Ziltener HJ. Modification of CD43 and other lymphocyte O-glycoproteins by core 2 N-acetylglucosaminyltransferase. Glycobiology. 1997;7(1):129–36. doi: 10.1093/glycob/7.1.129. [DOI] [PubMed] [Google Scholar]
- 25.Suzuki A, Andrew DP, Gonzalo JA, Fukumoto M, Spellberg J, Hashiyama M, Suda T, Takimoto H, Gerwin N, Webb J, Gutierrez-Ramos JC, Molineux G, McNiece I, Ley K, Butcher EC, May WS, Greaves MF, Amakawa R, Tada Y, Wakcham A, Mak TW. CD34 deficient mice have reduced eosinophil accumulation after allergen exposure and reveal a novel crossreactive 90 kD protein. Blood. 1996;87(9):3550–62. [PubMed] [Google Scholar]
- 26.Carlow DA, Ziltener HJ. CD43 deficiency has no impact in competitive in vivo assays of neutrophil or activated T cell recruitment efficiency. J Immunol. 2006;177(9):6450–9. doi: 10.4049/jimmunol.177.9.6450. [DOI] [PubMed] [Google Scholar]
- 27.Roque M, Fallon JT, Badimon JJ, Zhang WX, Taubman MB, Reis ED. Mouse model of femoral artery denudation injury associated with the rapid accumulation of adhesion molecules on the luminal surface and recruitment of neutrophils. Arterioscler Thromb Vasc Biol. 2000;20(2):335–42. doi: 10.1161/01.atv.20.2.335. [DOI] [PubMed] [Google Scholar]
- 28.Osaka M, Hagita S, Haraguchi M, Kajimura M, Suematsu M, Yoshida M. Real-time imaging of mechanically injured femoral artery in mice reveals a biphasic pattern of leukocyte accumulation. Am J Physiol Heart Circ Physiol. 2007;292(4):H1876–H1882. doi: 10.1152/ajpheart.00708.2006. [DOI] [PubMed] [Google Scholar]
- 29.Shimazawa M, Watanabe S, Kondo K, Hara H, Nakashima M, Umemura K. Neutrophil accumulation promotes intimal hyperplasia after photochemically induced arterial injury in mice. Eur J Pharmacol. 2005;520(13):156–63. doi: 10.1016/j.ejphar.2005.07.031. [DOI] [PubMed] [Google Scholar]
- 30.Welt FG, Edelman ER, Simon DI, Rogers C. Neutrophil, not macrophage, infiltration precedes neointimal thickening in balloon-injured arteries. Arterioscler Thromb Vasc Biol. 2000;20(12):2553–8. doi: 10.1161/01.atv.20.12.2553. [DOI] [PubMed] [Google Scholar]
- 31.Losordo DW, Isner JM, az-Sandoval LJ. Endothelial recovery: the next target in restenosis prevention. Circulation. 2003;107(21):2635–7. doi: 10.1161/01.CIR.0000071083.31270.C3. [DOI] [PubMed] [Google Scholar]
- 32.Maione TE, Gray GS, Petro J, Hunt AJ, Donner AL, Bauer SI, Carson HF, Sharpe RJ. Inhibition of angiogenesis by recombinant human platelet factor-4 and related peptides. Science. 1990;247(4938):77–9. doi: 10.1126/science.1688470. [DOI] [PubMed] [Google Scholar]
- 33.Zucker MB, Katz IR. Platelet factor 4: production, structure, and physiologic and immunologic action. Proc Soc Exp Biol Med. 1991;198(2):693–702. doi: 10.3181/00379727-198-43309. [DOI] [PubMed] [Google Scholar]
- 34.Smyth SS, Reis ED, Zhang W, Fallon JT, Gordon RE, Coller BS. Beta(3)-integrin-deficient mice but not P-selectin-deficient mice develop intimal hyperplasia after vascular injury: correlation with leukocyte recruitment to adherent platelets 1 hour after injury. Circulation. 2001;103(20):2501–7. doi: 10.1161/01.cir.103.20.2501. [DOI] [PubMed] [Google Scholar]
- 35.LaRosa CA, Rohrer MJ, Benoit SE, Rodino LJ, Barnard MR, Michelson AD. Human neutrophil cathepsin G is a potent platelet activator. J Vasc Surg. 1994;19(2):306–18. doi: 10.1016/s0741-5214(94)70106-7. [DOI] [PubMed] [Google Scholar]
- 36.Gupta SK, Hassel T, Singh JP. A potent inhibitor of endothelial cell proliferation is generated by proteolytic cleavage of the chemokine platelet factor 4. Proc Natl Acad Sci U S A. 1995;92(17):7799–803. doi: 10.1073/pnas.92.17.7799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Clark-Lewis I, Dewald B, Geiser T, Moser B, Baggiolini M. Platelet factor 4 binds to interleukin 8 receptors and activates neutrophils when its N terminus is modified with Glu-Leu- Arg. Proc Natl Acad Sci USA. 1993;90:3574–7. doi: 10.1073/pnas.90.8.3574. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.von HP, Koenen RR, Sack M, Mause SF, Adriaens W, Proudfoot AE, Hackeng TM, Weber C. Heterophilic interactions of platelet factor 4 and RANTES promote monocyte arrest on endothelium. Blood. 2005;105(3):924–30. doi: 10.1182/blood-2004-06-2475. [DOI] [PubMed] [Google Scholar]
- 39.Koenen RR, von HP, Nesmelova IV, Zernecke A, Liehn EA, Sarabi A, Kramp BK, Piccinini AM, Paludan SR, Kowalska MA, Kungl AJ, Hackeng TM, Mayo KH, Weber C. Disrupting functional interactions between platelet chemokines inhibits atherosclerosis in hyperlipidemic mice. Nat Med. 2009;15(1):97–103. doi: 10.1038/nm.1898. [DOI] [PubMed] [Google Scholar]
- 40.Yu G, Rux AH, Ma P, Bdeir K, Sachais BS. Endothelial expression of E-selectin is induced by the platelet-specific chemokine platelet factor 4 through LRP in an NF-kappaB-dependent manner. Blood. 2005;105(9):3545–51. doi: 10.1182/blood-2004-07-2617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Sachais BS, Turrentine T, wicki McKenna JM, Rux AH, Rader D, Kowalska MA. Elimination of platelet factor 4 (PF4) from platelets reduces atherosclerosis in C57Bl/6 and apoE-/- mice. Thromb Haemost. 2007;98(5):1108–13. [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.