

Abstract
Background and Objective
Elevated C-reactive protein (CRP) levels are associated with increased cardiovascular events and endothelial dysfunction. We have previously shown that CRP decreases endothelial nitric oxide synthase (eNOS) activity in endothelial cells and inhibits endothelium-dependent nitric oxide (NO)-mediated vasodilation in-vitro. Herein, we examined the effect of in-vivo administration of CRP on endothelial function and underlying mechanisms in a valid animal model.
Methods
Sprague-Dawley rats were injected intraperitoneally daily for 3 days with human CRP or human serum albumin (HuSA) at 20 mg/kg body weight. On day 4, mesenteric arterioles were isolated and pressurized for vasomotor study and aortic tissue was subjected to biochemical and molecular analysis.
Results
Dilation of mesenteric arterioles to acetylcholine but not to sodium nitroprusside was significantly reduced following CRP treatment. The eNOS activity, eNOS dimer/monomer ratio, tetrahydrobiopterin levels, and protein expression of GTPCH1 were significantly lower in aortic tissue homogenates from CRP-treated than HuSA-treated rats. CRP treatment also resulted in increased dihydroethidium staining for superoxide in aortic endothelium and membrane translocation of p47phox, a regulatory subunit of NADPH oxidase.
Conclusion
Our data provide novel evidence for the detrimental action of CRP in vivo by impairing eNOS-dependent vasodilation and uncoupling of eNOS.
Keywords: CRP, vasoreactivity, endothelial nitric oxide synthase, tetrahydrobiopterin, NADPH oxidase
Introduction
Inflammation plays a critical role in atherogenesis. C-reactive protein (CRP) is a prototypic marker of inflammation, and has been shown in numerous prospective studies, to predict cardiovascular events (1,2). While CRP is a risk marker, much data are evolving to suggest that CRP also promotes atherothrombosis (3). To date, numerous groups have shown pro-inflammatory and pro-atherogenic effects of CRP in-vitro (4–6). Although, the data with respect to the promotion of atherosclerosis in CRP transgenic (Tg) mice is conflicting, it appears that following certain perturbations, vasoreactivity is impaired (7,8). In addition, two groups (9,10) have shown that CRP causes down regulation of endothelial nitric oxide synthase (eNOS) by decreasing eNOS activity and nitric oxide (NO) bioactivity in endothelial cells (ECs). Also, very recently, we showed that CRP uncouples eNOS in human aortic ECs (HAECs) (11). Largely, the reports in the literature corroborate the inverse relationship between CRP and endothelium-dependent vasoreactivity in human subjects (12–14).
Despite all the above stated reports in-vitro, there is a paucity of in-vivo data along with plausible mechanistic insights for the inhibitory effect of CRP on eNOS in a valid animal model under steady state conditions. The reports of human CRP effects in mice are conflicting (6). However, human CRP administration in rats has been reported to promote myocardial infarct size (15) and cerebral infarct (16) in coronary artery ligation and cerebral artery occlusion models, respectively. Furthermore, Pepys et al (17) have recently validated the rat as an appropriate model to test the effect of human CRP by blocking CRP’s effects on infarct size with a small molecular weight inhibitor of CRP. We also recently showed the rat to be a valid model to test various pro-inflammatory effects of CRP in terms of increased Ox-LDL uptake, MMP-9 release, superoxide and tissue factor release in macrophages (18,19). Based on aforementioned studies, we explored the in-vivo effect of CRP on vascular reactivity and eNOS inhibition as well as the possible underlying mechanisms following intraperitoneal administration of human CRP in a rat model.
Materials and Methods
Acetylcholine, sodium nitroprusside, Nω-nitro-L-arginine-methyl ester (L-NAME) and human serum albumin were purchased from Sigma-Aldrich. The antibodies to eNOS, p47phox, guanosine triphosphate cyclohydrolase 1 (GTPCH1), and β-actin were obtained from Santa Cruz Biotechnology. CRP was purified from human ascitic/pleural fluids as described (18,19). LPS contamination was 0.125 endotoxin units/ml (~12.5 pg/ml) by the Limulus Assay (Cambrex). Recently, we have shown that our in-house purified, dialyzed CRP mediates its inflammatory effects in TLR4 knocked down cells providing further cogent data that CRP-mediated effects are not due to endotoxin contamination (20).
Animal Treatment
Male Sprague-Dawley rats (weighing 125–150 gm) were obtained from Charles River Lab. The protocol was approved by the animal committee of University of California at Davis and Texas A&M Health Science Center at Temple. The rats were divided into 2 groups; Group 1 (n = 5) - human serum albumin [HuSA], Group 2 (n = 7) - human CRP [CRP]. Additionally, experiments were performed in-vitro on mesenteric arterioles isolated from control rats (no treatment, n = 8). HuSA/CRP (20 mg/kg b.w) was injected intraperitoneally daily for 3 days as reported previously (18,19). The rats were sacrificed on the 4th day by overdose of pentobarbital following an overnight fast and the blood samples were collected for serum. The small intestine was removed following a median incision of the abdomen and immediately placed on iced (5 C) saline for isolation of the mesenteric arterioles for vasodilation/functional studies. Human CRP levels were measured in rat serum, which was done by a high sensitive assay (21) that does not recognize rat CRP. The abdominal aorta was excised, adventitial layer was removed quickly and the remaining aorta was snap-frozen in liquid nitrogen and then stored at − 80°C until use for various biochemical and molecular analysis.
Functional Assessment of Isolated Mesenteric Arterioles
Single third-order arterioles (~1 mm in length; 40–80 μm in internal diameter in situ) were carefully isolated from the mesentery and surrounding adipose tissue using microdissection scissors and forceps (Fine Science Tools, Foster City, CA) with the aid of a stereomicroscope (model SZX12, Olympus). The arteriole was then transferred for cannulation to a Lucite vessel chamber containing physiological salt solution (PSS; in mM: NaCl 145.0, KCl 4.7, CaCl2 2.0, MgSO4 1.17, NaH2PO4 1.2, glucose 5.0, pyruvate 2.0, EDTA 0.02, and MOPS 3.0) with 1% albumin equilibrated with room air at ambient temperature. One end of the arteriole was cannulated using a glass micropipette (tip outer diameter of 30–40 μm) filled with PSS-albumin solution, and the outside of the arteriole was securely tied to the pipette with 11-0 ophthalmic suture (Alcon). The other end of the vessel was cannulated with a second micropipette and also secured with suture. After cannulation, the vessel and pipettes were transferred to the stage of an inverted microscope (model CKX41, Olympus) coupled to a video camera (Sony DXC-190, Labtek) and video micrometer (Cardiovascular Research Institute, Texas A&M Health Science Center) for continuous measurement of the internal diameter throughout the experiment (22). The micropipettes were connected to independent pressure reservoirs. By adjusting the height of the reservoirs, the vessel was pressurized to 60 cm H2O intraluminal pressure without flow.
Experimental Protocols for Mesenteric Arterioles
Cannulated arterioles from HuSA-treated, CRP-treated, or nontreated rats were bathed in PSS-albumin at 36–37 °C to allow development of basal tone. After vessels obtained stable basal tone (~60 min), the concentration-dependent responses to acetylcholine (ACh, 0.1 nM to 0.1 mM) and sodium nitroprusside (SNP, 0.1 nM to 10 μM) were examined. In vessels from control rats without HuSA or CRP treatment, vasodilations to ACh and SNP were evaluated before and after incubation with NOS inhibitor L-NAME (10 μM) (5,23,24) for 30 min or following intraluminal incubation with CRP (7 μg/ml) or boiled CRP (7 μg/ml) for 60 min. Mesenteric arterioles were exposed to each dose of the vasodilator agents for 1–2 min until a stable diameter was established. Recent evidence has shown in rat mesenteric arteries that L-NAME-sensitive NO mediates the stable, sustained component of the dilation to acetylcholine (25). All drugs were dissolved in PSS. At the end of each functional experiment, the vessel was relaxed with 0.1 mM SNP in EDTA (1 mM)-calcium-free PSS to obtain its maximal diameter at 60 cmH2O intraluminal pressure. All diameter changes in response to agonists were normalized to this maximal vasodilation and expressed as a percentage of maximal dilation as previously reported (26).
Tissue Homogenates
The abdominal aortic tissue was homogenized in 0.3 ml of ice cold lysis buffer (Cell Signaling Technology) containing 1% Triton X-100, 25 mM sodium deoxycholate, 150 mM NaCl, 20 mM Tris, pH 7.4, 1 mM EDTA, 200 μM sodium orthovanadate, 2.5 mM sodium pyrophosphate, 1 mM NaF, 1 mM phenylmethysulfonyl fluoride and 2.5% protease inhibitor cocktail. After homogenization, tissue homogenate was kept on ice for 30 min, followed by centrifugation at 10,000×g for 10 min at 4 °C. Cytosolic and membrane fractions were prepared by the method used by Wenzel et al (27) to determine translocation of p47phox from cytosol to membrane.
eNOS Enzymatic Activity
The aortic homogenates were used for the measurement of eNOS enzymatic activity, by assessing the conversion of 14C-L-arginine to 14C-L-citrulline as previously reported (10).
Assay of eNOS Dimer/Monomer
eNOS dimer (active state) and monomer (inactive state) forms were assayed as a measure of eNOS uncoupling in aortic homogenates. The low temperature SDS-PAGE (6% mini gels) was run at 70V using non-boiled cell lysates and reducing sample buffer as described (11).
Detection of Superoxide
Superoxide production in aorta was evaluated with the fluorescent dye dihydroethidium (DHE) (28). Abdominal aortic tissues isolated from HuSA/CRP treated rats in-vivo were stained with DHE (4 μM) for 30 min, washed, and then embedded in OCT compound (Tissue-Tek) for cryostat sections. The embedded tissue was cut into sections 10-μm-thick and placed on glass slides. Images were taken with a fluorescence microscope. Settings for image acquisition were identical for both control and experimental tissues.
CRP and p47phox Translocation
We have previously reported that pharmacologic inhibition (by using apocynin - an inhibitor of p47phox translocation from cytosol to membrane) as well as RNA interference (siRNA to p47phox or to p22phox and not control siRNA) of NADPH oxidase reversed CRP-mediated inhibition of eNOS in HAECs in-vitro (11). In the present study, we assayed for p47phox translocation from cytosol to membrane fractions of abdominal aorta from rats treated with HuSA or CRP in-vivo. Membrane and cytosolic fractions isolated from aortic tissue homogenates were electrophoresed and immunoblotted for p47phox.
CRP, eNOS Uncoupling, BH4 Levels and GTPCH1 Expression
eNOS uncoupling has been linked to reduced tetrahydrobiopterin (BH4) availability (29), thus we tested the hypothesis that CRP-mediated eNOS uncoupling results from decreased BH4 levels. BH4 quantification was performed by high performance liquid chromatography (HPLC) with electrochemical detection (EC-HPLC) as previously described (30). Intracellular concentrations of BH4 were determined using authentic BH4 (10–100 nM) as standards and then normalized to protein content. Since BH4 is synthesized in a de novo fashion from guanosine triphosphate (GTP) via GTPCH1 we also examined the protein expression of GTPCH1 in aortic homogenates by western blotting using goat anti-rat GTPCH1.
For all western blot experiments, the membranes were blocked with 5% milk and then incubated with respective primary antibody (anti-eNOS, anti-GTPCH1, anti-p47phox or anti-β-actin [which served as loading control]) in 5% non-fat dry milk powder/TBS. After washing and incubating with specific HRP-conjugated secondary antibodies, the membranes were developed with enhanced chemiluminescence (Amersham-Pharmacia).
Statistical Analysis
Experimental results are presented as the mean ± SEM. Statistical comparisons of data were performed by Student’s t test or by two-way analysis of variance followed by the Bonferroni multiple-range test, as appropriate. The EC50 values are presented as the negative logarithm (pEC50) and calculated by fitting concentration-dependent response curves to a sigmoidal model of the form log-concentrations versus response using GraphPad Prism software. A value of P < 0.05 was considered significant.
Results
In mesenteric arterioles isolated from control rats, both ACh (Fig 1A) and SNP (Fig 1B) produced concentration-dependent dilation. To assess the involvement of NOS in the vasodilation to these agents, vessels were treated with NOS inhibitor L-NAME. In the presence of L-NAME (10 μM), the basal vascular tone was slightly increased but did not reach statistical significance (before L-NAME: 64±3% of maximal diameter; after L-NAME: 60±4% of maximal diameter; P = 0.08). However, the dilation of these vessels to ACh but not SNP was significantly inhibited (Figure 1), indicating the involvement of endothelial released NO in the ACh-induced vasomotor response. To determine whether CRP can directly influence NO-mediated dilation, the vascular responses to ACh and SNP were assessed before and after intraluminal incubation with CRP (7 μg/ml) for 60 min. Exposure of vessels to CRP did not alter basal tone (Control: 70±7 μm resting diameter, 65±1% of maximal diameter; CRP: 69±9 μm resting diameter; 63±5% of maximal diameter; P = 0.57) or the sensitivity (pEC50) to ACh (Control = 6.86±0.12 vs. CRP = 6.99±0.50; P = 0.99). However, the maximal vasodilation to ACh but not to SNP was significantly attenuated after incubation with CRP (Fig 1A), suggesting the direct inhibitory action of luminal CRP on the efficacy of the ACh-stimulated endothelial NO signaling pathway. Incubation with boiled CRP did not alter resting tone (Control: 76±6 μm resting diameter, 66±4% of maximal diameter; CRP: 80±8 μm resting diameter, 69±4% of maximal diameter; P = 0.08) or arteriolar dilation of to ACh (Fig 1A), supporting the specific inhibitory action of intact CRP on vasodilator function.
Figure 1.
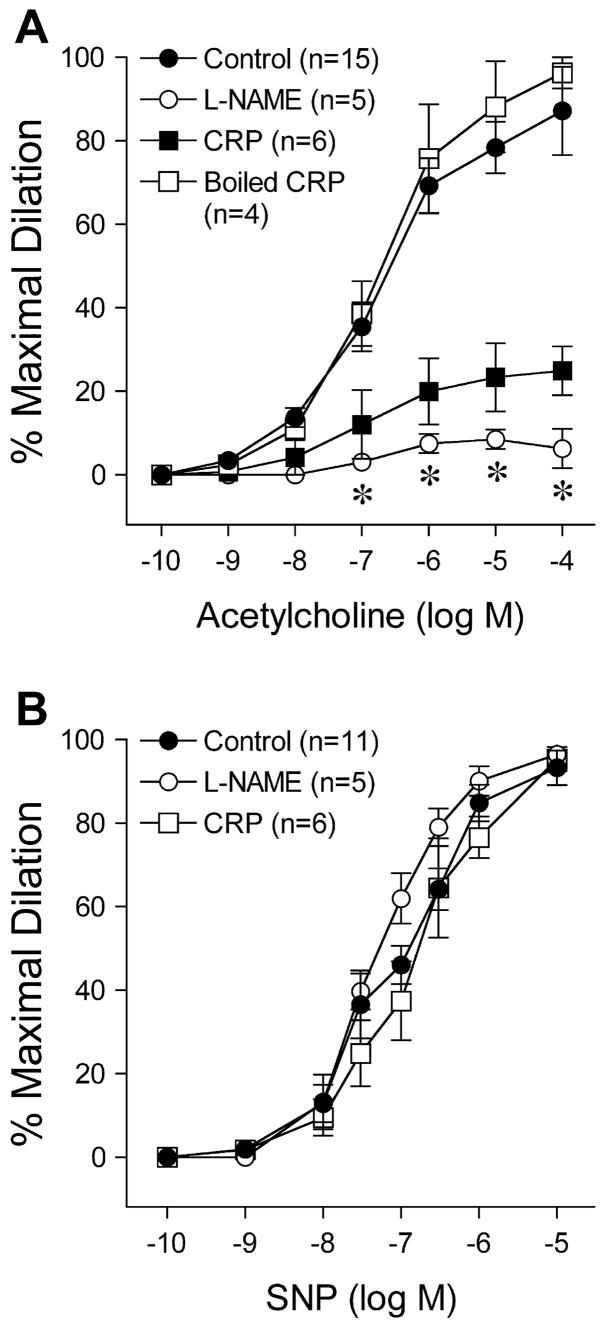
Effect of in-vitro CRP treatment on vascular reactivity. a) Dilation of mesenteric arterioles isolated from control rats to acetylcholine was examined before and after incubation with NOS inhibitor L-NAME (10 μM) for 30 min or with CRP (7 μg/ml) or boiled CRP (7 μg/ml) for 60 min. b) Dilation of mesenteric arterioles to sodium nitroprusside was examined before and after incubation with L-NAME (10 μM) or CRP (7 μg/ml). n = number of vessels/1 per rat. *P<0.05 L-NAME or CRP vs. Control.
Serum levels of CRP were higher in rats following 3-day administration of CRP than HuSA (17±6 μg/ml vs. undetected, CRP and HuSA groups, n = 5 and 7 respectively, P<0.001). The impact of in-vivo CRP treatment on ACh-stimulated vasodilator function was evaluated in mesenteric arterioles isolated from CRP-treated and HuSA-treated rats. Both groups of vessels developed a comparable level of resting basal tone (HuSA: 86±14 μm resting diameter, 71±4% of maximal diameter; CRP: 70±6 μm, 72±2% of maximal diameter; P = 0.86). Similar to the decreased dilations to ACh following L-NAME or CRP treatment in-vitro of control vessels, the maximal vasodilator response (HuSA, 10−4 M ACh: 81±8 % vs. CRP, 10−5 M ACh: 35±5%) but not sensitivity (HuSA: pEC50 = 6.98±0.17 vs. CRP: pEC50 = 7.10±0.32; P = 0.76) to ACh was significantly attenuated in vessels isolated from CRP-treated rats (Fig 2A). In-vivo CRP or HuSA treatment did not alter SNP-induced vasodilation (Fig 2B), indicating that the CRP does not alter ability of the vascular smooth muscle to respond to NO. Overall, our results indicate that CRP treatment in-vivo significantly attenuates endothelium-dependent NO-mediated vasoreactivity.
Figure 2.
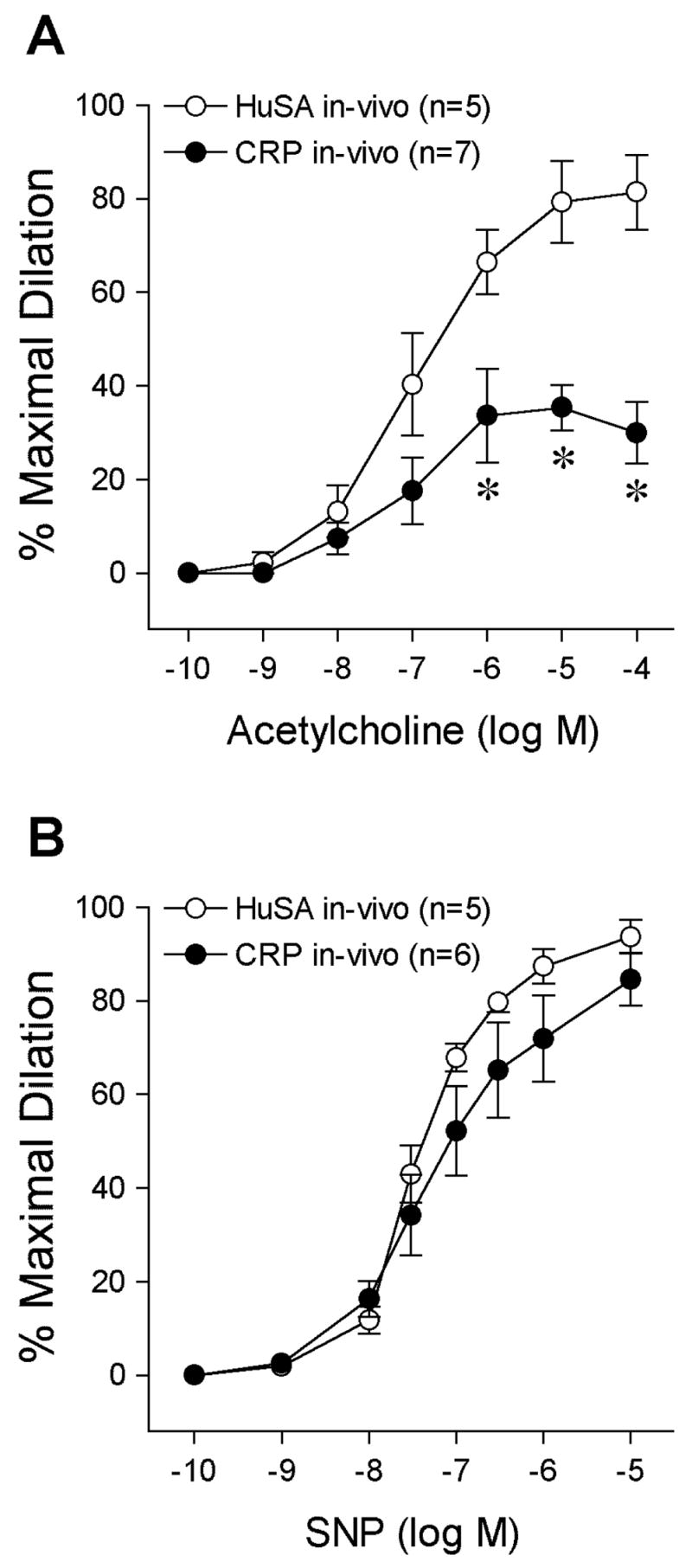
Effect of in-vivo CRP treatment on vascular reactivity. Concentration-dependent dilation of mesenteric arterioles isolated from HuSA-treated or CRP-treated Sprague-Dawley rats to (a) acetylcholine and (b) sodium nitroprusside was examined. n = number of vessels/1 per rat. *P<0.05 vs. HuSA in-vivo.
CRP treatment resulted in significant inhibition of eNOS activity in rat aortic tissue homogenates (P<0.05 compared to HuSA, Fig 3A). Since we recently reported that CRP uncouples eNOS in-vitro in HAEC, we also studied eNOS uncoupling following CRP administration in-vivo. To confirm this, we assayed eNOS dimerization, superoxide production, BH4 levels and GTPCH1 expression in aortic tissues. Endothelial NOS dimerization was altered in aortic tissue following CRP treatment. As depicted in Fig 3B, the ratio of eNOS dimers to monomers was significantly lower (P<0.03) in aortic tissues of CRP-treated than in HuSA-treated rats.
Figure 3.
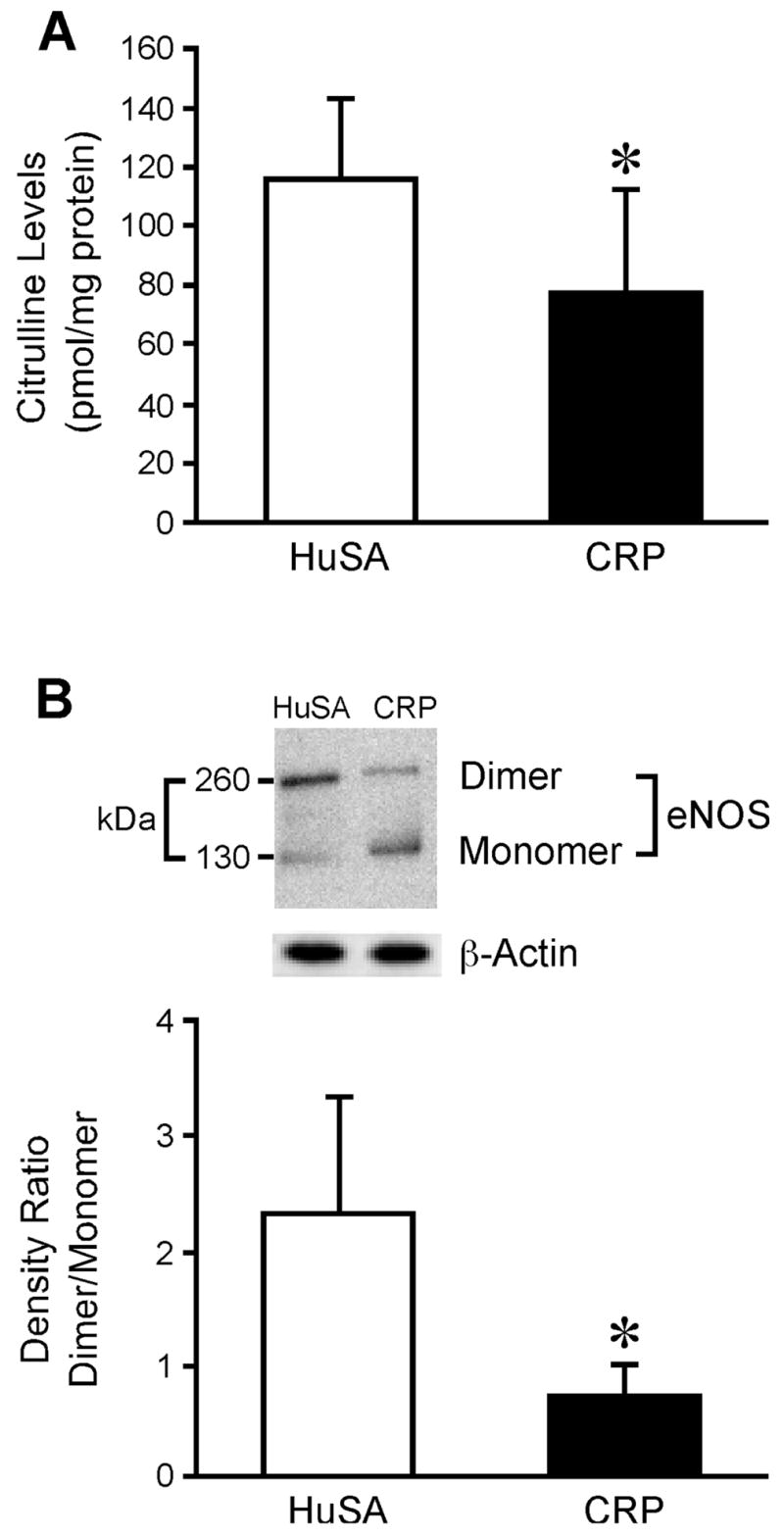
Effect of in-vivo CRP treatment CRP on eNOS activity and eNOS uncoupling. A) Abdominal aortic segments were isolated HuSA-treated or CRP-treated rats and eNOS activity was measured by assessing the conversion of 14C-L-arginine to 14C-L-citrulline in aortic homogenates. Data represent 5 rats per group. *P<0.05 vs. HuSA. B) Abdominal aortic segments were isolated HuSA-treated or CRP-treated rats for immunoblotting of eNOS dimer and monomer.β–actin served as loading control. The top panel shows a representative blot. The bottom panel shows the densitometric ratio of eNOS dimer/monomer. Data represent 4 rats per group. *P<0.03 vs. HuSA.
Since increased superoxide production via NADPH oxidase could result in eNOS inhibition, we explored whether CRP compared to HuSA administration in-vivo induces superoxide production in aortic tissue. In-vivo treatment with CRP markedly increased DHE staining in the endothelial layer, supporting increased vascular superoxide (Fig 4). The p47phox translocation is the key event in NADPH oxidase activation, so we next examined whether CRP treatment in-vivo affects p47phox translocation from cytosol to membrane fraction of abdominal aorta. As shown in Fig 5A, the p47phox level in aortic homogenates from CRP-treated compared to HuSA-treated rats was significantly increased in membrane fractions with concomitant decrease in cytosolic fractions. This translocation supports activation of vascular NADPH oxidase with exposure to CRP in-vivo as evidenced by increased DHE staining.
Figure 4.

Effect of in-vivo CRP treatment on vascular superoxide production. Abdominal aortic segments isolated from HuSA-treated or CRP-treated rats were incubated with oxidative fluorescent dye DHE and the luminal surface of the vessels was imaged using fluorescence microscopy. Endothelial cells are denoted by arrows. Data shown are representative of 4 separate experiments.
Figure 5.
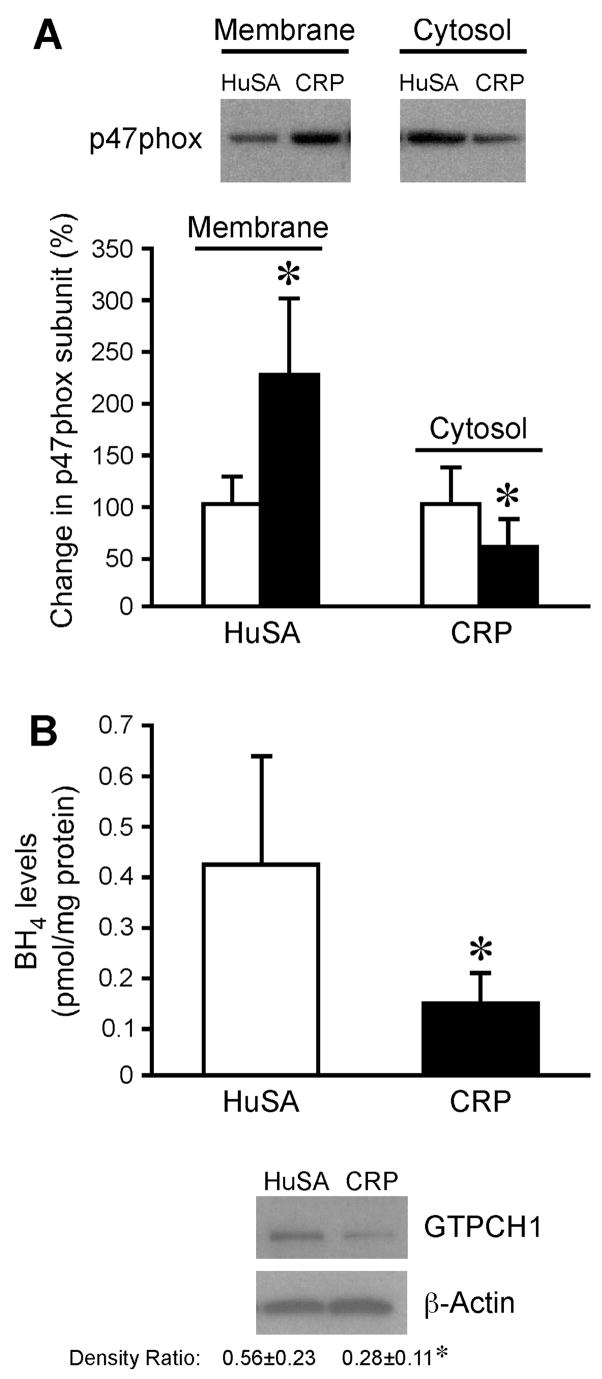
A) Effect of in-vivo CRP treatment on p47phox translocation. Aortic segments were isolated from HuSA-treated and CRP-treated rats and relative protein levels of p47 phox subunit in membrane and cytosol fractions of tissue homogenates were measured. The top panel shows a representative immunoblot using p47phox antibody and the bottom panel shows the densitometric analysis. *P<0.05 vs. HuSA. Data represent 4 rats per group. B) Effect of in-vivo CRP treatment on BH4 levels and GTPCH1 expression. The top panel shows BH4 levels in aortic homogenates isolated from HuSA-treated and CRP-treated rats. The bottom panel shows immunoblots for GTPCH1 and β–actin from these aortic homogenates. The densitometric ratio is provided. Data represent 4 rats per group for both BH4 and GTPCH1 analysis. *P<0.05 vs. HuSA.
The availability of BH4 has been reported to regulate the coupling/uncoupling of eNOS activity. CRP treatment compared to HuSA treatment in-vivo resulted in significant reduction of both BH4 (Fig 5B) and GTPCH1 protein expression in aortic tissue (Fig 5B).
Discussion
CRP induces endothelial dysfunction including inhibition of eNOS. Previously, we and others (9,10) have shown that CRP causes downregulation of eNOS in human aortic and coronary artery ECs. Recently, we identified multiple mechanistic events involved in CRP-mediated eNOS inhibition and documented that NADPH oxidase activation and GTPCH1 downregulation are associated with CRP-mediated eNOS uncoupling in HAECs in-vitro (11). Our present results appear to provide the first evidence for impairment of eNOS-dependent vasodilation following exposure to clinically relevant concentration of human CRP in vivo
Although various investigators have shown in-vivo that CRP Tg mice (7,8) or mice injected with human CRP (28,32) exhibit endothelial dysfunction, these studies had limitations as discussed below. Grad et al (7) have reported suppression of eNOS expression and bioactivity in femoral arteries of CRP Tg mice at 24 hrs following local arterial wire injury, as well as in distant heart and lung tissues, when CRP levels of ~58 μg/ml were achieved. Importantly, they did not reveal any significant effect on baseline eNOS levels between wild type (WT) and CRP Tg mice, although CRP levels reported at baseline were undetected and clinically relevant at ~12 μg/ml, respectively, in these groups of mice. However, endothelium-dependent vasoreactivity was not examined in this study. Teoh et al (8) reported impaired endothelial function, including reduction in vasoreactivity, nitric oxide release, and phosphorylated eNOS protein expression, as well as increase in expression of adhesion molecules in CRP Tg mice. However, these effects were observed only after administration of pro-inflammatory turpentine compared to vehicle control in CRP Tg mice. There was no difference in any of the parameters tested between WT and CRP Tg mice (i.e. at steady state in CRP Tg mice compared to WT mice). Turpentine treatment increased circulating CRP levels to 276 μg/ml. We would like to point out that the CRP levels achieved in this study, which resulted in endothelial dysfunction, are extremely high and can only be achieved with major inflammatory episodes. Furthermore, Schwedler et al (28) have reported that human CRP (2.5 mg/kg, subcutaneous; weekly for 8 weeks) compared to saline (control group) administration in ApoE knockout mice impairs endothelium-dependent vasorelaxation of isolated aortic rings. Since these authors have reported the presence of anti-CRP antibodies in mice following subcutaneous treatment with human CRP, it is speculated that the pathogenic effects observed in their study might be due to the presence of antigen-antibody complexes and not to a CRP effect. The pathophysiologic significance also remains unclear since circulating CRP levels were not reported. Impaired endothelial function in vivo was demonstrated by Mineo et al in C57BL/6 mice (32). Intraperitoneal administration of CRP (250 μg) compared to vehicle control resulted in 50% reduction of ACh-induced increase in carotid artery vascular conductance. Collectively, these earlier studies support the detrimental influence of CRP administration in-vivo on endothelial function. However, no detailed mechanisms have been provided for the CRP-mediated endothelial dysfunction, except that these effects appear to be mediated via FCγ receptors (32).
A valid animal model to test the effect of human CRP has been an issue in the literature in the past. As reviewed recently (6), CRP is not a major acute phase protein in mice. Furthermore, human CRP has been reported to generate immune responses in mice (33) and its impact on atherosclerosis in mice is conflicting (34,35). Thus, testing the effects of human CRP in the mouse model does not seem to be of great relevance. On the other hand, the rat has been used as an alternate model to explore the effects of human CRP in-vivo as recently validated by Pepys et al (17). As described earlier, we have demonstrated increased Ox-LDL uptake, MMP-9 release, and superoxide and tissue factor release in macrophages in Wistar rats following human CRP treatment in-vivo (18,19). These attributes further support the rat rather than the mouse as a suitable model to test effects of human CRP in-vivo. In the present study, we explored the effects of human CRP vis-à-vis HuSA administration in Sprague-Dawley rats on endothelial vasoreactivity and dysfunction with further delineation of underlying mechanisms.
We first tested the effect of human CRP by in-vitro treatment on vasoreactivity of mesenteric arterioles isolated from control rats. It is worthwhile to note that the we have previously reported significant inhibition of endothelium-dependent NO-mediated dilation in porcine coronary (28) and retinal (5) arterioles by CRP treatment in-vitro. In the present study, the vasodilation to ACh but not SNP was significantly attenuated in mesenteric arterioles following incubation with human CRP (7 ug/ml), suggesting the direct inhibitory action of luminal CRP on the endothelial NO signaling pathway. Incubation with boiled CRP failed to have any effect, supporting the action of intact CRP per se on vasodilator function. It is worth noting that CRP levels in the range of 3–10 μg/ml indicate high risk (1,2,6). Our findings suggest that CRP levels known to predict adverse cardiovascular events could also impair endothelial function of mesenteric resistance vessels, and thus influence peripheral blood flow.
Much clinical evidence demonstrates an inverse correlation between CRP levels and endothelial vascular reactivity in human subjects (2,13,14). In the present study, we translated our in-vitro findings with human CRP administration in-vivo resulting in circulating CRP levels of 17 μg/ml. These levels are achieved in human patients. We demonstrated significant attenuation of the endothelium-dependent NO-mediated dilation in mesenteric arterioles. Our findings indicate that the efficacy of the maximal response to acetylcholine is reduced following CRP treatment in-vivo, but the sensitivity or potency remains constant. It is likely that the uncoupling of the receptor-effector signaling cascade for NO production, as delineated in our molecular findings below, contributes to the reduction in maximal vasodilator capacity. Earlier, Ridker and Cook (1) have shown that CRP levels ~20 μg/ml predict future cardiovascular events. Additionally, CRP levels up to 50 μg/ml have been reported in patients with myocardial infarction (36). Thus, the levels of CRP achieved in the circulation by intraperitoneal administration of CRP in rats showing an adverse effect on vasoreactivity can be possibly attained in patients. It appears that CRP did not exert a general impairment of vasodilator function because vasodilation to endothelium-independent pharmacological agent SNP was unaffected. Thus, in the present study, we demonstrate for the first time that in-vivo treatment with human CRP attenuates endothelium-dependent NO-mediated dilation in a rat model at baseline.
Endothelial dysfunction has been linked to uncoupling of eNOS via reduction in BH4 availability (29,37) and/or dimerization of the enzyme with resultant decrease in its functional activity. Administration of BH4 improves endothelial dysfunction in conditions characterized by reduced availability of NO and increased production of reactive oxygen species (14,38–40). Importantly, BH4 availability is regulated by GTPCH1 (29). An earlier report by Cai et al (41) has shown that GTPCH1 gene transfer augments intracellular BH4, as well as eNOS activity, in cultured human endothelial cells. Recently, Takaya et al (42) showed that augmenting BH4 levels in the endothelium by GTPCH1 overexpression reduces atherosclerosis in ApoE KO/eNOS Tg mice. Based on these findings, GTPCH1 is reported to be a potential and rational target to augment endothelial BH4 in reversing eNOS activity in endothelial dysfunctional states. In the present study, we demonstrated a significant reduction in GTPCH1 expression and BH4 levels in rat aortic tissues following CRP administration in-vivo. These findings are consistent with our previous report, where we showed significant reduction in GTPCH1 protein and mRNA with CRP treatment and that sepiapterin, a BH4 analog, rescues the uncoupling of eNOS by CRP in-vitro (11). Overall, it appears that CRP-mediated decreased expression of GTPCH1 underlies the explanation for the deficiency of BH4 induced by CRP. Dimerization is also required for catalytic activity of functional eNOS (43). Our present findings showing significantly decreased eNOS dimer/monomer ratio by CRP administration in-vivo further supports CRP-mediated eNOS uncoupling. Taken together, diminution in both BH4 availability and eNOS dimerization appear to contribute to the endothelial dysfunction following CRP treatment in-vivo.
A growing body of evidence suggests a pivotal role of NADPH oxidase in vascular oxidant stress resulting in eNOS uncoupling (44). We have previously reported that CRP induces the production of superoxide anions in HAECs via upregulation of NADPH oxidase, as evidenced by increased p22phox and p47phox (11) and also inhibits endothelium dependent NO-mediated dilatation by activating p38 kinase and NADPH oxidase in coronary arterioles in-vitro (28). In the present study, we report that CRP administration in-vivo resulted in increased DHE fluorescence (superoxide production) as well as translocation of p47phox from cytosol to membrane, supporting activation of NADPH oxidase in aortic tissue. Overall, these results indicate that CRP administration in-vivo causes NADPH oxidase activation resulting in superoxide production and eNOS uncoupling. Different sources for reactive oxygen species production in vascular cells are NADPH oxidase, mitochondrial respiratory chain enzymes, xanthine oxidase and uncoupled eNOS (44). Earlier, we have delineated that NADPH oxidase and uncoupled eNOS rather than mitochondrial respiratory chain enzymes or xanthine oxidase mediate CRP-induced superoxide production in HAECs in-vitro (11). We have also recently shown in an air pouch model that CRP administration stimulates superoxide release via NADPH oxidase in macrophages harvested from pouch exudates and that this is reversed by pretreatment with apocynin (18). Nonetheless, we document NADPH oxidase activation in intact blood vessels, which might be responsible for the increased superoxide production in the present study. In support of our findings, there is documented link between oxidative stress, NADPH oxidase, BH4 levels and eNOS uncoupling with published evidence that activation of NADPH oxidase-derived superoxide leads to oxidation of BH4 and uncoupling of eNOS (45). Our results are also supported by a recently published report (46) demonstrating that NADPH oxidase plays a very important role in eNOS uncoupling mediated by hypochlorous acid, the major oxidant of leukocyte-derived myeloperoxidase. A limitation of the present study is that we did not measure prostacyclin and endothelin-1 in the arterial tissues.
We have demonstrated a novel effect of human CRP in-vivo in rats without any additional perturbation as reported in studies in mice. CRP specifically impairs endothelium-dependent vasoreactivity at concentrations achievable in patients. Endothelial dysfunction induced by CRP appears linked with uncoupling of eNOS due to reduction in dimerization of the enzyme, as well as inhibition of GTPCH1 and decrease in BH4 levels. CRP also causes the activation of NADPH oxidase resulting in eNOS uncoupling directly or via inhibition of GTPCH1 or oxidation of BH4. Thus, given the importance of CRP-induced pro-oxidative effects and resultant eNOS inhibition, CRP appears to be a key molecule to accentuate endothelial dysfunction. Also, since statins lower both low-density lipoproteins and CRP, both factors that decrease eNOS activity, this might explain in part the significant benefit of statins for cardiovascular events. In future studies, we will test the in-vivo effects of sepiapterin and apocynin on the uncoupling of eNOS by CRP since it is beyond the scope of this study. To conclude, strategies aimed at decreasing CRP (17) may prove to be beneficial in preventing endothelial dysfunction and reducing atherosclerosis-related events.
Acknowledgments
This work was supported by National Institutes of Health Grant K24 AT00596 as well as R01 HL074360 to IJ, R01 HL71761 to L. Kuo, R01 EY018420 to TWH, and R01 HL67244 to JVV. The authors are also grateful to Dr A. Afify for his help rendered in getting cryostat sectioning done for aortic tissues, and to Dr. Xin Xu and Ms. Natalie Xu for their technical assistance.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Ridker PM, Cook N. Clinical usefulness of very high and very low levels of CRP across the full range of Framingham Risk Scores. Circulation. 2004;109:1955–1959. doi: 10.1161/01.CIR.0000125690.80303.A8. [DOI] [PubMed] [Google Scholar]
- 2.Ridker PM. Clinical application of C-reactive protein for cardiovascular disease detection and prevention. Circulation. 2003;107:363–369. doi: 10.1161/01.cir.0000053730.47739.3c. [DOI] [PubMed] [Google Scholar]
- 3.Jialal I, Devaraj S, Venugopal SK. C-reactive protein: risk marker or mediator in atherothrombosis? Hypertension. 2004;44:6–11. doi: 10.1161/01.HYP.0000130484.20501.df. [DOI] [PubMed] [Google Scholar]
- 4.Ballou SP, Lozanski G. Induction of inflammatory cytokine release from cultured human monocytes by CRP. Cytokine. 1992;4:361–368. doi: 10.1016/1043-4666(92)90079-7. [DOI] [PubMed] [Google Scholar]
- 5.Nagaoka T, Kuo L, Ren Y, et al. C-reactive protein inhibits endothelium-dependent nitric oxide-mediated dilation of retinal arterioles via enhanced superoxide production. Invest Ophthalmol Vis Sci. 2008;49:2053–2060. doi: 10.1167/iovs.07-1387. [DOI] [PubMed] [Google Scholar]
- 6.Verma S, Devaraj S, Jialal I. Is C-reactive protein an innocent bystander or proatherogenic culprit? C-reactive protein promotes atherothrombosis. Circulation. 2006;113:2135–2150. [PubMed] [Google Scholar]
- 7.Grad E, Golomb M, Mor Yosef I, et al. Transgenic expression of human CRP suppresses endothelial nitric oxide synthase expression and bioactivity following vascular injury. Am J Physiol Heart Circ Physiol. 2007;293:H489–H495. doi: 10.1152/ajpheart.01418.2006. [DOI] [PubMed] [Google Scholar]
- 8.Teoh H, Quan A, Lovren F, et al. Impaired endothelial function in C-reactive protein overexpressing mice. Atherosclerosis. 2008 doi: 10.1016/j.atherosclerosis.2008.02.034. [Epub ahead of print] [DOI] [PubMed] [Google Scholar]
- 9.Verma S, Wang CH, Li SH, et al. A self-fulfilling prophecy: CRP attenuates nitric Oxide production and inhibits angiogenesis. Circulation. 2002;106:913–919. doi: 10.1161/01.cir.0000029802.88087.5e. [DOI] [PubMed] [Google Scholar]
- 10.Venugopal SK, Devaraj S, Yuhanna I, et al. Demonstration that CRP decreases eNOS expression and bioactivity in human aortic endothelial cells. Circulation. 2002;106:1439–1441. doi: 10.1161/01.cir.0000033116.22237.f9. [DOI] [PubMed] [Google Scholar]
- 11.Singh U, Devaraj S, Vasquez-Vivar J, et al. C-reactive protein decreases endothelial nitric oxide synthase activity via uncoupling. J Mol Cell Cardiol. 2007;43:780–791. doi: 10.1016/j.yjmcc.2007.08.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Cleland SJ, Sattar N, Petrie JR, et al. Endothelial dysfunction as a possible link between C-reactive protein levels and cardiovascular disease. Clin Sci. 2000;98:531–535. [PubMed] [Google Scholar]
- 13.Fichtlscherer S, Rosenberger G, Walter DH, et al. Elevated CRP and impaired endothelial reactivity in patients with CAD. Circulation. 2000;102:1000–1006. doi: 10.1161/01.cir.102.9.1000. [DOI] [PubMed] [Google Scholar]
- 14.Tomai F, Crea F, Gaspardone A, et al. Unstable angina and elevated CRP levels predict enhanced vasoreactivity of the culprit lesion. Circulation. 2001;104:1471–1476. doi: 10.1161/hc3801.096354. [DOI] [PubMed] [Google Scholar]
- 15.Griselli M, Herbert J, Hutchinson WL, et al. C-reactive protein and complement are important mediators of tissue damage in acute myocardial infarction. J Exp Med. 1999;190:1733–1740. doi: 10.1084/jem.190.12.1733. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Gill R, Kemp JA, Sabin C, et al. Human CRP increases cerebral infarct size after middle cerebral artery occlusion in adult rats. J Cereb Blood Flow Metab. 2004;24:1214–1218. doi: 10.1097/01.WCB.0000136517.61642.99. [DOI] [PubMed] [Google Scholar]
- 17.Pepys MB, Hirschfield GM, Tennent GA, et al. Targeting CRP for the treatment of CVD. Nature. 2006;440:1217–1221. doi: 10.1038/nature04672. [DOI] [PubMed] [Google Scholar]
- 18.Devaraj S, Dasu MR, Singh U, et al. C-reactive protein stimulates superoxide anion release and tissue factor activity in vivo. Atherosclerosis. 2008 doi: 10.1016/j.atherosclerosis.2008.05.060. [Epub ahead of print] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Singh U, Dasu MR, Yancey PG, et al. Human C-reactive protein promotes oxidized low density lipoprotein uptake and matrix metalloproteinase-9 release in Wistar rats. J Lipid Res. 2008;49:1015–1023. doi: 10.1194/jlr.M700535-JLR200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Dasu MR, Devaraj S, Du Clos, et al. Biological effects of CRP are not due to endotoxin contamination: evidence from toll like receptor 4 knock-down human aortic endothelial cells. J Lipid Res. 2006;48:509–512. doi: 10.1194/jlr.C600020-JLR200. [DOI] [PubMed] [Google Scholar]
- 21.Jialal I, Stein D, Balis D, et al. Effect of hydroxymethyl glutaryl coenzyme a reductase inhibitor therapy on high sensitive C-reactive protein levels. Circulation. 2001;103:1933–1935. doi: 10.1161/01.cir.103.15.1933. [DOI] [PubMed] [Google Scholar]
- 22.Kuo L, Chilian WM, Davis MJ. Interaction of pressure- and flow-induced responses in porcine coronary resistance vessels. Am J Physiol. 1991;261:H1706–H1715. doi: 10.1152/ajpheart.1991.261.6.H1706. [DOI] [PubMed] [Google Scholar]
- 23.Hein TW, Kuo L. cAMP-independent dilation of coronary arterioles to adenosine: role of nitric oxide, G proteins, and KATP channels. Circ Res. 1999;85:634–642. doi: 10.1161/01.res.85.7.634. [DOI] [PubMed] [Google Scholar]
- 24.Dubois-Aubecq V, Davvy M, Midol-Monnet M, Cohen Y. cGMP release in rat mesenteric arterioles and in conduit mesenteric artery. J Auton Pharmacol. 1996;16:7–11. doi: 10.1111/j.1474-8673.1996.tb00350.x. [DOI] [PubMed] [Google Scholar]
- 25.Harrington LS, Carrier MJ, Gallagher N, Gilroy D, Garland CJ, Mitchell JA. Elucidation of the temporal relationship between endothelial-derived NO and EDHF in mesenteric vessels. Am J Physiol Heart Circ Physiol. 2007;293:H1682–H1688. doi: 10.1152/ajpheart.00389.2007. [DOI] [PubMed] [Google Scholar]
- 26.Hein TW, Kuo L. LDLs impair vasomotor function of the coronary microcirculation: role of superoxide anions. Circ Res. 1998;83:404–414. doi: 10.1161/01.res.83.4.404. [DOI] [PubMed] [Google Scholar]
- 27.Wenzel P, Daiber A, Oelze M, et al. Mechanisms underlying recoupling of eNOS by HMG-CoA reductase inhibition in a rat model of streptozotocin-induced diabetes mellitus. Atherosclerosis. 2008;198:65–76. doi: 10.1016/j.atherosclerosis.2007.10.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Qamirani E, Ren Y, Kuo L, et al. CRP inhibits endothelium-dependent NO-mediated dilation in coronary arterioles by activating p38 kinase and NAD(P)H oxidase. Arterioscler Thromb Vasc Biol. 2005;25:995–1001. doi: 10.1161/01.ATV.0000159890.10526.1e. [DOI] [PubMed] [Google Scholar]
- 29.Vasquez-Vivar J, Kalyanaraman B, Martasek P, et al. Superoxide generation by endothelial nitric oxide synthase: the influence of cofactors. Proc Natl Acad Sci USA. 1998;95:9220–9225. doi: 10.1073/pnas.95.16.9220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Whitsett J, Martasek P, Zhao H, et al. Endothelial cell superoxide anion radical generation is not dependent on eNOS-serine 1179 phosphorylation and eNOS dimer/monomer distribution. Free Radic Biol Med. 2006;40:2056–2068. doi: 10.1016/j.freeradbiomed.2006.02.001. [DOI] [PubMed] [Google Scholar]
- 31.Schwedler SB, Kuhlencordt PJ, Ponnuswamy PP, et al. Native CRP induces endothelial dysfunction in apoE−/− mice: implications for iNOS and reactive oxygen species. Atherosclerosis. 2007;195:e76–84. doi: 10.1016/j.atherosclerosis.2007.06.013. [DOI] [PubMed] [Google Scholar]
- 32.Mineo C, Gormley AK, Yuhanna IS, et al. Fc gamma RIIB mediates CRP inhibition of endothelial NO synthase. Circ Res. 2005;97:1124–1131. doi: 10.1161/01.RES.0000194323.77203.fe. [DOI] [PubMed] [Google Scholar]
- 33.Klein TC, Döffinger R, Pepys MB, et al. Tolerance and immunity to the inducible self antigen C-reactive protein in transgenic mice. Eur J Immunol. 1995;25:3489–3495. doi: 10.1002/eji.1830251242. [DOI] [PubMed] [Google Scholar]
- 34.Hirschfield GM, Gallimore JR, Kahan MC, et al. Transgenic human CRP is not proatherogenic in apolipoprotein E-deficient mice. Proc Natl Acad Sci USA. 2005;102:8309–8314. doi: 10.1073/pnas.0503202102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Paul A, Ko KW, Li L, et al. C-reactive protein accelerates the progression of atherosclerosis in apolipoprotein E-deficient mice. Circulation. 2004;109:647–655. doi: 10.1161/01.CIR.0000114526.50618.24. [DOI] [PubMed] [Google Scholar]
- 36.Pietila KO, Harmoinen AP, Jokinitty J, Pasternack AI. Serum CRP concentration in acute myocardial infarction and its relationship to mortality during 24 months of follow-up in patients under thrombolytic treatment. Eur Heart J. 1996;17:1345–1349. doi: 10.1093/oxfordjournals.eurheartj.a015068. [DOI] [PubMed] [Google Scholar]
- 37.Topal G, Brunet A, Millanvoye E, et al. Homocysteine induces oxidative stress by uncoupling of NO synthase activity through reduction of tetrahydrobiopterin. Free Radic Biol Med. 2004;36:1532–1541. doi: 10.1016/j.freeradbiomed.2004.03.019. [DOI] [PubMed] [Google Scholar]
- 38.Heitzer T, Brockhoff C, Mayer B, et al. Tetrahydrobiopterin improves endothelium dependent vasodilation in chronic smokers: evidence for a dysfunctional nitric oxide synthase. Circ Res. 2000;86:e36–41. doi: 10.1161/01.res.86.2.e36. [DOI] [PubMed] [Google Scholar]
- 39.Maier W, Cosentino F, Lutolf R, et al. Tetrahydrobiopterin improves endothelial function in patients with coronary artery disease. J Cardiovasc Pharmacol. 2000;35:173–178. doi: 10.1097/00005344-200002000-00001. [DOI] [PubMed] [Google Scholar]
- 40.Stroes E, Kastelein J, Cosentino F, et al. Tetrahydrobiopterin restores endothelial function in hypercholesterolemia. J Clin Invest. 1997;99:41–46. doi: 10.1172/JCI119131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Cai S, Alp NJ, McDonald D, et al. GTP cyclohydrolase I gene transfer augments intracellular tetrahydrobiopterin in human endothelial cells: effects on nitric oxide synthase activity, protein levels and dimerisation. Cardiovasc Res. 2002;55:838–849. doi: 10.1016/s0008-6363(02)00460-1. [DOI] [PubMed] [Google Scholar]
- 42.Takaya T, Hirata K, Yamashita T, et al. A specific role for eNOS-derived reactive oxygen species in atherosclerosis progression. Arterioscler Thromb Vasc Biol. 2007;27:1632–1637. doi: 10.1161/ATVBAHA.107.142182. [DOI] [PubMed] [Google Scholar]
- 43.Klatt P, Schmidt K, Lehner D, et al. Structural analysis of porcine brain nitric oxide synthase reveals a role for tetrahydrobiopterin and L-arginine in the formation of an SDS-resistant dimer. EMBO J. 1995;14:3687–3695. doi: 10.1002/j.1460-2075.1995.tb00038.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Griendling KK, Sorescu D, Ushio-Fukai M. NAD(P)H oxidase: role in cardiovascular biology and disease. Circ Res. 2000;86:494–501. doi: 10.1161/01.res.86.5.494. [DOI] [PubMed] [Google Scholar]
- 45.Landmesser U, Dikalov S, Price SR, et al. Oxidation of tetrahydrobiopterin leads to uncoupling of endothelial cell nitric oxide synthase in hypertension. J Clin Invest. 2003;111:1201–1209. doi: 10.1172/JCI14172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Xu J, Xie Z, Reece R, et al. Uncoupling of endothelial nitric oxidase synthase by hypochlorous acid: role of NAD(P)H oxidase-derived superoxide and peroxynitrite. Arterioscler Thromb Vasc Biol. 2006;26:2688–2695. doi: 10.1161/01.ATV.0000249394.94588.82. [DOI] [PubMed] [Google Scholar]