

Abstract
The D405N and Y546F mutations of the human lutropin receptor (hLHR) have previously been shown to partially attenuate hCG-stimulated cAMP synthesis despite normal cell surface expression and hCG binding affinity (Min, L. and Ascoli, M. Mol. Endocrinol. 14:1797–1810, 2000). We now show that these mutations each stabilize a resting state of the hLHR. A combined mutant D405N,Y546F is similarly expressed at the cell surface and exhibits normal ligand binding, but is profoundly signaling impaired. Introduction of hLHR(wt) into cells stably expressing the signaling inactive D405N,Y546F resulted in attenuation of hCG-stimulated cAMP production by hLHR(wt) even if excess Gs is co-expressed. Similarly, co-expression of D405N,Y546F with hLHR constitutively active mutants (CAMs) attenuated their constitutive activity. Quantitative bioluminescence resonance energy transfer (BRET) analyses demonstrated that D405N,Y546F formed heterodimers with both wt and CAM hLHR. In contrast hLHR(D405N,Y546F) did not heterodimerize with the melanocortin 3 receptor (MC3R) and agonist-stimulated cAMP production through the MC3R was not attenuated when these two receptors were co-expressed. Taken altogether, our data demonstrate that a signaling inactive hLHR mutant (that is trafficked normally to the plasma membrane) attenuates the signaling of cell surface localized wt or constitutively active hLHR due to receptor heterodimerization. Our studies, therefore, suggest a novel ramification of GPCR signaling resulting from receptor dimerization.
Keywords: lutropin receptor, G protein coupled receptor, dimerization, bioluminescence resonance energy transfer
1. Introduction
The human lutropin receptor (hLHR) is a member of the rhodopsin-like Family A group of G protein-coupled receptors (GPCRs) expressed primarily in the gonads which mediates the effects of pituitary LH (males and females) or placental hCG (pregnant females) in reproductive physiology. It is composed of the prototypical GPCR serpentine region attached to a relatively large extracellular domain that confers high affinity hormone binding [1].
In the context of current models describing receptor activation [2], the cell surface hLHR is postulated to be in equilibrium between conformations representing different inactive and active states, with agonist binding stabilizing the hLHR in an active conformation. Although other signaling pathways may also be invoked by activation of the hLHR [3–5], the primary second messenger pathway activated by the hLHR is the Gs/adenylyl cyclase/PKA pathway. Numerous naturally occurring constitutively activating mutants (CAMs) of the hLHR have been described that result in gonadotropin-independent precocious puberty in young boys [6, 7]. Although the degree of activation of hLHR CAMs is lower than that observed when wild-type (wt) hLHR is maximally stimulated by hormone [8], CAMs nonetheless can provide important insights into structural alterations of the hLHR that mark its active state. Proteolysis experiments demonstrated a more rapid loss of hormone binding activity from cells expressing hLHR CAMs as compared to wild-type hLHR, indicating structural differences between the wt receptor and CAMs [9]. Molecular dynamics simulations of the wt hLHR and hLHR CAMs suggest that the active conformations of the hLHR are best described by an opening of the cytosolic crevice between intracellular loops 2 and 3 and between helices 5 and 6 [8, 10–15]. Potential exposure of the cytoplasmic end of helix 6 upon activation of the hLHR is further suggested by studies showing that a peptide corresponding to the cytoplasmic end of the rat LHR directly stimulated Gs [16]. The predicted structural alterations accompanying activation of hLHR are in general agreement with recent crystallographic studies on the active state of opsin bound to a peptide corresponding to the C-terminal portion of Gα, which was observed to interact extensively with residues in the cytoplasmic end of TM6 [17].
Naturally occurring loss-of-function mutations of the hLHR have also been described, most of which, as with similar mutations in other GPCRs, result from improper receptor folding and retention (to varying degrees depending on the extent of misfolding) in the endoplasmic reticulum, thus limiting cell surface expression [6, 7, 18–20]. Only one naturally occurring hLHR mutant has been shown to be signaling-impaired [21]. However, this mutant receptor also shows severely decreased cell surface expression such that, even if it were signaling competent, a loss-of-function phenotype would still be observed. Therefore, to study hLHR mutations resulting solely in impaired Gs activation, without the confounding effects of changes in cell surface expression, it has been necessary to utilize mutants of laboratory design. The goal of the present study was to examine the mechanisms underlying such intrinsically signaling impaired hLHR mutants. Our results suggest that the mutations D405N and Y546F stabilize the inactive state of the hLHR. Our data further show that a profoundly signaling impaired hLHR(D405N,Y546F) mutant, when co-expressed with either wt or CAM hLHRs, attenuates signaling of the latter and this is due to the formation of receptor heterodimers.
2. Materials and Methods
2.1. Plasmids and hormones
The cDNA for wt hLHR was kindly given to us by Ares Advanced Technology (Ares-Serono Group, Randolph, MA) and inserted into pcDNA3.1(neo). Mutations were introduced using standard methodologies. Where indicated, an epitope tag (HA) was placed at the N-terminus of the mature hLHR protein. This modification did not affect hLHR cell surface expression, binding or signaling properties. 3HA-tagged human melanocortin 3 receptor (MC3R), Gαs, Gβ1, and Gγ2, each in pcDNA3.1(neo), were purchased from Missouri S&T cDNA Resource Center (Rolla, MO; www.cdna.org). Plasmids were prepared using Qiagen maxiprep kits (Qiagen, Valencia, CA) and sequences were confirmed by automated DNA sequencing (performed by the DNA Core Facility of the University of Iowa Carver College of Medicine). Highly purified recombinant hCG was purchased from Dr. A. Parlow and the NIDDK National Hormone and Pituitary Program. A crude preparation of hCG, used only for the determination of non-specific binding in hormone binding assays, was purchased from Sigma (St. Louis, MO). NDP-MSH was purchased from Phoenix Pharmaceuticals (Burlingame, CA).
2.2. Cells and transfections
Human embryonic kidney (HEK) 293 and 293T cells were obtained from the American Type Tissue Collection and were maintained, plated for experiments, and transiently transfected as previously described [22]. When transfecting cells with varying concentrations of plasmid containing receptor cDNA, the total amount of plasmid was kept constant using empty vector. Stably transfected cells were established by transfecting cells with vectors containing a neo-selectable marker. After removing the transfection cocktail, the cells were cultured in growth media supplemented with 700 µg/ml G418 (selection media). Cells were grown in selection media for at least two weeks prior to use in experiments and were maintained in selection media.
2.3. 125I-hCG binding assays to evaluate cell surface hLHR expression
Maximal cell surface binding capacity was determined as previously described using intact cells incubated with 125I-hCG (500 ng/ml final concentration) with or without an excess of unlabeled crude hCG (50 IU/ml final concentration) [8].
2.4. Flow cytometry to evaluate cell surface receptor expression
On the day of the experiment, HEK293 cells were washed once with filtered PBS for immunohistochemistry (PBS-IH, 137 mM NaCl, 2.7 mM KCl, 1.4 mM KH2PO4, 4.3 mM Na2HPO4, pH 7.4). Cells were then detached by washing with PBS-IH, collected by gentle centrifugation, and resuspended in PBS-IH. They were incubated 1 hour at 4C with or without HA.11 monoclonal anti-HA antibody from Covance (Princeton, NJ) diluted 1:200 with PBS-IH containing 0.5% BSA (PBS-IH/BSA), washed with PBS-IH/BSA, and then incubated 1 hour at 4C with phycoerythrin-conjugated goat anti-mouse IgG from BD Biosciences (San Jose, CA) diluted 1:250 (Figure 4–Figure 6) or FITC-conjugated goat anti-mouse IgG (1:350) from Sigma (St. Louis, MO) (Figure 9) in PBS-IH/BSA. Cells were washed with PBS-IH/BSA, resuspended in 1 ml PBS-IH/BSA, and filtered through a 70 µm BD Falcon cell strainer into a clean test tube on ice. A BD DiVa fluorescence-activated cell sorter with a 488 nm laser was then used to quantify cell surface expression in 10,000 cells from each transfection. Control gating was set using cells transfected with empty vector and stained with antibody as described. Arbitrary fluorescence units describing total cell surface fluorescence were determined as the product of the percent cells gated and the geometric mean fluorescence of the sample minus the respective product in the control (empty vector) group.
Figure 4. Transient co-expression of hLHR(D405N) or hLHR(Y546F) with hLHR(wt) results in a slight attenuation of hormone-stimulated cAMP at low concentrations of hCG and a potentiation of cAMP at high concentrations of hCG.
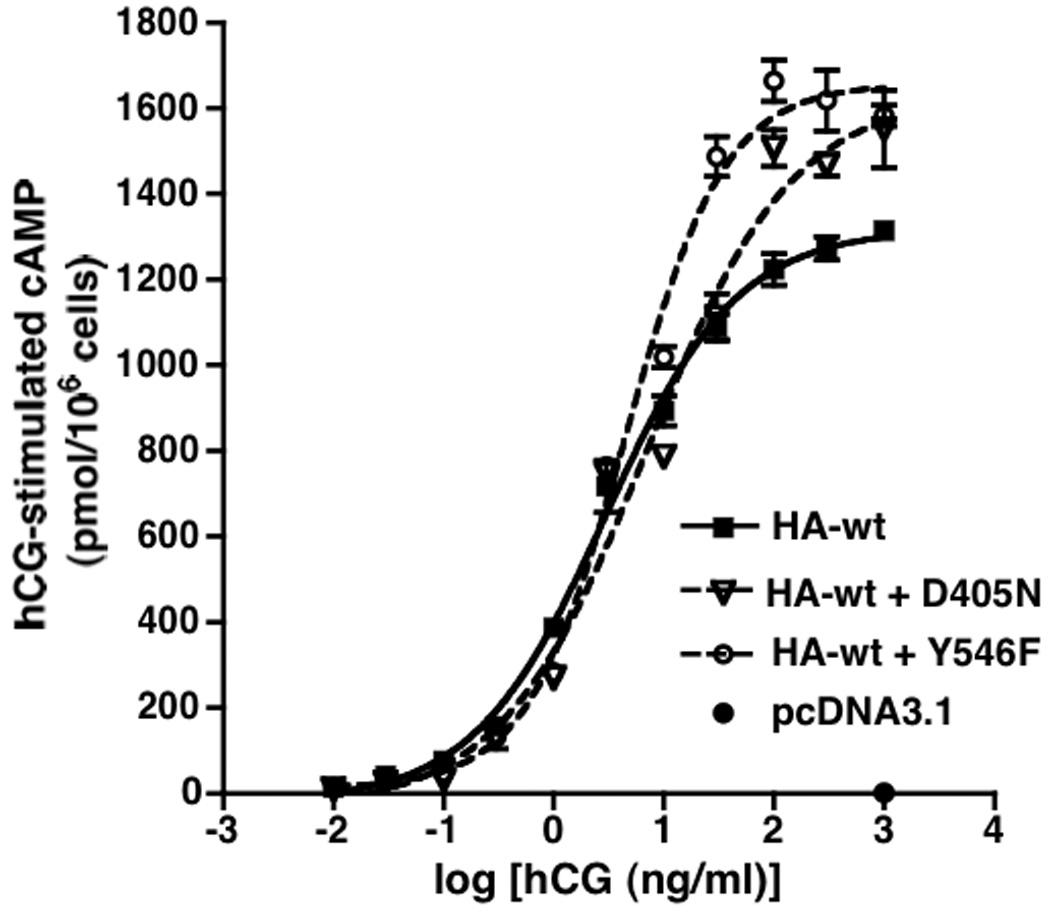
HEK293 cells were transiently transfected with HA-hLHR(wt) alone, or co-transfected with HA-hLHR(wt) plus hLHR(D405N) or HA-hLHR(wt) plus hLHR(Y546F) under conditions where the cell surface expression of HA-hLHR(wt) in each group of cells, as determined in the same experiment by flow cytometry, was similar. The accumulation of intracellular cAMP in response to different concentrations of hCG was determined. Data shown are the mean ± SEM of triplicate determinations within a single experiment that is representative of two independent experiments. In the experiment shown, the flow cytometric data for cell surface HA-hLHR(wt) expression in cells transfected with HA-hLHR(wt) alone, HA-hLHR(wt) plus hLHR(D405N), and HA-hLHR(wt) plus hLHR(Y546F) were 1170, 1153, and 1057 relative light units, respectively.
Figure 6. Attenuation of hCG-stimulated cAMP by hLHR(wt) when co-expressed with the signaling inactive hLHR(D405N,Y546F) is associated with heterodimerization.
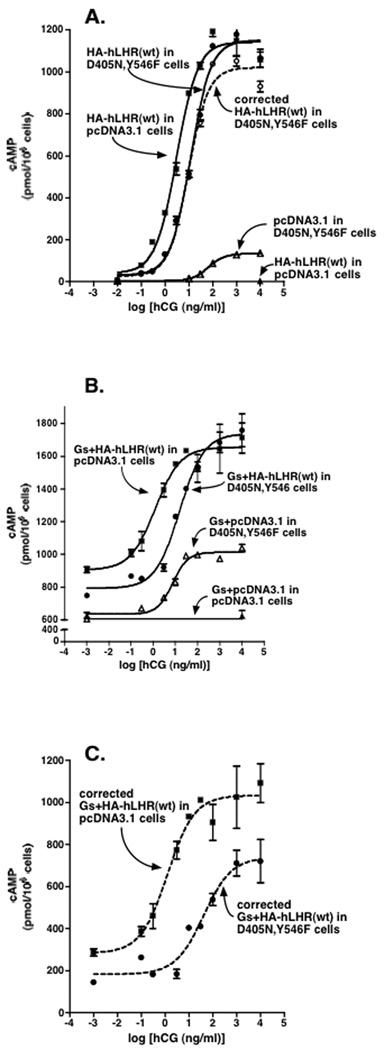
In the same experiment, HEK293 cells stably expressing hLHR(D405N,Y546F) or empty vector pcDNA3.1 were each transfected with HA-hLHR(wt) without (Panel A) or with (Panel B) Gαs, Gβ1, and Gγ2 referred to in the text as Gs). Flow cytometry data for cell surface HA-hLHR(wt) expressed in the hLHR(D405N,Y546) cells vs. empty vector cells were 227 and 240, respectively, in Panel A and 340 and 360, respectively, in Panel B. Accumulation of intracellular cAMP in response to increasing concentrations of hCG was determined and is shown by the solid symbols and solid lines as indicated. Panel A: Solid symbols and lines depict raw data. The open circles and dashed line depict corrected hCG-stimulated cAMP values for D405N,Y546F cells transfected with HA-hLHR(wt) in which the values obtained for hCG-stimulated cAMP in D405N,Y546F cells transfected with empty vector were subtracted. Panel B: Raw data are shown. Note the break in the y-axis and the elevated levels of cAMP under all conditions due to expression of recombinant Gs. Panel C: The data in Panel C represent those from Panel B that have been corrected as follows. The cAMP values for Gs+pcDNA3.1 in pcDNA3.1 cells were subtracted from Gs+HA-hLHR(wt) in pcDNA3.1 cells and the cAMP values for Gs+pcDNA3.1 in D405N,Y546F cells were subtracted from Gs+HA-hLHR(wt) in D405N,Y546F cells. Data shown are the mean ± SEM of triplicate determinations within a single experiment, representative of three independent experiments.
Figure 9. hLHR(D405N,Y546F) does not form heterodimers with the MC3R and does attenuate signaling activity of the MC3R.
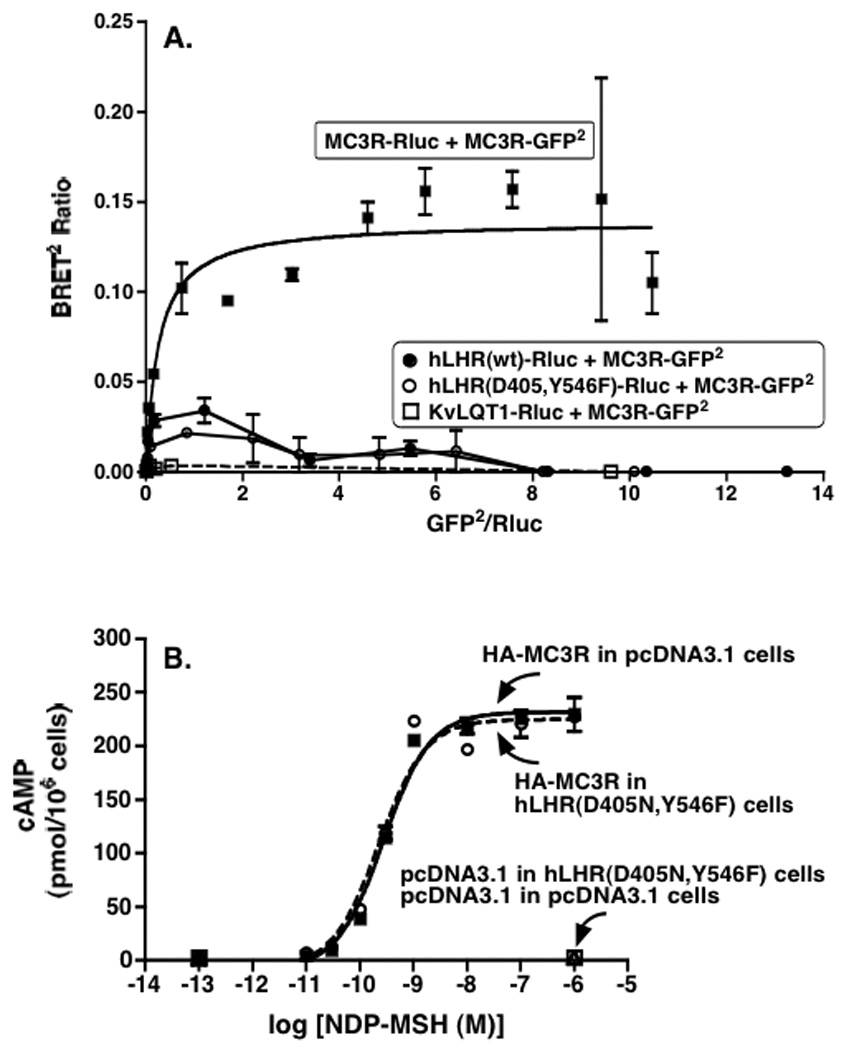
Panel A: HEK293T cells were transiently co-transfected with the indicated pairs of Rluc and GFP2 fusion proteins. For each set, the concentration of the Rluc fusion protein was held constant and the concentrations of the GFP2 fusion protein were increased. The concentrations of the Rluc fusion proteins were chosen so that their expression levels (as determined by luciferase activity) in the absence of any GFP2 fusion protein were the same. The co-expression of KvLQT1 with the MC3R was included as a negative control. Data shown are the mean ± range of duplicate net BRET ratios determined as a function of Rluc/GFP2 expression taken from one experiment that is representative of two independent experiments. Panel B: HEK293 cells stably expressing hLHR(D405N,Y546F) or empty pcDNA3.1 were each transiently transfected with HA-MC3R under conditions where the cell surface expression of MC3R in both groups (as determined in the same experiment by flow cytometry) was similar. Accumulation of intracellular cAMP in response to increasing concentrations of NDP-MSH was determined. Data shown are the mean ± SEM of triplicate determinations within a single experiment, representative of four independent experiments. In the experiment shown, the flow cytometric data for cell surface HA-MC3R when expressed in the hLHR(D405N,Y546F) cells vs. the empty vector cells were 768 and 772 relative light units, respectively.
2.5. Determination of intracellular cAMP production
Intracellular cAMP in response to hCG (100 ng/ml final concentration) was measured as described previously [8].
2.6. BRET assays
HEK293T cells were transiently co-transfected with vectors encoding Rluc-fusion or GFP2-fusion proteins. In a given experiment, the total amount of plasmid transfected was made constant by the addition of empty vector. On the day of the experiment, cells were washed twice with calcium and magnesium free D-PBS, and then detached from the well in 1ml D-PBS. Detached cells were collected by gentle centrifugation and the cell pellet was resuspended in a small volume of D-PBS. Cells were transferred to a white-bottomed 96 well microplate (white Optiplate; Perki[8]nElmer Life and Analytical Sciences, Waltham, MA) such that all samples were of equal volume and protein concentration. Total fluorescence of the cell suspensions was measured using a POLARstar Optima plate reader (BMG LABTECH, Offenburg, Germany) with an excitation filter at 485 nm and an emission filter at 520 nm and was corrected for fluorescence measured in cells transfected with empty vector only. The substrate Coelenterazine 400a (Biosynth; Zurich, Switzerland) was then added at a final concentration of 5 µM and readings at 410/80 nm (reflecting bioluminescence catalyzed by Rluc) and 515/30 nm (reflecting resonance energy transfer from Rluc to GFP2) were measured simultaneously. Bioluminescence readings were corrected for those obtained from cells transfected with empty vector only. BRET ratios were calculated as the ratio of the light emitted by the receptor-GFP2 (515/30 nm) over the light emitted by the receptor-Rluc (410/80 nm). The BRET ratios reported were corrected by subtracting the ratios obtained when receptor-Rluc was expressed alone. For saturation curves, cells were co-transfected with a fixed concentration of cDNA for Rluc fusion protein and increasing concentrations of a GFP2 fusion protein. When more than one curve was generated in a given experiment, the concentrations of plasmids encoding the Rluc fusion proteins were adjusted so that, after substrate addition, luminescence values of the Rluc fusion proteins expressed alone were similar. Data are expressed as the net BRET ratio relative to the ratio of acceptor to donor plotted using GraphPad Prism (San Diego, CA).
3. Results
3.1. hLHR mutations that stabilize an inactive state of the receptor
hLHR(D405N) and hLHR(Y546F) are mutations of laboratory design that result in normal levels of cell surface expression and hormone binding, but attenuated signaling responses to hCG [23]. To initially confirm that under our experimental conditions we would observe similar results, HEK293 cells were transfected with hLHR(wt), hLHR(D405N) or hLHR(Y546F) under conditions yielding similar cell surface receptor expression levels. Consistent with previously published results [23], we observed that D405N and Y546F each exhibited a partial attenuation of hCG-stimulated cAMP production, characterized both by increases in the EC50 and reductions in maximal responsiveness (Figure 1).
Figure 1. hLHR(D405N) and hLHR(Y546F) are signaling impaired mutants.
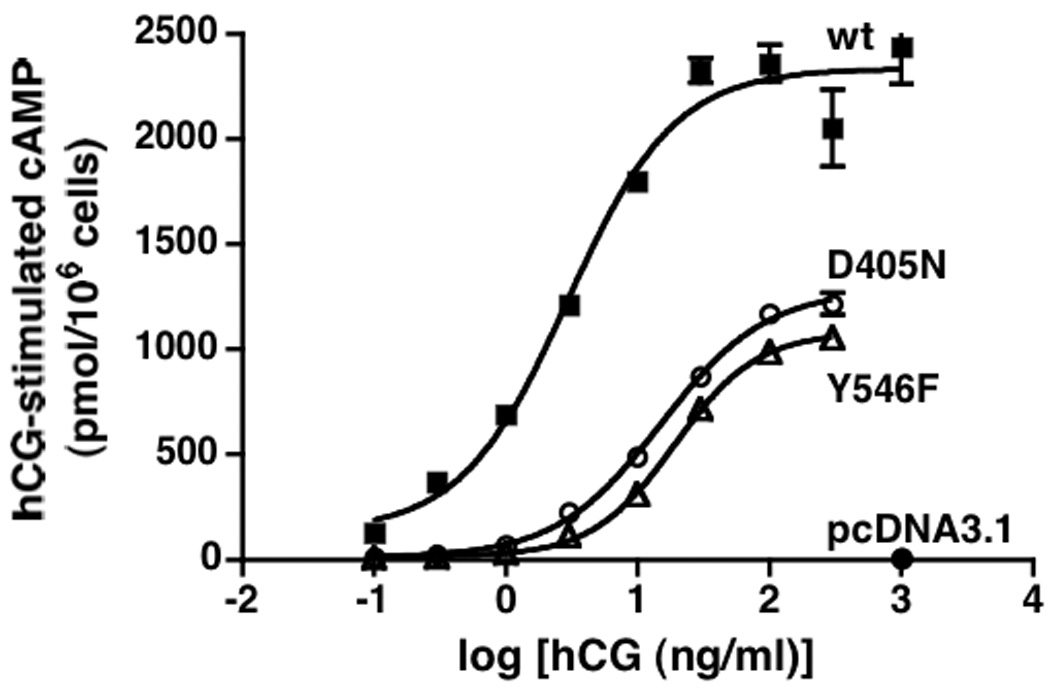
HEK293 cells were transiently transfected with hLHR(wt), hLHR(D405N), or hLHR(Y546F) under conditions where the cell surface expression of each, as determined in the same experiment by 125I-hCG binding to intact cells, was the same. Accumulation of intracellular cAMP in response to increasing concentrations of hCG was determined. Data shown are the mean ± SEM of triplicate determinations within a single experiment, which is representative of at least three independent experiments. In the experiment shown, the cells expressing hLHR(wt), hLHR(D405N), and hLHR(Y546F) bound 18.9, 20.4, and 22.6 ng hCG/106 cells, respectively.
The properties of the signaling-impaired mutant hLHR(D405N) were examined further over a range of cell surface receptor densities and in the context of both basal and hormone-stimulated activity. Within the same experiment, we also examined the activities of three different hLHR CAMs (A373V, L457R, and D578Y) alone and with the D405N mutation also introduced into each construct. As we had reported previously [8, 14] and is observed in the data shown in Figure 2, hormone-stimulated cAMP by the wt hLHR and the basal activities of some hLHR CAMs are not linear with respect to cell surface hLHR expression, preventing data reduction calculating a ratio of cAMP produced as a function of receptor expression, and confirming the rationale for performing experiments over a range of receptor densities. This experiment shows that, at all receptor densities examined, the maximal hormone responsiveness of D405N was significantly reduced compared to wt hLHR (Figure 2A). Notably, over the same range of receptor levels, the basal activity of the hLHR was also significantly reduced by introduction of the D405N mutation (Figure 2B). Furthermore, the introduction of the D405N mutation into three different hLHR CAMs also resulted in an attenuation of constitutive activity (Panels C, D, and E of Figure 2). The most profound effects were on A373V and D578Y, which exhibited little or no constitutive activity when D405N was introduced. In contrast, while the L457R CAM showed a marked attenuation in a background of D405N, it nonetheless still retained significant constitutive activity. The same experimental protocol was used to examine the effects of the Y546F mutation on basal and hormone-stimulated signaling of wt hLHR over a range of receptor levels and also on the activities of the same hLHR CAMs. The results, shown in Figure 3, are remarkably similar to those observed for the D405N mutation. Taken altogether, the data in Figure 2 and Figure 3 suggest D405N and Y546F do not specifically inhibit the efficacy of hCG, but rather stabilize a resting state conformation of the hLHR, thus inhibiting not only agonist-stimulated receptor activity, but also basal activities of wt hLHR and hLHR CAMs.
Figure 2. hLHR(D405N) exhibits a partial attenuation of both basal and hormone-stimulated cAMP over a range of receptor densities and, when introduced into hLHR CAMs, attenuates their constitutive activities.
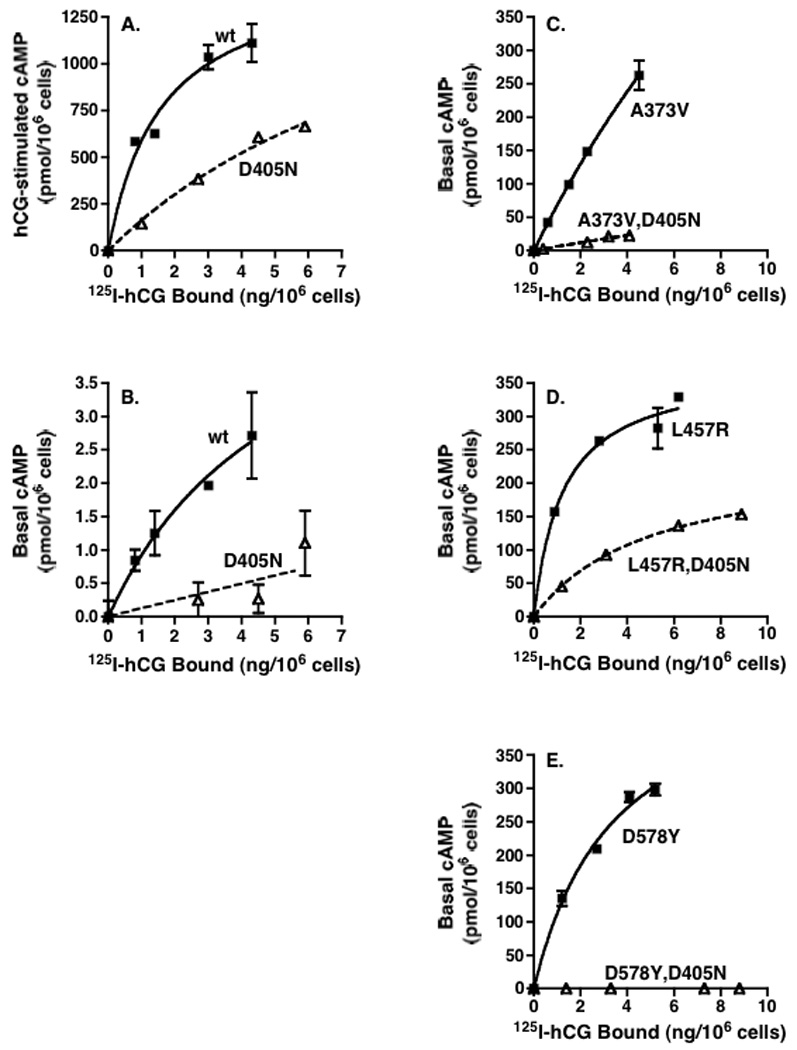
HEK293 cells were transiently transfected with varying plasmid concentrations of the indicated constructs. Intracellular cAMP concentrations as a function of increasing cell surface receptor density (determined in the same experiment by 125I-hCG binding to intact cells) were measured in response to a saturating concentration of hCG (Panel A) and under basal conditions (Panel B, C, D, and E). Data shown are the mean ± SEM of triplicate determinations within a single experiment that contained all panels. The experiment shown is representative of two independent experiments.
Figure 3. hLHR(Y546F) exhibits a partial attenuation of both basal and hormone-stimulated cAMP over a range of receptor densities and, when introduced into hLHR CAMs, attenuates their constitutive activities.
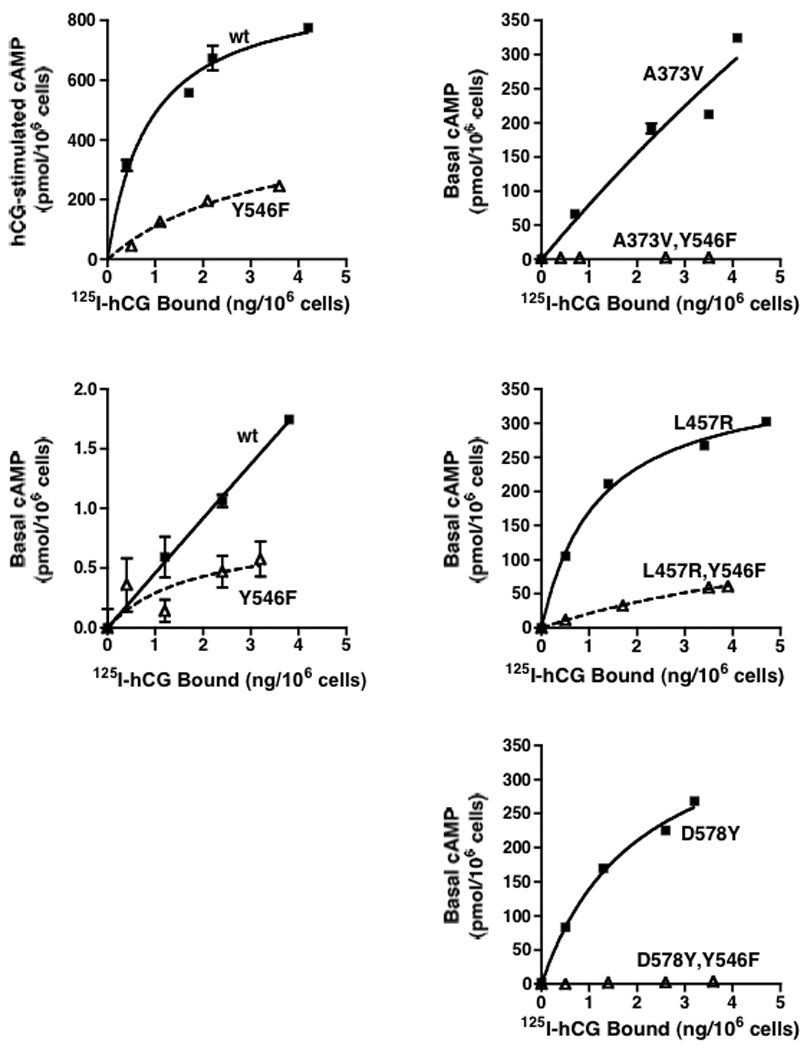
HEK293 cells were transiently transfected with varying plasmid concentrations of the indicated constructs. Intracellular cAMP concentrations as a function of increasing cell surface receptor density (determined in the same experiment by 125I-hCG binding to intact cells) were measured in response to a saturating concentration of hCG (Panel A) and under basal conditions (Panel B, C, D, and E). Data shown are the mean ± SEM of triplicate determinations within a single experiment that contained all panels. The experiment shown is representative of two independent experiments.
3.2. Attenuation of signaling of a wt or CAM hLHR by co-expression of a cell surface localized signaling inactive hLHR
Because the hLHR can constitutively self-associate into dimers and higher order oligomers (referred to hereafter simply as dimers) ([19, 22, 24], we wished to ascertain if a signaling impaired hLHR mutant could modify the signaling properties of co-expressed wt hLHR. To initially address this question, we transiently co-transfected HEK293 cells with D405N or Y546F mutants and HA-tagged hLHR(wt). The hCG responsiveness of these cells was compared to a parallel group of cells in the same experiment co-transfected with HA-hLHR(wt) and empty vector. Flow cytometry was used to verify similar levels of HA-hLHR(wt) cell surface expression in the two groups of cells. Under these conditions, a slight attenuation of hCG-stimulated cAMP production was observed at low hCG concentrations and a potentiation of hCG-stimulated cAMP production was seen at high concentrations of hCG (Figure 4). It is likely that the greater stimulation of cAMP production provoked by high concentrations of hCG when the signaling impaired mutants were co-expressed is due to the ability of the D405N and Y456F mutants to respond to high concentrations of hCG (see Figure 1). However, the attenuation observed at low hCG concentrations suggested an inhibitory effect of the signaling impaired mutants on the wt receptor.
A potential inhibitory effect on the wt receptor would be expected to be greater if the signaling-impaired mutant were completely devoid of activity. Towards that end, we tested hLHR(K605R), which had been reported to be incapable of signaling [25]. However, in our studies, hCG-stimulated cAMP dose response curves in cells expressing comparable levels of cell surface K605R were very similar to those observed for the D405N and Y546F mutants, suggesting that the signaling properties of K605R were only partially impaired (data not shown). An hLHR mutant that is nearly devoid of signaling activity was created though, by combining the D405N and Y546F mutations. As shown in Figure 5, when the D405N,Y546F mutant was expressed at the same cell surface density as the hLHR(wt) it exhibited little or no hCG stimulation of cAMP formation even at high concentrations of hormone. It should be noted that, in addition to being expressed normally at the cell surface, hLHR(D405N,Y546F) also bound hCG with the same affinity as the wt receptor (data not shown). Therefore, hLHR(D405N,Y546F) is a trafficking competing but signaling inactive mutant.
Figure 5. hLHR(D405N,Y546F) is a signaling inactive mutant with little or no responsiveness to hCG.
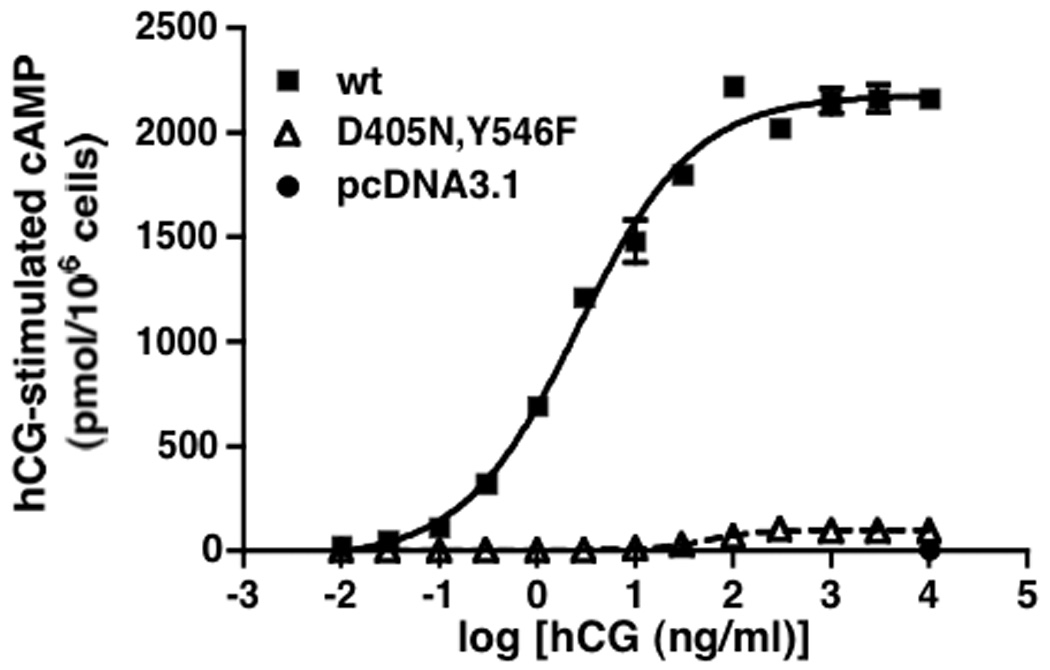
HEK293 cells were transiently transfected with the hLHR(wt), hLHR(D405N,Y546F), or empty vector under conditions where the cell surface expression of each, as determined in the same experiment by 125I-hCG binding to intact cells, was the same. Accumulation of intracellular cAMP in response to increasing concentrations of hCG was determined. Data shown are the mean ± SEM of triplicate determinations within a single experiment, which is representative of three independent experiments. In the experiment shown, the cells expressing hLHR(wt) and hLHR(D405N,Y546F) bound 8.9 and 8.7 ng hCG/106 cells, respectively.
The experiment shown in Figure 6A was then performed to examine if co-expression of the signaling inactive D405N,Y546F mutant with the wt hLHR would have any effect on signaling of the wt receptor. To achieve a higher probability of the wt receptor being co-expressed with the signaling inactive mutant, we created a line of cells stably expressing D405N,Y546F and, as a control, a line stably expressing the empty vector only. HA-hLHR(wt) was then transiently transfected into each of these two stable cell lines and flow cytometry was used to confirm that the levels of cell surface wt receptor were similar in the two cell lines. In addition, transfection of the wt hLHR was performed so that its cell surface expression would be 5-10-fold less than that of the signaling inactive mutant. Hormone binding assays with the untransfected D405N,Y546F stable cell line and to the pcDNA3.1 stable cell line transfected with wt hLHR were used to independently determine cell surface expression levels of wt hLHR and D405N,Y546F within a given experiment.) Because the D405N,Y546F stable cells respond weakly to high concentrations of hCG, hCG-provoked cAMP responses of D405N,Y546F cells transfected with wt hLHR are shown with and without subtraction of hCG-stimulated cAMP formation elicited in untransfected D405N,Y546F cells (Figure 6A). Regardless of the data correction, we observed a 3–4 fold increase in the EC50 for hCG stimulated cAMP production in D405N,Y546F stable cells transfected with wt hLHR as compared to pcDNA3.1 stable cells transfected with wt hLHR.
One possible explanation for the above results is that the signaling inactive mutant engages Gs in a non-productive manner, thus sequestering it. If so, then one would predict that attenuation of wt receptor signaling by D405N,Y546F would not be observed if heterotrimeric Gs were overexpressed. To test this, we performed the same experiment as above, but under the conditions where the cells stably transfected with empty vector or D405N,Y546F were transiently co-transfected with Gαs, Gβ, and Gγ in addition to wt hLHR or empty vector (Figure 6B). As would be expected if Gs were over-expressed, basal and hormone-stimulated levels of cAMP were markedly increased. While Figure 6B depicts the actual levels of cAMP measured in the two stable cell lines transiently transfected with hLHR and Gs, Figure 6C reflects corrected cAMP values after subtracting those values obtained in the stable cell lines transiently transfected with empty vector and Gs. Regardless of whether or not the data are corrected, the results indicate that, even in the presence of additional Gs, one observes an attenuation of signaling through the hLHR(wt) when co-expressed with D405N,Y546F (Figure 6, Panels B and C). The above experiments show that co-expression of D405N,Y546F with the wt hLHR causes an attenuation of signaling by the wt receptor without affecting cell surface expression of the wt hLHR and this attenuation is not due to a sequestration of Gs by D405N,Y546F.
Additional studies were performed to ascertain if the activities of hLHR CAMs would be similarly attenuated by D405N,Y546F. To address this question, increasing amounts of myc-labeled hLHR CAMs (A373V, L457R, or D578Y) were transiently transfected into cell lines stably expressing D405N,Y546F or empty vector and cell surface expression of the myc-hLHR CAM was determined using flow cytometry. As shown in Figure 7, an attenuation of constitutive activity of each of the CAMs was observed over a range of cell surface receptor levels when they were co-expressed with D405N,Y546F.
Figure 7. Co-expression of the signaling inactive hLHR(D405N,Y546F) with hLHR CAMs causes an attenuation of their constitutive activities.
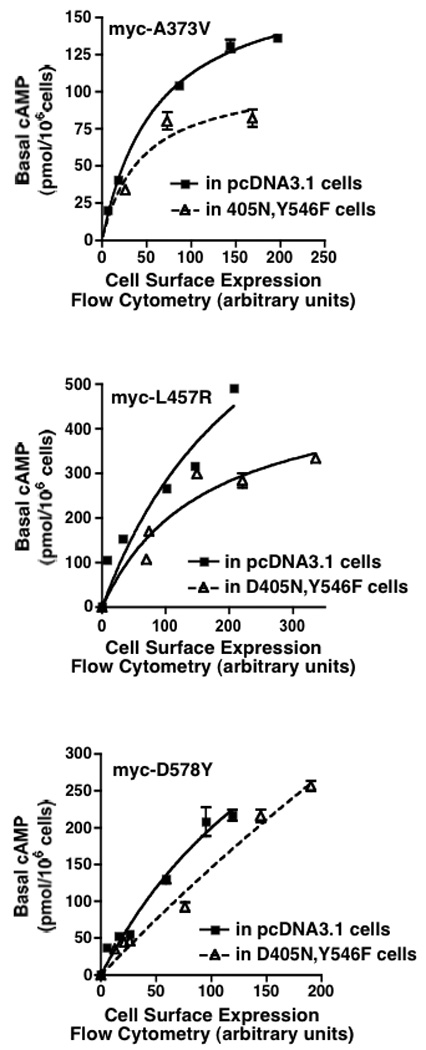
HEK293 cells stably expressing hLHR(D405N,Y546F) or empty vector were each transiently transfected with increasing concentrations of myc-hLHR(A373V) (Panel A), myc-hLHR(L457R) (Panel B), or with myc-hLHR(D578Y) (Panel C). Basal levels of cAMP were measured as a function of cell surface receptor density of the CAM (determined by flow cytometry). Data shown are the mean ± SEM of triplicate determinations within a single experiment, representative of three independent experiments.
3.3. Heterodimerization of the signaling inactive hLHR with wt or CAM hLHR
To determine if the attenuating effects of the D405N,Y546F signaling inactive mutant on the wt or CAM hLHR may be due to dimerization between the signaling inactive and signaling active forms of the hLHR, we utilized BRET to determine if, in living cells, physical interactions between the receptors could be detected. First examining the potential interaction of D405N,Y546F with wt hLHR, cells were transiently co-transfected with a fixed concentration of the energy donor hLHR(wt)-Rluc and increasing concentrations of either the energy acceptor hLHR(wt)-GFP2 or hLHR(D405N,Y546F)-GFP2. As shown in Figure 8A, in both cases, as the ratio of Rluc to GFP2 expression increased, the BRET ratio increased and then reached a plateau, indicative of a saturable process that results from a specific interaction between energy donor and acceptor [26, 27]. As a negative control, cells were co-transfected with the plasma membrane-localized, voltage-gated K+ channel KvLQT1 fused to Rluc and hLHR(wt)-GFP2. In the latter case, little or no BRET signal was detected. These data demonstrate that, in addition to the wt hLHR forming homodimers, it can also heterodimerize with the signaling inactive D405N,Y546F (Figure 8A). Each of the hLHR CAMs was similarly shown to be capable of heterodimerizing with D405N,Y546F as determined by BRET saturation experiments (Figure 8B). It should be noted that, under the conditions used, the BRET ratios reflect only dimeric receptor associations. The data do not, however, rule out higher order receptor associations that may also be occurring.
Figure 8. hLHR(D405N,Y546F) forms heterodimers with wt and CAM hLHRs as determined by BRET.
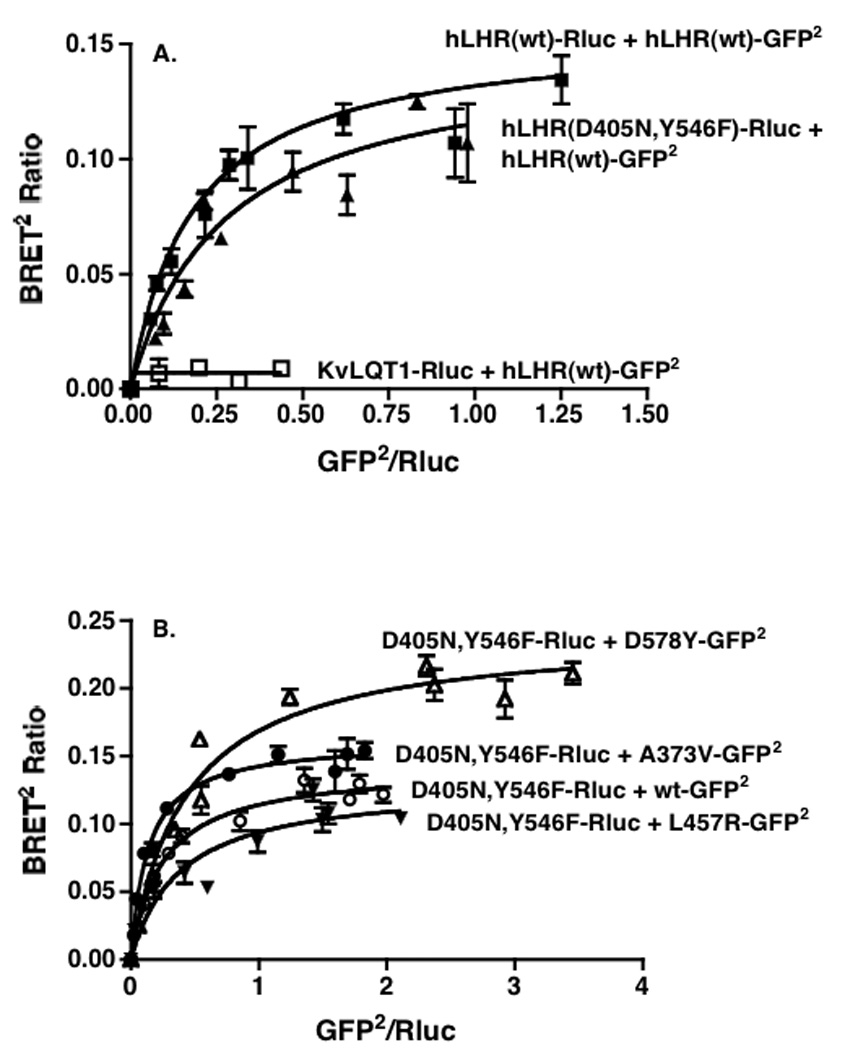
HEK293T cells were transiently co-transfected with the indicated pairs of Rluc and GFP2 fusion proteins. For each pair, the concentration of the Rluc fusion protein was held constant and the concentrations of the GFP2 fusion protein were increased. Within the pairs shown in a given panel, the concentrations of the Rluc fusion proteins were chosen so that their expression levels (as determined by luciferase activity) in the absence of any GFP2 fusion protein were the same. The data in Panels A and B are from different experiments. Data shown are the mean ± range of duplicate net BRET ratios determined as a function of Rluc/GFP2 expression taken from one experiment that is representative of three independent experiments.
If the attenuating effects of D405N,Y546F on the signaling properties of wt and CAM hLHRs was attributable to receptor heterodimerization, and not to changes in downstream signaling events due to expression of D405N,Y546F, then we would not expect to observe an attenuation of signaling through a Gs-coupled receptor that did not heterodimerize with D405N,Y546F. Towards that end, we screened several Gs-coupled receptors in BRET assays and determined that the MC3R did not show specific heterodimerization with either the wt hLHR or the signaling inactive D405N,Y546F (Figure 9A). Although the experiment shown was performed with MC3R as the energy acceptor, we similarly did not observe specific heterodimerization when MC3R was the energy donor (data not shown). In preliminary experiments (not shown) performed in D405N,Y546F cells, we determined the plasmid concentration of MC3R that yielded the same cell surface expression as the wt hLHR under the conditions shown in Figure 6. When MC3R was expressed at those cell surface levels in hLHR(D405N,Y546F) cells, we did not observe an attenuation of agonist-stimulated cAMP as compared to the empty vector cells expressing the same cell surface levels of MC3R (Figure 9B). These data suggest, therefore, that the ability of the signaling inactive hLHR(D405N,Y546F) mutant to exert an attenuating effect on the signaling of a co-expressed GPCR is dependent on its ability to heterodimerize with the GPCR and not due to downstream effects mediated by the signaling inactive mutant.
4. Discussion
The partially inactivating hLHR mutants D405N and Y546F, as previously reported [28] and confirmed herein, cause a partial attenuation of hCG-stimulated cAMP production in spite of normal cell surface receptor expression and normal hCG binding affinity. This is in marked contrast to most naturally occurring inactivating hLHR mutations, as well as inactivating mutations of other GPCRs, which result from intracellular retention of misfolded receptors by cellular quality control systems and, therefore, decreased cell surface expression [20]. As we have now shown, the D405N and Y546F mutants also inhibit the basal activity of the wt hLHR. Furthermore, the introduction of either mutation into hLHR CAMs results in a significant decrease in constitutive activity. A direct interaction of Gs with Asp405 can be ruled out since this residue is located centrally in TM2. Although Tyr546 is located at the predicted cytoplasmic end of TM5, structural studies of opsin in its G protein-interacting conformation also suggest that this tyrosine residue does not directly interact with G protein [17]. Therefore, the attenuating effects of the D405N and Y546F mutations on basal, hormone-stimulated, as well as mutation-induced constitutive activity of the hLHR can best be explained by their stabilizing an inactive conformation of the hLHR. Although there are no known inverse agonists of the hLHR, the effects of these mutations would be consistent with the predicted actions of an inverse agonist [29]. Consistent with decreased signaling activity of the D405N and Y546F hLHR mutants, it had previously been shown that they each exhibited decreased association with arrestin-3 [28] and a decreased rate of hCG-induced receptor internalization as compared to the wt receptor [23].
It has become increasingly accepted that GPCR dimerization is a general phenomenon, with the physiological ramifications of GPCR homo- and heterodimerization under intense investigation (see refs. [30–32] for recent reviews). With respect to the hLHR, studies from our laboratory [19, 22] and from Urizar et al. [24] have shown that the hLHR constitutively self-associates. Under basal conditions, hLHR dimers and oligomers have been detected by co-immunoprecipitation [19] and by BRET in living cells [22, 24]. We have further shown that hLHR dimerization can be detected in the plasma membrane as well as the ER, suggesting that they are formed early in the biosynthetic pathway and transported to the cell surface as dimers or oligomers [22]. Indeed, misfolded mutants of the hLHR, retained in the ER can nonetheless still be observed to specifically dimerize. Consequently, co-expression of a misfolded hLHR mutant with wt receptor resulted in decreased hormone responsiveness due to heterodimerization of the misfolded mutant with the wt receptor in the ER and the consequent reduction in cell surface expression [19, 22]. In this respect, many so-called inactivating or loss-of-function mutants of other GPCRs have similarly been shown to be a result of protein misfolding and subsequent intracellular retention [20].
The D405N and Y546F mutants are thus quite distinct from GPCR folding mutants because these mutants are expressed normally at the cell surface and have normal binding affinity. Despite this, they remain signaling impaired, having decreased basal activities and attenuated responses to hCG as compared to wt hLHR. Because they also attenuated constitutive activities of hLHR CAMs when introduced into those receptors, it appears that the D405N and Y546F mutations stabilize the hLHR in an inactive state. A much more profoundly signaling inactive receptor was created by simultaneously introducing these two mutations into the hLHR. Importantly, the D405N,Y546 mutant was similarly expressed at the cell surface and it also exhibited normal binding affinity. Therefore, unlike other inactivating hLHR mutants that cause a loss-of-function cellular phenotype due to misfolding and intracellular retention, hLHR(D405N,Y546F) is a trafficking competent mutant that is intrinsically signaling inactive.
Remarkably, co-transfection of wt hLHR into cells stably expressing the signaling inactive D405N,Y546F mutant resulted in an attenuation of hCG-stimulated signaling by the wt receptor as compared to the same cell surface levels of wt hLHR expressed in cell lines stably transfected with only empty vector. Importantly, these data show an attenuation of wt hLHR activity in the absence of any change in cell surface wt receptor expression, indicating that it is the activity of the cell surface receptor per se that is being affected by the signaling inactive mutant. Constitutive activities of hLHR CAMs were also shown to be attenuated when they were expressed in the D405N,Y546F background. Again, the attenuation of CAM activity could not be attributed to decreased cell surface CAM expression. Therefore, co-expression of the signaling inactive D405N,Y546F with wt or constitutively active forms of the hLHR results in decreased signaling activity of the cell surface active receptor. BRET titration curves indicate specific heterodimerization of the signaling inactive mutant with the wt hLHR and with the hLHR CAMs. In contrast, no heterodimerization was observed between hLHR(D405N,Y546F) and the MC3R and consequently no attenuation of MC3R signaling by the signaling inactive hLHR was detected.
One possible explanation for the attenuating effects of D405N,Y546F on a co-expressed wt or CAM hLHRs is that Gs might have been be non-productively engaged by D405N,Y546F, essentially sequestering Gs from the wt receptor. Our data argue against this because (i) attenuation of signaling by co-expressed wt hLHR is observed even in the presence of an excess of co-expressed Gs, and (ii) attenuation of signaling by co-expressed MC3R, another Gs-coupled receptor, was not observed. Another possibility is that the attenuating effects of D405N,Y546F on wt or CAM hLHRs were due to alterations in one or more signaling molecules affecting signaling through Gs-coupled receptors in general. The lack of attenuation of MC3R signaling argues against this possibility as well. These findings, therefore, strongly suggest that the basis for the attenuating effects of the signaling inactive mutant on the active hLHRs is due to the formation of receptor dimers with the wt hLHR.
Recent studies suggest that two GPCRs physically associate with one G protein and that activation of only one of the GPCR protomers is sufficient to activate the G protein [33–36]. However, even within a GPCR homodimer, there are data to suggest that the protomers may not be fully equivalant with respect to G protein activation, implying a functional asymmetry of the GPCR protomers within a dimeric complex [35–37]. Interestingly, when a GPCR is forced to be monomeric, it is still capable of activating G protein [37–39]. While these findings indicate that a dimeric form of the GPCR is not a priori required for GPCR activation, they do not refute the findings that suggest that physiologically the dimeric form of the GPCR most likely associates with and activates G proteins. Therefore, as we consider the possible mechanisms by which the heterodimerization of D405N,Y546F with a wt or CAM hLHR attenuates the active receptor, we do so thinking within the conceptual framework of the hLHR heterodimer associating with a single Gs. Because we deliberately maintained a much higher ratio of signaling inactive mutant to active receptor, it is reasonable to assume that all or most of the wt or CAM receptor would be heterodimerized with the signaling inactive mutant.
Two different scenarios, which are not mutually exclusive, are consistent with the data presented. One scenario to consider is if, within heterodimers of signaling inactive hLHR and wt or CAM hLHR, there was an equal probability of Gs interacting with the inactive mutant as with the other receptor. If so, then 50% of the heterodimerized wt or CAM hLHRs would not be engaged with Gs. Therefore, although the actual concentration of cell surface wt or CAM hLHR protein would be unchanged, the number of cell surface wt or CAM receptor available to activate Gs would be decreased. Thus, there would be reduction in functional, but not absolute, numbers of wt or CAM hLHRs. Another possible scenario is that, due to receptor heterodimerization, there may be allosteric interactions between the signaling active hLHR and the physically associated signaling inactive D405N,Y546F that cause the wt or CAM receptor to be stabilized in less active conformations. There is no reason to exclude the possibility that the allosteric effects could be mutual, with the signaling inactive mutant stabilizing the wt or CAM in a less active state, while the wt or CAM conversely stabilizing the signaling inactive mutant in a somewhat more active state. Either situation would be consistent with the observed results. Along these lines, it may be useful to consider the Family C of GPCRs. Although the glycoprotein hormone receptors are not closely related to the Family C receptors, they do share a similar structural organization by virtue of having a large extracellulular, ligand-binding domain attached to a serpentine region that activates G proteins. Within a dimeric complex there are, therefore, four structural and functional subunits, two extracellular domains and two serpentine regions. Activation of metabotropic glutamate receptors and GABAB receptors is thought to involve allosterically-induced subunit rearrangements allowing for both cis and trans activation of the receptor protomers [40, 41]. It will be relevant to determine if similar subunit rearrangements in the context of a receptor dimer accompany activation of the hLHR. Other studies supporting allosteric effects between receptor protomers within a GPCR dimer come from the opioid receptors, where it has been shown that 6-guanidinonaltrindole is an agonist specific for heterodimers, but not homodimers, of the opioid receptors [42]. These findings are compatible with a model of allosterically-induced conformational changes that occur specifically as a result of heterodimerization, allowing for the activation of the heterodimer by this agonist. The signaling impairment of wt or CAM hLHRs when heterodimerized with signaling inactive receptors may reflect allosteric effects between the hLHR protomers and provide the basis for further investigation in this area.
In summary, the data presented demonstrate an attenuation of wt or CAM hLHR signaling by co-expression of a trafficking competent, but intrinsically signaling inactive, hLHR mutant D405N,Y546F. The attenuating effects of the signaling inactive D405N,Y546F represent a novel functional modulation of signaling within a dimeric (or oligomeric) receptor complex that is independent of any effects on receptor folding during receptor biosynthesis or trafficking.
Acknowledgements
We thank Dr. Francesca Fanelli for helpful discussions.
Role of the funding source
These studies were supported by NIH grants DK068614 and HD22196 (D.L.S.) and from CIHR (T.E.H.). T.E.H. is a Chercheur National of the Fonds de la Recherche en Santé du Québec. The funding sources did not participate in the study design; in the collection, analysis, and interpretation of the data; in the writing of the report; or in the decision to submit the paper for publication.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Disclosure Statement
The authors have no conflicts of interest to report.
References
- 1.Ascoli M, Fanelli F, Segaloff DL. Endocr. Rev. 2002;23:141–174. doi: 10.1210/edrv.23.2.0462. [DOI] [PubMed] [Google Scholar]
- 2.Kenakin T. Nature Reviews Drug Discovery. 2002;1:103–110. doi: 10.1038/nrd722. [DOI] [PubMed] [Google Scholar]
- 3.Hirakawa T, Galet C, Ascoli M. Endocrinology. 2002;143:1026–1035. doi: 10.1210/endo.143.3.8702. [DOI] [PubMed] [Google Scholar]
- 4.Hirakawa T, Ascoli M. Endocrinology. 2003;144:3872–3878. doi: 10.1210/en.2003-0365. [DOI] [PubMed] [Google Scholar]
- 5.Ascoli M. Mol. Cell. Endocrinol. 2007;260–262:244–248. doi: 10.1016/j.mce.2005.09.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Latronico A, Segaloff D. Am. J. Hum. Genet. 1999;65:949–958. doi: 10.1086/302602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Themmen APN, Huhtaniemi IT. Endocr. Rev. 2000;21:551–583. doi: 10.1210/edrv.21.5.0409. [DOI] [PubMed] [Google Scholar]
- 8.Zhang M, Tao YX, Ryan GL, Feng X, Fanelli F, Segaloff DL. J. Biol. Chem. 2007;282:25527–25539. doi: 10.1074/jbc.M703500200. [DOI] [PubMed] [Google Scholar]
- 9.Jaquette J, Segaloff DL. Mol. Cell. Endocrinol. 2002;194:211–215. doi: 10.1016/s0303-7207(02)00220-4. [DOI] [PubMed] [Google Scholar]
- 10.Fanelli F. J. Mol. Biol. 2000;296:1333–1351. doi: 10.1006/jmbi.2000.3516. [DOI] [PubMed] [Google Scholar]
- 11.Fanelli F, Themmen AP, Puett D. IUBMB Life. 2001;51:149–155. doi: 10.1080/152165401753544214. [DOI] [PubMed] [Google Scholar]
- 12.Angelova K, Fanelli F, Puett D. J. Biol. Chem. 2002;277:32202–32213. doi: 10.1074/jbc.M203272200. [DOI] [PubMed] [Google Scholar]
- 13.Fanelli F, Verhoef-Post M, Timmerman M, Zeilemaker A, Martens JW, Themmen AP. Mol. Endocrinol. 2004;18:1499–1508. doi: 10.1210/me.2003-0050. [DOI] [PubMed] [Google Scholar]
- 14.Zhang M, Mizrachi D, Fanelli F, Segaloff DL. J. Biol. Chem. 2005;280:26169–26176. doi: 10.1074/jbc.M502102200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Feng X, Muller T, Mizrachi D, Fanelli F, Segaloff DL. Endocrinology. 2008;149:1705–1717. doi: 10.1210/en.2007-1341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Abell AN, Segaloff DL. J. Biol. Chem. 1997;272:14586–14591. doi: 10.1074/jbc.272.23.14586. [DOI] [PubMed] [Google Scholar]
- 17.Scheerer P, Park JH, Hildebrand PW, Kim YJ, Krauss N, Choe HW, Hofmann KP, Ernst OP. Nature. 2008;455:497–502. doi: 10.1038/nature07330. [DOI] [PubMed] [Google Scholar]
- 18.Mizrachi D, Segaloff DL. Mol. Endocrinol. 2004;18:1768–1777. doi: 10.1210/me.2003-0406. [DOI] [PubMed] [Google Scholar]
- 19.Tao YX, Johnson NB, Segaloff DL. J. Biol. Chem. 2004;279:5904–5914. doi: 10.1074/jbc.M311162200. [DOI] [PubMed] [Google Scholar]
- 20.Tao YX. Pharmacol. Ther. 2006;111:949–973. doi: 10.1016/j.pharmthera.2006.02.008. [DOI] [PubMed] [Google Scholar]
- 21.Latronico AC, Chai Y, Arnhold IJP, Liu X, Mendonca BB, Segaloff DL. Mol. Endocrinol. 1998;12:442–450. doi: 10.1210/mend.12.3.0077. [DOI] [PubMed] [Google Scholar]
- 22.Guan R, Feng X, Wu X, Zhang M, Zhang X, Hebert TE, Segaloff DL. J. Biol. Chem. 2009;284:7483–7494. doi: 10.1074/jbc.M809150200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Min L, Ascoli M. Mol. Endocrinol. 2000;14:1797–1810. doi: 10.1210/mend.14.11.0555. [DOI] [PubMed] [Google Scholar]
- 24.Urizar E, Montanelli L, Loy T, Bonomi M, Swillens S, Gales C, Bouvier M, Smits G, Vassart G, Costagliola S. EMBO J. 2005;24:1954–1964. doi: 10.1038/sj.emboj.7600686. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Ji I, Lee CW, Song YS, Conn PM, Ji TH. Mol. Endocrinol. 2002;16:1299–1308. doi: 10.1210/mend.16.6.0852. [DOI] [PubMed] [Google Scholar]
- 26.Mercier J-F, Salahpour A, Angers S, Breit A, Bouvier M. J. Biol. Chem. 2002;277:44925–44931. doi: 10.1074/jbc.M205767200. [DOI] [PubMed] [Google Scholar]
- 27.Pfleger KD, Eidne KA. Nat Methods. 2006;3:165–174. doi: 10.1038/nmeth841. [DOI] [PubMed] [Google Scholar]
- 28.Min L, Galet C, Ascoli M. J. Biol. Chem. 2002;277:702–710. doi: 10.1074/jbc.M106082200. [DOI] [PubMed] [Google Scholar]
- 29.Kenakin T. FASEB J. 2001;15:598–611. doi: 10.1096/fj.00-0438rev. [DOI] [PubMed] [Google Scholar]
- 30.Milligan G. Biochim. Biophys. Acta. 2007;1768:825–835. doi: 10.1016/j.bbamem.2006.09.021. [DOI] [PubMed] [Google Scholar]
- 31.Bulenger S, Marullo S, Bouvier M. Trends Pharmacol. Sci. 2005;26:131–137. doi: 10.1016/j.tips.2005.01.004. [DOI] [PubMed] [Google Scholar]
- 32.Dalrymple MB, Pfleger KD, Eidne KA. Pharmacol. Ther. 2008;118:359–371. doi: 10.1016/j.pharmthera.2008.03.004. [DOI] [PubMed] [Google Scholar]
- 33.Baneres JL, Parello J. J. Mol. Biol. 2003;329:815–829. doi: 10.1016/s0022-2836(03)00439-x. [DOI] [PubMed] [Google Scholar]
- 34.Pascal G, Milligan G. Mol. Pharmacol. 2005;68:905–915. doi: 10.1124/mol.105.013847. [DOI] [PubMed] [Google Scholar]
- 35.Damian M, Martin A, Mesnier D, Pin JP, Baneres JL. EMBO J. 2006;25:5693–5702. doi: 10.1038/sj.emboj.7601449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Bayburt TH, Leitz AJ, Xie G, Oprian DD, Sligar SG. J. Biol. Chem. 2007;282:14875–14881. doi: 10.1074/jbc.M701433200. [DOI] [PubMed] [Google Scholar]
- 37.White JF, Grodnitzky J, Louis JM, Trinh LB, Shiloach J, Gutierrez J, Northup JK, Grisshammer R. Proc. Natl. Acad. Sci. U. S. A. 2007;104:12199–12204. doi: 10.1073/pnas.0705312104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Whorton MR, Bokoch MP, Rasmussen SG, Huang B, Zare RN, Kobilka B, Sunahara RK. Proc. Natl. Acad. Sci. U. S. A. 2007;104:7682–7687. doi: 10.1073/pnas.0611448104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Ernst OP, Gramse V, Kolbe M, Hofmann KP, Heck M. Proc. Natl. Acad. Sci. U. S. A. 2007;104:10859–10864. doi: 10.1073/pnas.0701967104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Brock C, Oueslati N, Soler S, Boudier L, Rondard P, Pin JP. J. Biol. Chem. 2007;282:33000–33008. doi: 10.1074/jbc.M702542200. [DOI] [PubMed] [Google Scholar]
- 41.Rondard P, Huang S, Monnier C, Tu H, Blanchard B, Oueslati N, Malhaire F, Li Y, Trinquet E, Labesse G, Pin JP, Liu J. EMBO J. 2008;27:1321–1332. doi: 10.1038/emboj.2008.64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Waldhoer M, Fong J, Jones RM, Lunzer MM, Sharma SK, Kostenis E, Portoghese PS, Whistler JL. Proc. Natl. Acad. Sci. U. S. A. 2005;102:9050–9055. doi: 10.1073/pnas.0501112102. [DOI] [PMC free article] [PubMed] [Google Scholar]