

Abstract
We have previously shown that voluntary exercise upregulates brain-derived neurotrophic factor (BDNF) within the hippocampus and is associated with an enhancement of cognitive recovery after a lateral fluid-percussion injury (FPI). In order to determine if BDNF is critical to this effect we used an immunoadhesin chimera (TrkB-IgG) that inactivates free BDNF. This BDNF inhibitor was administered to adult male rats two weeks after they had received a mild fluid percussion injury (FPI) or sham surgery. These animals were then housed with or without access to a running wheel (RW) from post-injury-day (PID) 14 to 20. On PID 21, rats were tested for spatial learning in a Morris Water Maze. Results showed that exercise counteracted the cognitive deficits associated with the injury. However this exercise-induced cognitive improvement was attenuated in the FPI-RW rats that were treated with TrkB-IgG. Molecules important for synaptic plasticity and learning were measured in a separate group of rats that were sacrificed immediately after exercise (PID 21). Western blot analyses showed that exercise increased the mature form of BDNF, synapsin I and cyclic-AMP response-element-binding protein (CREB) in the vehicle treated Sham-RW group. However, only the mature form of BDNF and CREB were increased in the vehicle treated FPI-RW group. Blocking BDNF (pre administration of TrkB-IgG) greatly reduced the molecular effects of exercise in that exercise-induced increases of BDNF, synapsin I and CREB were not observed. These studies provide evidence that BDNF has a major role in exercise's cognitive effects in traumatically injured brain.
Keywords: TBI, hippocampus, fluid-percussion-injury, Synapsin I and CREB
Introduction
Cognitive and neurological impairments are prevalent features of traumatic brain injury (TBI) and unfortunately there are no scientifically established effective treatments (Ashman et al., 2006; Binder et al., 2005). Cognitive deficits are frequently related to impaired hippocampal function (Wilde et al., 2007), and have been reproduced in animals models of TBI (Fujimoto et al., 2004; Hamm et al., 1992; Hicks et al., 1993). Based on evidence that voluntary exercise activates neuroplasticity mechanisms within the hippocampus and counteracts cognitive deficits that are typically exhibited after experimental TBI (Griesbach et al., 2004b), we hypothesize that voluntary exercise programs could be implemented to enhance recovery of function.
Voluntary exercise has been found to increase brain derived neurotrophic factor (BDNF) within the hippocampus (Cotman and Berchtold, 2002; Neeper et al., 1995) and this exercise-induced increase in BDNF has been proposed as one of the main mechanisms for the effects of exercise on cognition. However, this association between exercise-induced up-regulation of BDNF and improvement in cognition has yet to be established after TBI.
Both human and animal studies have demonstrated the effects of exercise supporting cognitive function (Hillman et al., 2008). Furthermore, BDNF blockade diminishes the cognitive benefits of voluntary exercise in intact rats (Vaynman et al., 2004). The proposed effect of BDNF on learning and memory appears to be in agreement with the role of BDNF promoting synaptic facilitation (Tyler and Pozzo-Miller, 2001; Tyler et al., 2006) and neurotransmitter release (Albensi, 2001; Levine et al., 1995; Levine et al., 1998; Takei et al., 1997). BDNF's effects on improved cognition are also associated with several downstream systems to BDNF including synapsin I and cyclic-AMP response-element-binding protein (CREB). Synapsin I facilitates synaptic transmission by controlling the amount of synaptic vesicles and consequentially regulating neurotransmitter release (Greengard et al., 1993). CREB, which also increases with voluntary exercise, is a transcriptional regulator that has been linked to long-term potentiation (LTP), a physiological correlate of learning and memory (Abel and Kandel, 1998; Silva et al., 1998).
In spite of substantial evidence arguing for a role of BDNF on learning and memory, an action of BDNF on enhancing recovery of cognitive function after TBI remains controversial. Previous studies, based on intracerebral infusion of BDNF into rats that have sustained TBI, have failed to demonstrate a reduction in cognitive impairments following TBI (Blaha et al., 2000; Conte et al., 2008). The results of these studies using exogenous BDNF contrast with evidence associating increasing levels of endogenous BDNF via voluntary exercise with improved cognitive performance after TBI (Griesbach et al., 2004b). The present study was designed to determine if in fact BDNF underlies the basic mechanism by which cognitive enhancement occurs with voluntary exercise after TBI in rats.
We have utilized a mild lateral fluid-percussion injury (FPI) model of TBI that, in our hands, results in cognitive impairment on the absence of significant gross morphological cell death (Griesbach et al., 2004b; Prins et al., 1996), and has shown to be responsive to voluntary running wheel exercise (RW). Within the current study, we blocked the function of BDNF by preventing activation of the high affinity receptor for BDNF. This was accomplished by using a specific immunoadhesin chimera of the tyrosine kinase B receptor (TrkB-IgG) (Esper and Loeb, 2004; Ghiani et al., 2007; Rex et al., 2007; Urfer et al., 1995; Vaynman et al., 2006). TrkB-IgG was injected into directly the dorsal hippocampus two weeks following FPI just before rats were exposed to voluntary exercise for one week. Following exercise, cognitive performance was evaluated and protein levels of BDNF, synapsin I and CREB were determined.
Results
Subjects
Injured rats had a mean (± SEM) unconsciousness time of 92 ± 4.9 s and a mean apnea time of 12 ± 1.5 s. This length of unconsciousness and apnea are characteristic of a mild level of injury. Due to complications, two animals were deleted from the study after FPI given the severity of injury was determined to be too high. No significant differences in weight gain were observed between the injured and sham groups. Also there were no gross motor impairments of ambulatory ability observed in any of the injured rats as well as no differences in the amount of voluntary exercise exhibited between sham injured and FPI groups.
Cognitive Performance
Across all groups of animals there were no significant differences in swim-speed. Indicating that at 21 days after FPI animals did not exhibit general swimming deficit. Both exercise and injury influenced Morris Water Maze (MWM) performance during PIDs 21-25. During this time rats had one MWM session per day, resulting in a total of 5 sessions. Each session had 2 consecutive trials.
A significant group effect was found for latency reaching the hidden platform (F4, 35 = 3.48, p < 0.05). These group differences were particularly present on the last day of training (session 5) (F4, 35 = 5.73, p<0.005). Bonferroni corrected pairwise comparisons indicated that the non-exercised (Sed) and vehicle (Sal) treated FPI rats needed significantly more time to reach the platform compared to the FPI-Sed/Sal rats (p<0.05). Pairwise comparisons also indicated that FPI-RW/TrkB-IgG rats took significantly longer to reach the hidden platform, compared to FPI-RW/Sal (p<0.05) and Sham-RW/Sal (p<0.05). In effect, both FPI and Sham Sal treated rats treated benefited from exercise, compared to sedentary counterparts, as indicated by a significant contrast (F1, 35 = 5.03, p<0.05). In spite of these differences all groups showed learning. This was supported by significant within effect indicative of a decrease in latency for all groups across sessions (F4= 37.74, P<0.0005) (Figure 1). An injury effect indicating that FPI-Sed/Sal rats needed significantly mote time to reach the platform on day 5 compared to the Sham.Sed/Sal was only observed through a ttest (p<0.05).
Fig. 1. Swimming latency in the Morris Water Maze task.
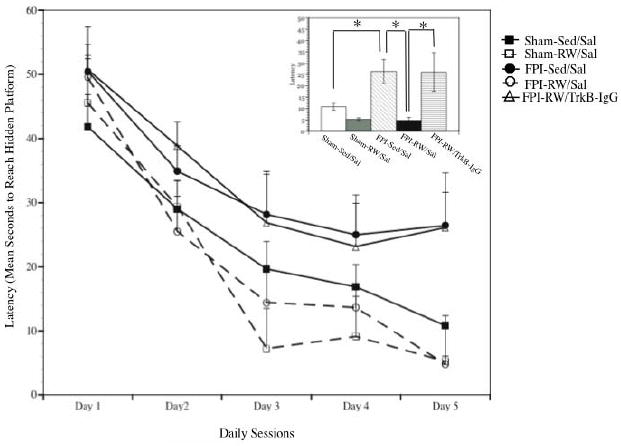
Rats were tested for one session daily beginning on post-injury day 21. Time between sessions was 24 hours. A significant decrease in latency across sessions was observed in fluid-percussion injury (FPI) and sham groups (*p<0.05). The latency to reach the hidden platform, across all days, was significantly longer in the vehicle treated (Sal) sedentary (Sed) groups compared to the exercised (RW) groups. FPI rats that received TrkB-IgG did not benefit from exercise. Each value in line plot represents the mean and upper SEM. Insert illustrates the latency to reach the hidden platform on the last session. Each value within the insert represents the mean ± SEM.
The beneficial effect of exercise was also evident when criteria scores were analyzed. A significant main effect for groups was found (F4, 35 = 13.9, p<0.0005). Bonferroni corrected multiple comparisons indicated again that exercise improved performance in Sham (p<0.005) and FPI (p<0.005) saline treated rats compared to sedentary counterparts. Further analyses through Bonferroni corrected pairwise comparisons indicated that the Sham-RW/Sal had significantly higher scores for all criteria except the two least demanding criteria (p<0.05), compared to Sham-Sed/Sal. A higher score indicates that the learning criterion was reached earlier during training. FPI-RW/Sal rats, when compared to FPI-Sed/Sal, had significantly higher scores for each criterion (p<0.05), except the criterion that required reaching the platform in less than 4 s. In other words, the benefits of exercise in the FPI were not observed for the strictest criterion. A significant within effect for criteria scores was found, indicating these were dependent on the level of difficulty of each criterion (F6 = 2.99, p<0.05) (Figure 2). Similar effects were found when data was analyzed as overall percent of subjects reaching each of the learning criterions (F4, 35 = 48.39, p<0.0005). Again, Bonferroni corrected pairwise comparisons indicated that exercise improved performance in Sham (p<0.0005) and FPI (p<0.0005) saline treated rats compared to sedentary counterparts. Although pairwise comparisons did not show significant differences between FPI-Sed/Sal and Sham-Sed/Sal, it should be noted that the percentage of rats reaching each of the learning criterions was consistently lower in the FPI-Sed/Sal group compared to the Sham-Sed/Sal group. An injury effect indicating that FPI-Sed/Sal rats reached a lower percentage of learning criterions compared to the Sham.Sed/Sal was observed through a ttest (p<0.05) (Table 2).
Fig. 2. Effects of exercise on the rate of acquisition on the Morris Water Maze task.
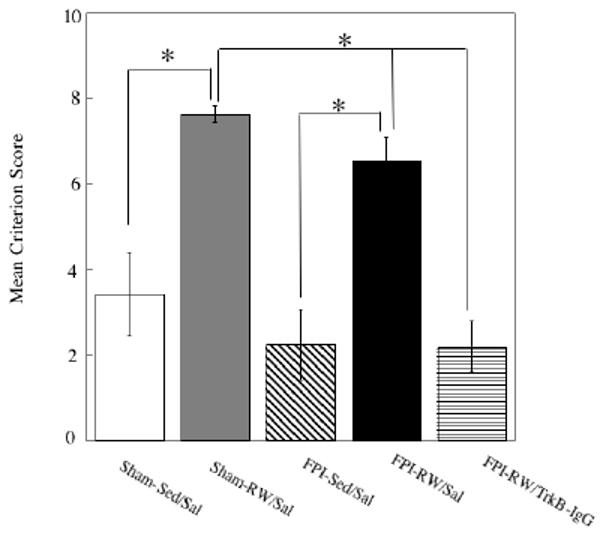
A high Criterion Score indicates that the rat was able to learn the task during the first days of training according to a strict learning criterion. Criterion scores were significantly higher in the saline treated (Sal) exercised (RW) groups compared to the sedentary (Sed) groups. Fluid-percussion injured (FPI) rats that received TrkB-IgG did not benefit from exercise. Each value represents the mean ± SEM,
Table 2. Percent of subjects reaching each criterion.
A fluid percussion injury (FPI) effect was observed. Exercise (RW) significantly improved Morris Water Maze performance. This improvement was absent when rats were treated with TrkB-IgG. *P<0.05 compared to Sham-Sed/Sal; #P<0.05 compared to FPI-Sed/Sal.
| 4 s or less | 5 s or less | 6 s or less | 7 s or less | 8 s or less | 9 s or less | 10 s or less | |
|---|---|---|---|---|---|---|---|
| Sham-Sed/Sal | 25% | 25% | 25% | 25% | 50% | 62.5% | 62.5% |
| Sham-RW/Sal | 87.5 % * | 100% * | 100% * | 100% * | 100% * | 100% | 100% |
| FPI-Sed/Sal | 12.5% | 12.5% | 12.5% | 12.5% | 25% | 25% | 37.5% |
| FPI-RW/Sal | 62.5% | 75% # | 87.5% # | 100% # | 100% # | 100% # | 100% # |
| FPI-RW/TrkB/IgG | none | 12.5% | 12.5% | 25% | 25% | 37.5% | 50% |
Hippocampal BDNF protein
Both the precursor (proBDNF) and mature form of BDNF (mBDNF) were analyzed. Exercise increased levels of mBDNF in sham and FPI groups. This was supported by a significant group effect (F4,27 = 7.68, P<0.0005). Further analysis indicated a significant increase in mBDNF in the Sham-RW/Sal compared to Sham-Sed/Sal (p < 0.05) and FPI-Sed/Sal (p < 0.005). Likewise, mBDNF was higher in the FPI-RW/Sal group compared to FPI-Sed/Sal (p < 0.05). Significant increases in the FPI-RW/Sal group compared to the Sham-Sed/Sal group were evident [Fishers LSD (p < 0.05)]. BDNF blockade with TrkB-IgG appeared to prevent the exercise–induced increases in mBDNF compared to FPI-RW/Sal (p < 0.005) and Sham-RW/Sal groups (p < 0.0005). A group main effect for the proBDNF approached statistical significance (p = 0.061). However, it should be noted that multiple comparisons analysis (Fishers LSD) indicated significantly higher levels of proBDNF in the FPI-RW/TrkB-IgG group compared to either FPI-Sed/Sal or FPI-RW/Sal (p<0.05).
Hippocampal synapsin I and CREB
Exercise was associated with increased levels of phosphorylated synapsin I in the shams. Analysis of phosphorylated synapsin I indicated a significant group effect (F4,26 = 4.55, P < 0.05). Further analysis indicated a significant increase in synapsin I in the Sham-RW/Sal compared to Sham-Sed/Sal (p < 0.05) or FPI-Sed/Sal (p < 0.05). Fishers LSD test revealed that FPI animals that were given voluntary exercise and treated with TrkB-IgG (FPI-RW/TrB-IgG) had lower levels of synapsin I compared to Sham-RW/Sal (p < 0.05).
Exercise was associated with increased levels of phosphorylated CREB in the sham (Sham-RW/Sal) and FPI (FPI-RW/Sal) groups (Figure 5). Analysis of phosphorylated CREB indicated a significant group effect (F4,27 = 5.49, P<0.005). Further analysis indicated lower levels of CREB in the FPI-Sed/Sal compared to FPI-RW/Sal (p < 0.05) and Sham-RW/Sal (p < 0.005). Fishers LSD testing revealed significant CREB increases in Sham-RW/Sal compared to Sham-Sed/Sal (p < 0.05), FPI-Sed/Sal (p < 0.0005) and FPI-RW/TrkB-IgG (p < 0.05). Fishers LSD testing also revealed a significant decrease in FPI-Sed/Sal compared to Sham-Sed/Sal (p < 0.05).
Fig. 5. Effects of exercise on hippocampal CREB.
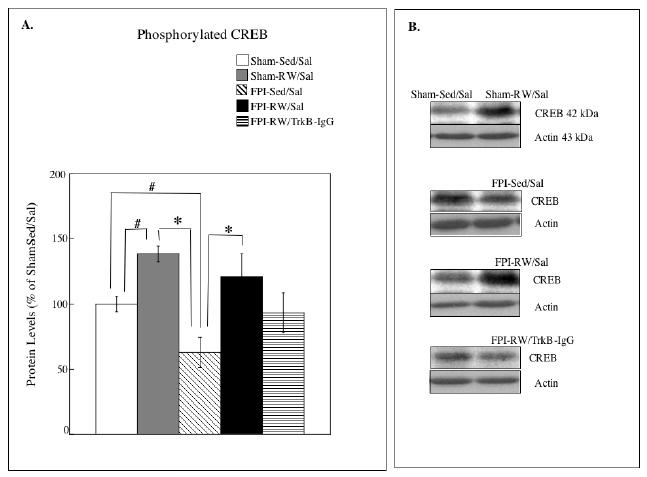
(A) Levels of phosphorylated CREB. The levels of CREB were significantly increased in the saline treated (Sal) exercised (RW) groups compared to the sedentary (Sed) groups. Fluid-percussion injured (FPI) rats that received TrkB-IgG did not benefit from exercise. * P<0.05 according to Bonferroni correction; # P<0.05 according to Fishers LSD test. Each value represents the mean ± S.E.M. (B) Representative blots measuring CREB I are shown for each experimental group compared to Sham-S-Sed/Sal controls (left side blots).
Discussion
Within this series of experiments, we evaluated the degree to which BDNF is responsible for the benefits of voluntary exercise on counteracting cognitive deficits after TBI. Our results show that blocking BDNF receptors within the left hippocampus, preventing BDNF function, impedes exercise-induced spatial learning enhancement after a lateral (left hemisphere) FPI. Inhibition of this exercise-induced spatial learning enhancement by blocking the BDNF receptor also prevented the exercise-induced increase of plasticity molecules linked to BDNF. The current study corroborates our previously published studies indicating that exercise alleviates the spatial learning impairment after FPI in rats. In addition, results from the current study indicate that blockade of BDNF receptors only in the left hippocampus is capable of preventing voluntary exercise–induced increases in BDNF and the corresponding benefit in recovery of function This suggests that the beneficial effect of post-injury exercise is dependent on bilateral BDNF up-regulation within the hippocampus.
Effects of mild FPI on learning and BDNF levels
Many studies addressing the effects of more severe FPI (Fujimoto et al., 2004; Hicks et al., 1993; Smith et al., 1991) report a more pronounced deficit on the Morris Water Maze, than the one shown here. This is most likely because the current subjects not only received a relatively mild level of injury, but also were tested on the MWM for spatial learning on the fourth post-injury week. This delay would allow substantial time for spontaneous recovery of function. In spite of these factors, the FPI-Sed/Sal group did not learn the MWM task at the same degree and rate as the Sham-Sed/Sal controls. Nevertheless, mild FPI reduced the capacity for learning a spatial memory task even when evaluated 3 weeks after injury, as evidenced by a long latency in the FPI-Sed/Sal group to find the platform on the fifth day (25 days after TBI) of MWM testing. In addition, in the comparisons of different criterions, not as many of the FPI-Sed/Sal rats were able to reach the learning criterions when compared to Sham-Sed/Sal. This further emphases that deficits in cognitive function may still be evident in rats following mild TBI even when tested weeks after injury.
Given that decreases in BDNF were not found at PID 21, it does not appear that low levels of BDNF are related to the cognitive deficits revealed in similarly treated animals tested on the MWM from 21-25 days post-injury. In contrast, we have recently reported that rats subjected to a controlled cortical impact injury (CCI) have a pronounced spatial learning impairment, in the MWM task, that is accompanied by a decrease in BDNF (Griesbach et al., 2009). The reductions of BDNF that are found after CCI may be related to the hippocampal cell loss typical of CCI (Baldwin et al., 1997; Colicos and Dash, 1996; Colicos et al., 1996; Smith et al., 1991). In contrast to the CCI model, no gross morphological hippocampal cell death has been observed in our model of mild FPI (Griesbach et al., 2004b; Prins et al., 1996). As in previous studies using the FPI model, injury itself did not result in significant decreases of BDNF and synapsin I within the dorsal hippocampus during the post acute period (Griesbach et al., 2004b; Griesbach et al., 2007; Hicks et al., 2002). However in this study, as well as in previous studies cited above, we have observed that synapsin's response to exercise is absent for several post-injury weeks, including the period of behavioral testing (Griesbach et al., 2004b). Synapsin I usually mirrors changes in BDNF regulation and is upregulated with exercise (Griesbach et al., 2004a; Jovanovic, 2000; Vaynman et al., 2003). Given synapsin's downstream nature to BDNF, it is possible that intracellular signaling mechanisms that lead to synapsin I phosphorylation are not fully recovered by PID 21.
Exercise after TBI enhances cognitive function and increases BDNF
All animals receiving vehicle injections, showed improved performance in a spatial learning and memory task when exercised for seven days, beginning at PID 14. In effect a learning acquisition deficit, as observed through MWM testing, of the FPI rats was not seen after exercise. TBI rats that performed exercise were able to reach stricter learning criterions, thus indicating that their rate and degree of learning was improved compared to TBI sedentary controls. This is in accordance with previous studies indicating that exercise can benefit TBI rats when it is administered at the appropriate post injury time (Griesbach et al., 2004b; Griesbach et al., 2007).
We have previously shown that voluntary exercise increases BDNF at the mRNA and protein level in both sham and TBI rats, providing the exposure to voluntary exercise occurs at least 14 days after injury (Griesbach et al., 2004a). Here we report that mainly the mature form of BDNF (mBDNF) is affected by exercise. The relationship between the “pro” and “mature” form of BDNF has received extensive study and is revealed in the synthesis of BDNF.
BDNF is first synthesized as a precursor that undergoes post-translational modifications (Meyer et al., 1996; Seidah et al., 1996) and is proteolytically cleaved to form mBDNF. It is the mBDNF that activates pre and post-synaptic TrkB receptors and enhances synaptic efficacy (Pang et al., 2004). In addition, evidence suggests that the mBDNF (along with associated CREB) is critical for the late phase of LTP (Barco et al., 2005; Lu, 2003). Given the function of mBDNF and the fact that it is found to increase with voluntary exercise after TBI, it seems likely that increases in mBDNF strongly contributed to the recovery that was observed in the form of enhanced MWM performance following exercise. The relationship between BDNF and enhanced spatial learning has also been found in intact rats. Although this study did not test for the effects of BDNF blockade during exercise in non-TBI rats, we have previously found similar results in non-injured animals. A unilateral intrahippocampal TrkB-IgG injection in intact rats prevented voluntary exercise-induced increases in BDNF and abolished the MWM learning enhancement that was observed after exercise, while showing no effects on sedentary animals (Vaynman et al., 2004).
Molecular effects of BDNF blockade
In the current study, TrkB-IgG markedly reduced the ability of voluntary exercise to increase mBDNF in animals subjected to FPI. As mentioned above, similar findings have been observed in intact rats where administration of TrkB-IgG blocked exercise-induced increases of BDNF protein (Vaynman et al., 2004). These reductions in BDNF may be related to a decrease in CREB, which has been shown to potentiate BDNF transcription (Fang et al., 2003; Vaynman et al., 2004). In effect, blocking the binding of BDNF to its receptor, not only inhibited exercise-induced increases in BDNF but also prevented the increases in synapsin I and CREB that were observed in the saline treated groups.
Contrasting with the effects on mBDNF, the levels of proBDNF were elevated in the FPI-RW rats treated with the TrkB-IgG. This increase was not related to the RW exposure given unchanged proBDNF levels in the exercised sham and FPI rats treated with saline. Although it remains unclear why this increase in proBDNF was seen in the FPI-RW/TrkB-IgG group, an increase in proBDNF may be a compensatory response to altered BDNF signaling due to TrkB blockade and incapacity for TrkB autophosphorylation (Mowla et al., 2001). It may also provide an increase in the storage of BDNF that could be later utilized when there is an increase in demand.
Increases in proBDNF were not accompanied by increases in mBDNF, suggesting that the conversion from proBDNF to mBDNF had been affected. In addition, the mechanism leading to proBDNF increases remains to be explored. For example, TrkB blockade may increase the synthesis of proBDNF instead of it secretion. A recent study states that it is not proBDNF, but mainly mBDNF that is secreted from neurons (Matsumoto et al., 2008). If so the increase in proBDNF may be indicative of an increase in its synthesis and not its secretion.
BDNF facilitates exercise-induced cognitive enhancement after TBI
Information about the molecular mechanisms involved in the effects of voluntary exercise on cognition can be used to understand the potential application of exercise to alleviate TBI related cognitive impairments. In particular, hippocampal-dependent learning and memory has been associated with BDNF function through multiple presynaptic (Kang, 1996; Levine et al., 1998; Pozzo-Miller, 1999; Tyler and Pozzo-Miller, 2001) and postsynaptic (Berninger, 1999; Lin, 1999; Suen, 1997) mechanisms. These findings provide evidence that BDNF facilitates learning and memory after TBI.
Administration of TrkB-IgG abolished the improvement of spatial learning and memory associated with voluntary exercise. Indeed the behavioral benefit from exercise observed in the current FPI-RW/Sal group was not found after blocking the BDNF receptor (FPI-RW/TrkB-IgG). Consequently, the FPI-RW/TrkB/IgG rats had longer escape latencies and were only capable of reaching those learning criterions that were the least stringent. These findings are consistent with other studies indicating that blockade of TrkB signaling abolishes LTP, (Kang et al., 1997). It is of interest that following FPI the voluntary exercise–induced benefit in MWM performance was prevented with unilateral (left) hippocampal TrkB-IgG administration. This would suggest that in order for voluntary exercise to have a therapeutic effect, the dorsal hippocampus must exhibit an increase in BDNF bilaterally. That affecting only one hippocampus is insufficient to produce a therapeutic effect.
These studies evaluated the effects of voluntary exercise and not those of forced exercise. Given the intrinsic characteristics of forced exercise, there should be caution in the extrapolation of these studies' findings to other forms of exercise. Although forced exercise has been shown to offer protection from stroke (Ding et al., 2006), it is likely to produce greater levels of stress. Stress inhibits BDNF (Schaaf et al., 1998; Smith, 1995; Ueyama, 1997) and decreases CREB phosphorylation (Gronli et al., 2006). Forced exercise is usually administered in the form of treadmill running or forced swimming thus being the result of an avoidance behavior. It usually requires pre-training. In contrast, voluntary exercise requires no pre-training and has been shown to counteract the effects of stress, in that stress-induced decreases in BDNF can be prevented with voluntary exercise (Adlard and Cotman, 2004).
It is intriguing that BDNF administration directly into the hippocampus has shown to be ineffective in promoting behavioral recovery after TBI (Blaha et al., 2000). In the current study re we show that voluntary exercise improves cognitive performance after TBI and that the mechanism behind this therapeutic effect is the action of BDNF. There are several differences in the mechanisms of actions between exogenous BDNF and exercise-induced BDNF. Importantly, the physiological character of exercise involves activation of an array of molecular systems using the pharmacology that is intrinsic to the brain, i.e., activating both ligand and receptor. On the contrary, most of the exogenous compound applications neglect the function of systems required to complement the action of the drug. Physiological activation through exercise is particularly advantageous given that it not only endogenously upregulates BDNF but also influences multiple systems that are likely to support BDNF functionality such as synapsin I and CREB. In addition, exercise circumvents many of the limitations of neurotrophin delivery such as inadequate delivery devices, BDNF half-life, BDNF intracellular transport, etc. Consequently, voluntary exercise is a non-invasive way to facilitate recovery after TBI that can be implemented in the rehabilitative setting.
Experimental Procedure
Subjects
A total of 72 male Sprague-Dawley rats (mean weight: 265 g ± 6.9 g SEM) were utilized in these experiments and randomly assigned to groups. Rats underwent surgery to induce either Sham or FPI injury. At PID 14 rats received an injection of microbeads embedded with TrkB-IgG or standard control. Rats were then housed with or without access to a RW until PID 20 (a total of 7 days). Following exercise (PID 21) rats were either tested in the MWM or sacrificed for tissue analysis. All animals were monitored and cared for by veterinary care staff upon arrival to UCLA. During the experiments, rats were housed in opaque plastic bins (50.8 × 25.4 × 25.4 cm), which were lined with bedding material. All procedures were performed in accordance with the United States National Institute of Health Guide for the Care and Use of Laboratory Animals (NIH Publications No. 80-23) revised 1996. Procedures were approved by the UCLA Chancellor's Animal Research Committee and can be provided upon request. The suffering and number of animals used was minimized without compromising the power of the experimental design.
Lateral Fluid-Percussion Injury
Rats were initially anesthetized with isofluorane (4% for induction, 2.0% for maintenance, in 100% O2). The level of anesthesia was monitored by level of respiration, muscular relaxation and the corneal and pedal reflexes. After loss of corneal and pedal reflexes the scalp and scapular regions were shaved, the animal was secured in a stereotaxic head frame, and the scalp was cleansed with ethanol and betadine. Rectal temperature was monitored and maintained between 36.5-38.0°C with a thermostatically controlled heating pad (Braintree Scientific Inc., Braintree, MA, USA). A midline sagittal incision was made with the aid of a microscope (Wild, Heerburg, Switzerland). The scalp and temporal muscle were reflected and a 3 mm diameter circular craniotomy was made over the left parietal cortex, centered at 3 mm posterior to bregma and 6 mm lateral to the midline. The bone flap was removed and the dura left intact in all animals to receive FPI. A plastic injury cap was placed over the craniotomy with silicone adhesive, cyanoacrylate and dental cement. When the dental cement hardened, the cap was filled with 0.9% saline solution. Anesthesia was discontinued and the animal was removed from the stereotaxic device. The injury cap was attached to the fluid percussion device. At the first sign of hind-limb withdrawal to a paw pinch, a mild fluid percussion pulse (1.5 atm) was administered. Apnea times were determined as the time from injury to the return of spontaneous breathing. Time of unconsciousness was determined from the time of injury until the return of the hind-limb withdrawal reflex. Sham animals underwent an identical preparation with the exception of the FPI. Immediately upon responding to a paw pinch, anesthesia was restored, the injury cap removed, and the scalp was sutured. Bupivacaine (0.25 mg) was injected into the margins of the scalp incision and triple antibiotic ointment was applied over the incision. The rat was placed in a recovery chamber for approximately one hour before returning to its cage.
Microbead Preparation
A microbead vehicle was utilized to allow for the inhibitory effect of TrkB-IgG to last during the week of RW exposure as determined in previous studies (Vaynman et al., 2004). Recombinant human TrkB-IgG chimera (R & D System, Inc., Minneapolis, M.N, USA) comprises the intracellular domain of human Trk-B and the Fc domain of IgG. This chimeric protein was expressed in mouse myeloma cell line, NSO. TrkB-IgG was infused into fluorescent latex microbeads (Lumaflour, Naples, FL, USA) by methods previously described that have demonstrated effective delivery of neurotrophins and other bio-agents (Lom, 1999; Quattrochi, 1989; Riddle et al., 1997). Briefly, microbeads were incubated overnight at 4°C with a 1:5 ratio of microbeads to TrkB-IgG (5 ug/uL in PBS with BSA), or saline as control vehicle. Microbeads were then centrifuged at 14,000 g for 30 min and resuspended in sterile water at 10% concentration.
Microbead Injection
Rats received a unilateral injection to the left dorsal hippocampus. Injections were administered during the morning, thus allowing rats to recover prior to being housed with access to the running-wheels. Due to the numerous connecting fibers (Amaral and Witter, 1989), a unilateral injection has been shown to suffice for effective BDNF inhibition within the hippocampus (Vaynman et al., 2004). Rats were anesthetized and monitored as described above. After loss of corneal and pedal reflexes the scalp and scapular regions were shaved, the animal was secured in a stereotaxic head frame, and the scalp was cleansed with ethanol and betadine. A midline sagittal incision was made and the sight for injection was measured. A 1.0 ul Hamilton syringe (Hamilton, Reno, NV, USA) was utilized to slowly infuse 2 ul of TrkB-IgG or Sal coated microbeads into the left hippocampus (3.8 mm posterior to Bregma, 1 mm from the midline and 3.7 mm vertically). The location of the microbead was verified by histological evaluation of selected brains as previously described (Quattrochi, 1989; Riddle et al., 1997; Vaynman et al., 2004). Microbead injections were also verified by fluorescence microscopy using an Olympus BX51 microscope.
Voluntary Wheel Exercise
Rats were individually caged with or without access to a RW from PID 14-20. The time length for exercise was based on previous studies indicating increases in BDNF and synapsin I after 7 days of voluntary exercise (Griesbach et al., 2004a; Griesbach et al., 2004b; Griesbach et al., 2008). Rats had ad-lib access to food and water and were maintained on a 12 hr light/dark cycle. Animals, provided the opportunity for spontaneous exercise, were placed in cages equipped with a RW (diameter = 31.8 cm, width = 10 cm; Nalge Nunc International, Rochester, NY, USA) that rotated against a resistance of 100 g. Non-exercised animals were left undisturbed in their home-cages. Exercise was quantified by recording the number of wheel revolutions per hour using VitalView Data Acquisition System software (Mini Mitter Company Inc., Sunriver, OR, USA). The mean number of revolutions was calculated for each night (7 PM to 7 AM), given that this was the most active period.
Behavioral Testing
Spatial navigation learning and memory were tested using a MWM task, beginning on PID 21. There were 8 animals in each group and the investigators were blinded to the experimental conditions. The water maze consisted of a 1.5-m-diameter, 0.6-m-height circular tank made opaque with white organic paint (Stechler, Albuquerque, NM, USA). The water level was kept at 2 cm above an escape platform (15 × 15 cm) and maintained at 20°C. The platform was fixed in position in the north-west quadrant of the tank. Rats received 1 session consisting of 2 training trials per day for 5 sequential days, with an inter-trial interval of 10 s. On each trial animals were released from one of 4 predetermined points around the water maze in random order and were given 60 s to locate the platform. It was insured that each session included one long (south or east release point) and one short (north or west release point) trial swim. Once they reached the platform, they remained there for 10 s before the second trial was initiated. If they failed to locate the platform, they were manually guided to it. Swimming latency was recorded with the SMART tracking system (San Diego Instruments, San Diego, CA, USA). Experimenters were blind to treatment conditions.
Western Blotting
Additional rats (included within the 72 subjects described above) were utilized for the molecular studies because animals exposed to the MWM would not allow us to distinguish between changes due to the behavior experience and those due to exercise. These rats were sacrificed by decapitation at PID 21 after exercise or sedentary housing. Hippocampal tissue within the injured hemisphere was dissected and homogenized in lysis buffer (137 mM NaCl, 20 mM Tris-HCl pH 8.0, 1% NP40, 10% glycerol, 1 mM PMSF, 10 ug/ml aprotinin, 0.1 mM benzothonium, 0.5 mM sodium vanadate). After centrifuging at 12,500 g for 15 min, supernatants were collected and immediately processed for total protein concentration determination according to the Micro BCA procedure (Pierce, Rockford, IL, USA), using bovine serum albumin as standard. All chemicals were obtained from Sigma (St. Louis, MO, USA) unless otherwise noted.
BDNF, synapsin I and CREB protein samples were analyzed by Western blot. Phosphorylated forms were measured. Separate gels, each including samples from one experimental group and control rats (Sham-Sed/Sal), were processed. Actin was utilized as an internal control, and each blot was standardized to its corresponding actin value. Protein samples were separated by electrophoresis on a 10% polyacrylamide gel and electrotransfered to a nitrocellulose membrane. Non-specific binding sites were blocked in TBS with 2% BSA and 0.1% Tween-20 for 1 h at room temperature. Membranes were rinsed in buffer (0.1% Tween-20 in TBS) and incubated at 4°C overnight, with anti-BDNF (1:1,000, Santa Cruz Biotechnology, Santa Cruz, CA, USA), followed by anti-Rabbit IgG horseradish peroxidase conjugate (1:100,000, Pierce Biotechnology, Rockford, IL, USA). Blots were processed using the SuperSignal West Femto Maximum Sensitivity Substrate kit (Pierce Biotechnology, Rockford, IL, USA) according to manufacturer's instructions. Anti-actin (1:1000, Santa Cruz Biotechnology Inc., Santa Cruz, CA, USA) was followed by anti-goat IgG horseradish peroxidase-conjugate (1:100,000 Santa Cruz Biotechnology Inc., CA, USA). Anti-phosphorylated-synapsin I (1:1000, Cell Signaling, Beverly, MA, USA) or anti-phosphorylated-CREB (1:500, Upstate Biotechnology, Lake Placid, NY, USA) was followed by anti-Rabbit IgG horseradish peroxidase-conjugate (1:100,000 Santa Cruz Biotechnology Inc., CA, USA). After rinsing in buffer four times for 10 min, immunocomplexes were analyzed by chemiluminescence using the ECL Plus kit (Amersham Pharmacia Biotech Inc., Piscataway, NJ, USA), according to manufacturer's instructions.
Optical densities for Sham-Sed/Sal blots were normalized across all gels for BDNF, synapsin I and CREB, and blots for each experimental group were normalized to Sham-Sed/Sal values within the same gel. The final data was expressed as the percent change from the mean Sham-Sed/Sal values.
Statistical Analysis
Group latencies to reach the hidden platform were analyzed with a repeated measures analysis of variance (ANOVA) with the average daily latencies as the dependent variables. Effects were further analyzed by performing Bonferroni corrected pairwise comparisons and means comparisons with desired contrast weights. Data were also analyzed by acquisition of criterions. Whereas swim latencies indicate performance across days of training, a high final MWM criterion score indicates that a rat is able to efficiently learn the task, by reaching a strict criterion, during the first days of training. Criterions were defined as the ability to locate the platform in determined amounts of time for both trials within a session. Similar measures have previously been utilized to measure behavioral outcome after TBI (Fineman et al., 2000; Giza et al., 2005; Griesbach et al., 2009; Gurkoff et al., 2006). Criterion times ranged from 4 s or less to 10 s or less to reach the platform. Different levels of criteria were utilized rather than arbitrarily choosing a single criterion score, which allowed us to assess the rate of MWM acquisition. Each criterion received a score, ranging from 1 to 10 s by determining the session in which that criterion was reached. A score of 10 was given if criterion was reached on the first session of training, a score of 9 if criterion was reached on the second session of training and so on. If a criterion was not reached a score of 1 would be given (Table 1). Criterion scores were analyzed with repeated measures ANOVA with criteria scores as the dependent variables. Effects were further analyzed by performing Bonferroni corrected corrected pairwise comparisons. Protein levels were analyzed through univariate ANOVA. Multiple comparisons are Bonferroni corrected unless indicated.
Table 1. Conversion to Criterion Scores.
Each criterion, ranging from 4s or less to 10s or less to reach the hidden platform, received a score that was determined by the session in that criterion was obtained. Table indicates the conversion from the Morris Water Maze session, in that criterion was achieved, to the Criterion Score.
| Crit Trial | 1 | 2 | 3 | 4 | 5 | never |
| Score | 10 | 9 | 8 | 7 | 6 | 1 |
Fig. 3. Effects of exercise on hippocampal brain derived neurotrophic factor (BDNF).
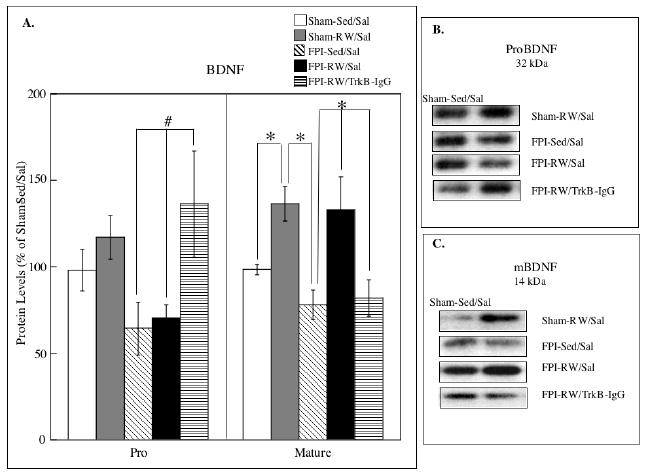
(A) Levels of precursor (proBDNF) and mature (mBDNF) forms of BDNF. The levels of mBDNF were significantly increased in the saline treated (Sal) exercised (RW) groups compared to the sedentary (Sed) groups. Fluid-percussion injured (FPI) rats that received TrkB-IgG did not benefit from exercise. * P<0.05 according to Bonferroni correction; # P<0.05 according to Fishers LSD test. Each value represents the mean ± S.E.M. (B) Representative blots measuring proBDNF are shown for each experimental group compared to Sham-S-Sed/Sal controls (left side blots). (C) Representative blots measuring mBDNF are shown for each experimental group compared to Sham-S-Sed/Sal controls (left side blots).
Fig. 4. Effects of exercise on hippocampal synapsin I.
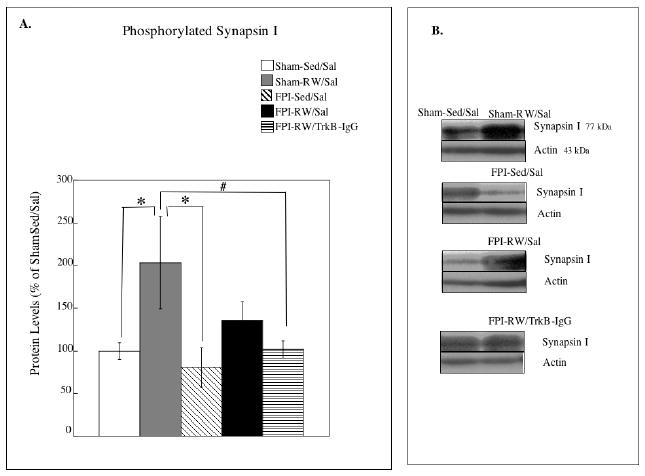
(A) Levels of phosphorylated synapsin I. The levels of synapsin I were significantly increased in the saline treated (Sal) exercised (RW) sham group compared to the sedentary (Sed) shams. The exercise-induced increase in synapsin was not observed after a fluid percussion injury (FPI). * P<0.05 according to Bonferroni correction; # P<0.05 according to Fishers LSD test. Each value represents the mean ± S.E.M. (B) Representative blots measuring syanpsin I are shown for each experimental group compared to Sham-S-Sed/Sal controls (left side blots).
Acknowledgments
This study was supported by NINDS awards NSO48535, NS27544, NS50465 and the UCLA Brain Injury Research Center. We would also wish to thank Yumei Chueng for her excellent technical help.
Abbreviations
- ANOVA
analysis of variance brain derived
- BDNF
neurotrophic factor
- CCI
controlled cortical impact
- CREB
cyclic-AMP response-element-binding protein
- FPI
fluid-percussion injury
- TrkB-IgG
immunoadhesin chimera of the tyrosine kinase B receptor
- LTD
long-term depression
- LTP
long-term potentiation
- mBDNF
mature form of BDNF
- MWM
Morris Water Maze
- Sed
Non-exercised
- PID
post-injury-day
- proBDNF
precursor form of BDNF
- RW
running wheel
- TBI
traumatic brain injury
- Sal
vehicle treated
- UCLA
University of California at Los Angeles
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Abel T, Kandel E. Positive and negative regulatory mechanisms that mediate long-term memory storage. Brain Res Brain Res Rev. 1998;26:360–78. doi: 10.1016/s0165-0173(97)00050-7. [DOI] [PubMed] [Google Scholar]
- Adlard PA, Cotman CW. Voluntary exercise protects against stress-induced decreases in brain-derived neurotrophic factor protein expression. Neuroscience. 2004;124:985–92. doi: 10.1016/j.neuroscience.2003.12.039. [DOI] [PubMed] [Google Scholar]
- Albensi BC. Models of brain injury and alterations in synaptic plasticity. J Neurosci Res. 2001;65:279–83. doi: 10.1002/jnr.1151. [DOI] [PubMed] [Google Scholar]
- Amaral DG, Witter MP. The three-dimensional organization of the hippocampal formation: a review of anatomical data. Neuroscience. 1989;31:571–91. doi: 10.1016/0306-4522(89)90424-7. [DOI] [PubMed] [Google Scholar]
- Ashman TA, Gordon WA, Cantor JB, Hibbard MR. Neurobehavioral consequences of traumatic brain injury. Mt Sinai J Med. 2006;73:999–1005. [PubMed] [Google Scholar]
- Baldwin SA, Gibson T, Callihan CT, Sullivan PG, Palmer E, Scheff SW. Neuronal cell loss in the CA3 subfield of the hippocampus following cortical contusion utilizing the optical disector method for cell counting. J Neurotrauma. 1997;14:385–98. doi: 10.1089/neu.1997.14.385. [DOI] [PubMed] [Google Scholar]
- Barco A, Patterson S, Alarcon JM, Gromova P, Mata-Roig M, Morozov A, Kandel ER. Gene expression profiling of facilitated L-LTP in VP16-CREB mice reveals that BDNF is critical for the maintenance of LTP and its synaptic capture. Neuron. 2005;48:123–37. doi: 10.1016/j.neuron.2005.09.005. [DOI] [PubMed] [Google Scholar]
- Berninger B, Schinder AF, Poo MM. Synaptic reliability correlates with reduced suceptibility to synaptic potentiation by brain-derived neurotrfophic factor. Learning and Memory. 1999;6:232–242. [PMC free article] [PubMed] [Google Scholar]
- Binder S, Corrigan JD, Langlois JA. The public health approach to traumatic brain injury: an overview of CDC's research and programs. J Head Trauma Rehabil. 2005;20:189–95. doi: 10.1097/00001199-200505000-00002. [DOI] [PubMed] [Google Scholar]
- Blaha GR, Raghupathi R, Saatman KE, McIntosh TK. Brain-derived neurotrophic factor administration after traumatic brain injury in the rat does not protect against behavioral or histological deficits. Neuroscience. 2000;99:483–93. doi: 10.1016/s0306-4522(00)00214-1. [DOI] [PubMed] [Google Scholar]
- Colicos MA, Dash PK. Apoptotic morphology of dentate gyrus granule cells following experimental cortical impact injury in rats: possible role in spatial memory deficits. Brain Res. 1996;739:120–31. doi: 10.1016/s0006-8993(96)00824-4. [DOI] [PubMed] [Google Scholar]
- Colicos MA, Dixon CE, Dash PK. Delayed, selective neuronal death following experimental cortical impact injury in rats: possible role in memory deficits. Brain Res. 1996;739:111–9. doi: 10.1016/s0006-8993(96)00819-0. [DOI] [PubMed] [Google Scholar]
- Conte V, Raghupathi R, Watson DJ, Fujimoto S, Royo NC, Marklund N, Stocchetti N, McIntosh TK. TrkB gene transfer does not alter hippocampal neuronal loss and cognitive deficits following traumatic brain injury in mice. Restor Neurol Neurosci. 2008;26:45–56. [PMC free article] [PubMed] [Google Scholar]
- Cotman CW, Berchtold NC. Exercise: a behavioral intervention to enhance brain health and plasticity. Trends Neurosci. 2002;25:295–301. doi: 10.1016/s0166-2236(02)02143-4. [DOI] [PubMed] [Google Scholar]
- Ding YH, Li J, Yao WX, Rafols JA, Clark JC, Ding Y. Exercise preconditioning upregulates cerebral integrins and enhances cerebrovascular integrity in ischemic rats. Acta Neuropathol (Berl) 2006;112:74–84. doi: 10.1007/s00401-006-0076-6. [DOI] [PubMed] [Google Scholar]
- Esper RM, Loeb JA. Rapid axoglial signaling mediated by neuregulin and neurotrophic factors. J Neurosci. 2004;24:6218–27. doi: 10.1523/JNEUROSCI.1692-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fang H, Chartier J, Sodja C, Desbois A, Ribecco-Lutkiewicz M, Walker PR, Sikorska M. Transcriptional activation of the human brain-derived neurotrophic factor gene promoter III by dopamine signaling in NT2/N neurons. J Biol Chem. 2003;278:26401–9. doi: 10.1074/jbc.M211539200. [DOI] [PubMed] [Google Scholar]
- Fineman I, Giza CC, Nahed BV, Lee SM, Hovda DA. Inhibition of neocortical plasticity during development by a moderate concussive brain injury. J Neurotrauma. 2000;17:739–49. doi: 10.1089/neu.2000.17.739. [DOI] [PubMed] [Google Scholar]
- Fujimoto ST, Longhi L, Saatman KE, Conte V, Stocchetti N, McIntosh TK. Motor and cognitive function evaluation following experimental traumatic brain injury. Neurosci Biobehav Rev. 2004;28:365–78. doi: 10.1016/j.neubiorev.2004.06.002. [DOI] [PubMed] [Google Scholar]
- Ghiani CA, Ying Z, de Vellis J, Gomez-Pinilla F. Exercise decreases myelin-associated glycoprotein expression in the spinal cord and positively modulates neuronal growth. Glia. 2007;55:966–75. doi: 10.1002/glia.20521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Giza CC, Griesbach GS, Hovda DA. Experience-dependent behavioral plasticity is disturbed following traumatic injury to the immature brain. Behav Brain Res. 2005;157:11–22. doi: 10.1016/j.bbr.2004.06.003. [DOI] [PubMed] [Google Scholar]
- Greengard P, Valtorta F, Czernik AJ, Benfenati F. Synaptic vesicle phosphoproteins and regulation of synaptic function. Science. 1993;259:780–5. doi: 10.1126/science.8430330. [DOI] [PubMed] [Google Scholar]
- Griesbach GS, Gomez-Pinilla F, Hovda DA. The upregulation of plasticity-related proteins following TBI is disrupted with acute voluntary exercise. Brain Res. 2004a;1016:154–62. doi: 10.1016/j.brainres.2004.04.079. [DOI] [PubMed] [Google Scholar]
- Griesbach GS, Hovda DA, Molteni R, Wu A, Gomez-Pinilla F. Voluntary exercise following traumatic brain injury: brain-derived neurotrophic factor upregulation and recovery of function. Neuroscience. 2004b;125:129–39. doi: 10.1016/j.neuroscience.2004.01.030. [DOI] [PubMed] [Google Scholar]
- Griesbach GS, Gomez-Pinilla F, Hovda DA. Time window for voluntary exercise-induced increases in hippocampal neuroplasticity molecules after traumatic brain injury is severity dependent. J Neurotrauma. 2007;24:1161–71. doi: 10.1089/neu.2006.0255. [DOI] [PubMed] [Google Scholar]
- Griesbach GS, Hovda DA, Gomez-Pinilla F, Sutton RL. Voluntary exercise or amphetamine treatment, but not the combination, increases hippocampal brain-derived neurotrophic factor and synapsin I following cortical contusion injury in rats. Neuroscience. 2008;154:530–40. doi: 10.1016/j.neuroscience.2008.04.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Griesbach GS, Sutton RL, Hovda DA, Ying Z, Gomez-Pinilla F. Controlled contusion injury alters molecular systems associated with cognitive performance. J Neurosci Res. 2009;87:795–805. doi: 10.1002/jnr.21893. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gronli J, Bramham C, Murison R, Kanhema T, Fiske E, Bjorvatn B, Ursin R, Portas CM. Chronic mild stress inhibits BDNF protein expression and CREB activation in the dentate gyrus but not in the hippocampus proper. Pharmacol Biochem Behav. 2006;85:842–9. doi: 10.1016/j.pbb.2006.11.021. [DOI] [PubMed] [Google Scholar]
- Gurkoff GG, Giza CC, Hovda DA. Lateral fluid percussion injury in the developing rat causes an acute, mild behavioral dysfunction in the absence of significant cell death. Brain Res. 2006;1077:24–36. doi: 10.1016/j.brainres.2006.01.011. [DOI] [PubMed] [Google Scholar]
- Hamm RJ, Dixon CE, Gbadebo DM, Singha AK, Jenkins LW, Lyeth BG, Hayes RL. Cognitive deficits following traumatic brain injury produced by controlled cortical impact. J Neurotrauma. 1992;9:11–20. doi: 10.1089/neu.1992.9.11. [DOI] [PubMed] [Google Scholar]
- Hicks RR, Smith DH, Lowenstein DH, Saint Marie R, McIntosh TK. Mild experimental brain injury in the rat induces cognitive deficits associated with regional neuronal loss in the hippocampus. J Neurotrauma. 1993;10:405–14. doi: 10.1089/neu.1993.10.405. [DOI] [PubMed] [Google Scholar]
- Hicks RR, Zhang L, Atkinson A, Stevenon M, Veneracion M, Seroogy KB. Environmental enrichment attenuates cognitive deficits, but does not alter neurotrophin gene expression in the hippocampus following lateral fluid percussion brain injury. Neuroscience. 2002;112:631–7. doi: 10.1016/s0306-4522(02)00104-5. [DOI] [PubMed] [Google Scholar]
- Hillman CH, Erickson KI, Kramer AF. Be smart, exercise your heart: exercise effects on brain and cognition. Nat Rev Neurosci. 2008;9:58–65. doi: 10.1038/nrn2298. [DOI] [PubMed] [Google Scholar]
- Jovanovic JN, Czernik AJ, Fienberg AA, Greengard P, Sihra TS. Synapsins as mediators of BDNF-enhanced neurotransmitter release. Nature Neuroscience. 2000;3:323–329. doi: 10.1038/73888. [DOI] [PubMed] [Google Scholar]
- Kang H, Welcher AA, Shelton D, Schuman EM. Neurotrophins and time: different roles for TrkB signaling in hippocampal long-term potentiation. Neuron. 1997;19:653–64. doi: 10.1016/s0896-6273(00)80378-5. [DOI] [PubMed] [Google Scholar]
- Kang H, Shuman EM. A requirement for local protein synthesis in neurotrophin-induced hippocampal synaptic plasticity. Science. 1996;273:1402–1406. doi: 10.1126/science.273.5280.1402. [DOI] [PubMed] [Google Scholar]
- Levine ES, Dreyfus CF, Black IB, Plummer MR. Brain-derived neurotrophic factor rapidly enhances synaptic transmission in hippocampal neurons via postsynaptic tyrosine kinase receptors. Proc Natl Acad Sci U S A. 1995;92:8074–7. doi: 10.1073/pnas.92.17.8074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Levine ES, Crozier RA, Black IB, Plummer MR. Brain-derived neurotrophic factor modulates hippocampal synaptic transmission by increasing N-methyl-D-aspartic acid receptor activity. Proc Natl Acad Sci U S A. 1998;95:10235–9. doi: 10.1073/pnas.95.17.10235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin SY, Wu K, Len GW, Xu JL, Levine ES, Suen PC, Mount HT, Black IB. Brain-derived neurotrophic factor enhances association of protein tyrosine posphatase PTP1D with the NMDA receptopr subunit NR2B in the cortical postsynaptic density. Molecular Brain Research. 1999;70:18–25. doi: 10.1016/s0169-328x(99)00122-9. [DOI] [PubMed] [Google Scholar]
- Lom B, Cohen-Cory S. Brain-derived neurptrophic factor differentially regulates retinal ganglion cell dendritic and axonal arborization. The Journal of Neuroscience. 1999;19:9928–9938. doi: 10.1523/JNEUROSCI.19-22-09928.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu B. Pro-region of neurotrophins: role in synaptic modulation. Neuron. 2003;39:735–8. doi: 10.1016/s0896-6273(03)00538-5. [DOI] [PubMed] [Google Scholar]
- Matsumoto T, Rauskolb S, Polack M, Klose J, Kolbeck R, Korte M, Barde YA. Biosynthesis and processing of endogenous BDNF: CNS neurons store and secrete BDNF, not pro-BDNF. Nat Neurosci. 2008;11:131–3. doi: 10.1038/nn2038. [DOI] [PubMed] [Google Scholar]
- Meyer A, Chretien P, Massicotte G, Sargent C, Chretien M, Marcinkiewicz M. Kainic acid increases the expression of the prohormone convertases furin and PC1 in the mouse hippocampus. Brain Res. 1996;732:121–32. doi: 10.1016/0006-8993(96)00502-1. [DOI] [PubMed] [Google Scholar]
- Mowla SJ, Farhadi HF, Pareek S, Atwal JK, Morris SJ, Seidah NG, Murphy RA. Biosynthesis and post-translational processing of the precursor to brain-derived neurotrophic factor. J Biol Chem. 2001;276:12660–6. doi: 10.1074/jbc.M008104200. [DOI] [PubMed] [Google Scholar]
- Neeper SA, Gomez-Pinilla F, Choi J, Cotman C. Exercise and brain neurotrophins. Nature. 1995;373:109. doi: 10.1038/373109a0. [DOI] [PubMed] [Google Scholar]
- Pang PT, Teng HK, Zaitsev E, Woo NT, Sakata K, Zhen S, Teng KK, Yung WH, Hempstead BL, Lu B. Cleavage of proBDNF by tPA/plasmin is essential for long-term hippocampal plasticity. Science. 2004;306:487–91. doi: 10.1126/science.1100135. [DOI] [PubMed] [Google Scholar]
- Pozzo-Miller LD, Gottschalk W, Zhang L, McDermott K, Du J, Gopalakrishnan R, Oho C, Sheng ZH, Lu B. Impairments in high-frequency transmission, synaptic vesicle docking, and synaptic protein distribution in the hippocampus of BDNF knockout mice. Journal of Neuroscience. 1999;19:4972–4983. doi: 10.1523/JNEUROSCI.19-12-04972.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prins ML, Lee SM, Cheng CL, Becker DP, Hovda DA. Fluid percussion brain injury in the developing and adult rat: a comparative study of mortality, morphology, intracranial pressure and mean arterial blood pressure. Brain Res Dev Brain Res. 1996;95:272–82. doi: 10.1016/0165-3806(96)00098-3. [DOI] [PubMed] [Google Scholar]
- Quattrochi JJ, Mamelak AN, Madison RD, Macklis JD, Hobson A. Mapping neuronal inputs to REM sleep induction sites with carbachol-fluorescent microspheres. Science. 1989;245:984–986. doi: 10.1126/science.2475910. [DOI] [PubMed] [Google Scholar]
- Rex CS, Lin CY, Kramar EA, Chen LY, Gall CM, Lynch G. Brain-derived neurotrophic factor promotes long-term potentiation-related cytoskeletal changes in adult hippocampus. J Neurosci. 2007;27:3017–29. doi: 10.1523/JNEUROSCI.4037-06.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Riddle DR, Katz LC, Lo DC. Focal delivery of neurotrophins into the central nervous system using fluorescent latex microspheres. Biotechniques. 1997;23:928–34. 936–7. doi: 10.2144/97235rr02. [DOI] [PubMed] [Google Scholar]
- Schaaf MJ, de Jong J, de Kloet ER, Vreugdenhil E. Downregulation of BDNF mRNA and protein in the rat hippocampus by corticosterone. Brain Res. 1998;813:112–20. doi: 10.1016/s0006-8993(98)01010-5. [DOI] [PubMed] [Google Scholar]
- Seidah NG, Benjannet S, Pareek S, Chretien M, Murphy RA. Cellular processing of the neurotrophin precursors of NT3 and BDNF by the mammalian proprotein convertases. FEBS Lett. 1996;379:247–50. doi: 10.1016/0014-5793(95)01520-5. [DOI] [PubMed] [Google Scholar]
- Silva AJ, Kogan JH, Frankland PW, Kida S. CREB and memory. Annu Rev Neurosci. 1998;21:127–48. doi: 10.1146/annurev.neuro.21.1.127. [DOI] [PubMed] [Google Scholar]
- Smith DH, Okiyama K, Thomas MJ, Claussen B, McIntosh TK. Evaluation of memory dysfunction following experimental brain injury using the Morris water maze. J Neurotrauma. 1991;8:259–69. doi: 10.1089/neu.1991.8.259. [DOI] [PubMed] [Google Scholar]
- Smith MA, Makino S, Kvetnansky R, Post RM. Effects of stress on neurotrophic factor expression in the rat brain. Ann N Y Acad Sci. 1995;771:234–239. doi: 10.1111/j.1749-6632.1995.tb44684.x. [DOI] [PubMed] [Google Scholar]
- Suen PC, Wu K, Levine ES, Mount HTJ, Xu JL, Lin SY, Black IB. Brain-derived neurotrophic factor rapidly enhances phosphorylation of the postsynaptic N-methyl-D-aspartate receptor subunit 1. Proceedings from the National Academy of Sciences, USA. 1997;94:8191–8195. doi: 10.1073/pnas.94.15.8191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takei N, Sasaoka K, Inoue K, Takahashi M, Endo Y, Hatanaka H. Brain-derived neurotrophic factor increases the stimulation-evoked release of glutamate and the levels of exocytosis-associated proteins in cultured cortical neurons from embryonic rats. J Neurochem. 1997;68:370–5. doi: 10.1046/j.1471-4159.1997.68010370.x. [DOI] [PubMed] [Google Scholar]
- Tyler WJ, Pozzo-Miller LD. BDNF enhances quantal neurotransmitter release and increases the number of docked vesicles at the active zones of hippocampal excitatory synapses. J Neurosci. 2001;21:4249–58. doi: 10.1523/JNEUROSCI.21-12-04249.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tyler WJ, Zhang XL, Hartman K, Winterer J, Muller W, Stanton PK, Pozzo-Miller L. BDNF increases release probability and the size of a rapidly recycling vesicle pool within rat hippocampal excitatory synapses. J Physiol. 2006;574:787–803. doi: 10.1113/jphysiol.2006.111310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ueyama T, K Y, Nemoto K, Sekimoto M, Tone S, Senba E. Immobilization stress reduced the expression of neurotrohins and their receptors in the rat brain. Neuroscience Research. 1997;28:103–110. doi: 10.1016/s0168-0102(97)00030-8. [DOI] [PubMed] [Google Scholar]
- Urfer R, Tsoulfas P, O'Connell L, Shelton DL, Parada LF, Presta LG. An immunoglobulin-like domain determines the specificity of neurotrophin receptors. EMBO J. 1995;14:2795–805. doi: 10.1002/j.1460-2075.1995.tb07279.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vaynman S, Ying Z, Gomez-Pinilla F. Interplay between brain-derived neurotrophic factor and signal transduction modulators in the regulation of the effects of exercise on synaptic-plasticity. Neuroscience. 2003;122:647–57. doi: 10.1016/j.neuroscience.2003.08.001. [DOI] [PubMed] [Google Scholar]
- Vaynman S, Ying Z, Gomez-Pinilla F. Hippocampal BDNF mediates the efficacy of exercise on synaptic plasticity and cognition. Eur J Neurosci. 2004;20:2580–90. doi: 10.1111/j.1460-9568.2004.03720.x. [DOI] [PubMed] [Google Scholar]
- Vaynman SS, Ying Z, Yin D, Gomez-Pinilla F. Exercise differentially regulates synaptic proteins associated to the function of BDNF. Brain Res. 2006;1070:124–30. doi: 10.1016/j.brainres.2005.11.062. [DOI] [PubMed] [Google Scholar]
- Wilde MC, Castriotta RJ, Lai JM, Atanasov S, Masel BE, Kuna ST. Cognitive impairment in patients with traumatic brain injury and obstructive sleep apnea. Arch Phys Med Rehabil. 2007;88:1284–8. doi: 10.1016/j.apmr.2007.07.012. [DOI] [PubMed] [Google Scholar]