

Abstract
Earlier, we showed that streptozocin (STZ)-induced type 1 diabetes in rats leads to the development of painful peripheral diabetic neuropathy (PDN) manifested as thermal hyperalgesia and mechanical allodynia accompanied by significant enhancement of T-type calcium currents (T-currents) and cellular excitability in medium-sized dorsal root ganglion (DRG) neurons. Here, we studied the in-vivo and in-vitro effects of gene-silencing therapy specific for the Cav3.2 isoform of T-channels, on thermal and mechanical hypersensitivity, and T-current expression in small and medium-size DRG neurons of STZ-treated rats. We found that silencing of the T-channel Cav3.2 isoform using antisense oligonucleotides, had a profound and selective anti-hyperalgesic effect in diabetic rats and is accompanied by significant down-regulation of T-currents in DRG neurons. Anti-hyperalgesic effects of CaV3.2 antisense oligonucleotides in diabetic rats were similar in models of rapid and slow onset of hyperglycemia following intravenous and intraperitoneal injections of STZ, respectively. Furthermore, treatments of diabetic rats with daily insulin injections reversed T-current alterations in DRG neurons in parallel with reversal of thermal and mechanical hypersensitivity in-vivo. This confirms that CaV3.2 T-channels, important signal amplifiers in peripheral sensory neurons, may contribute to the cellular hyperexcitability that ultimately leads to the development of painful PDN.
Introduction
One of the most common complications of early-onset diabetes mellitus (type 1) is peripheral neuropathy, which occurs in about 66% of patients [11]. A prominent feature of peripheral diabetic neuropathy (PDN) is neuropathic pain, which typically involves the extremities and is characterized in its early phase by hyperalgesia and/or allodynia to mechanical or thermal stimuli. Current therapies for painful PDN are largely inadequate and often are associated with serious side effects.
Although the precise cellular mechanisms of hyperalgesia and allodynia in PDN remain poorly understood, some promising clues have emerged. It has been suggested, for example, that the remodeling of voltage- and ligand-gated ion channels, which can increase excitability of the sensory neurons, has a critical function in hyperalgesia and allodynia [7, 39, 5]. Of particular interest with regard to this study are findings suggesting that the neuronal T-type, low-voltage-activated Ca2+ channels (T-channels) are important in modulating both acute and chronic peripheral pain signals, thus contributing to the development of acute and chronic pain conditions in-vivo [36, 37, 30].
We have shown that type 1 diabetes induced in rats by streptozocin (STZ) leads to the development of painful PDN, which is manifested as thermal hypersensitivity and mechanical allodynia. PDN is accompanied by significant increases in T-currents and cellular excitability recorded from medium-size dorsal root ganglion (DRG) neurons [18]. Although an increase in the excitability of sensory neurons often leads to pathologically exaggerated pain perception, such as increased responses to painful stimuli (hyperalgesia) or responses to ordinarily innocuous stimuli as though they were painful (allodynia) [5], a possible relationship between PDN and the up-regulation of T-channels in sensory neurons remains to be established. To determine the importance of T-channel modulation in STZ-induced PDN, we examined the in-vivo and in-vitro effects of gene silencing therapy specific for the most prevalent form of T-channels in DRG, the Cav3.2 isoform, on thermal hyperalgesia and mechanical hypersensitivity, as well as T-current modulation in acutely dissociated DRG neurons.
Materials and Methods
I. Chemicals and animals
Sprague-Dawley adult female rats (retired breeders, 10–12 months old, weight 297 ± 8 g) were used in all experiments. STZ was purchased from Sigma, St. Louis, MO. Antisense oligonucleotides and mismatched oligonucleotides (using the sequence previously published) [4] were purchased from Invitrogen (Carlsbad, CA, USA). Antisense-CaV3.2 (AS): CCACCTTCTTACGCCAGCGG, which was used to knock down the T-type-channel pore-forming subunit of the gene encoding the α1H (Cav3.2) or Mismatch-CaV3.2 (Mis-CaV3.2; MIS): TACTGTACTTGCGAGGCCAC were dissolved in sterile neutral pH buffer solution. Similarly, antisense oligonucleotides for CaV3.1 (α1G), AS-CaV3.1: CGAGACCCATTGGCATCCCT or CaV3.3 (α1I), AS-CaV3.3, GCTGAGGGCGGCTTGTGTTT were used to probe the involvement of these isoforms of T-channels [4]. Vehicle experiments were performed using sterile saline neutral pH buffer solution (SAL).
II. Induction of PDN with streptozocin
All experimental protocols were approved by the University of Virginia Animal Care and Use Committee and were in accordance with the Guide for the Care and Use of Laboratory Animals (NIH). All possible efforts were made to minimize animals’ suffering and to minimize the number of animals used.
To induce PDN, we intravenously (i.v) injected freshly dissolved STZ solution at pH 5–6, using a dose of 50 mg/kg, which causes severe hyperglycemia and pain-like behavior within the first few days after injection without causing severe generalized sickness (e.g., ketoacidosis, malaise, wasting) [1]. Control rats received the same volume/kg of i.v sterile saline. The animals were studied for 10 days after the day of i.v. injection.
Three days after administering the injections of STZ (or saline), at which point STZ-injected rats had developed PDN, we intrathecally (i.t) injected into L5-6 region of spinal cord 12.5 μg/25 μl of either AS or MIS (or 25 μl of saline-SAL) every 12 h for 4 days (a total of 8 injections) to test the effects of oligonucleotides. All solutions were pH balanced to 7.4 to avoid spinal cord irritation. Rats were maintained in a surgical plane of anesthesia with isoflurane (2%–3% in oxygen delivered via nose cone) throughout the injection procedure.
In the separate set of experiments we injected rats with 50 mg/kg of STZ or saline intraperitoneally (i.p.) and began to study thermal and mechanical hypersensitivity during the 4-th week following injections. In this group of animals, we initiated i.t. injections of either AS-CaV3.2 or MIS-CaV3.2 when 3 consecutive daily measurements indicated stable thermal and mechanical hyperalgesia.
III. Behavioral studies
Assessment of thermal sensitivity
The nociceptive response to thermal stimulation was measured using a paw thermal stimulation system consisting of a clear plastic chamber (10 x 20 x 24 cm) that sits on a clear elevated glass floor and is temperature regulated at 30° C as we previously published [30, 36, 37]. Each animal is placed in the plastic chamber for 15 min to acclimate. A radiant heat source mounted on a movable holder beneath the glass floor is positioned to deliver a thermal stimulus to the plantar side of the hind paw. When the animal withdraws the paw, a photocell detects interruption of a light beam reflection and the automatic timer shuts off. This method has a precision of ± 0.05 sec for the measurement of paw withdrawal latency (PWL). To prevent thermal injury, the light beam is automatically discontinued at 20 sec if the rat fails to withdraw its paw. Pain testing was done before STZ or vehicle injection (day 0) and daily thereafter for up to 10 days. The stability of daily pain recordings was confirmed by saline-injected controls.
Assessment of mechanical sensitivity
The response to mechanical stimulation was measured by our standard method [18, 30, 36] which allows time for effective daily assessment of mechanical sensitivity while minimizing residual behavioral responses from repetitious testing (e.g., learning, habituation). Rats were placed in a clear plastic cage with a wire-mesh-bottom. The cage is divided into four compartments, permitting rats freedom of movement while allowing investigators access to their paws. Von Frey filaments (Stoelting, Wood Dale, IL) are used to assess the mechanical threshold for paw withdrawal. These filaments are designated as the log10 (milligram weight required to cause bending X10). We have found that applying the filaments 4.93 to the plantar surface of the foot causes a noxious response in female rats that results in an average of 3–4 paw withdrawal responses (PWRs) in 10 trials. Baseline withdrawal scores were determined in both paws before intravenous administration of either STZ or saline (day 0) and daily thereafter for up to 10 days. In all behavioral pain testing paradigms, the experimenter was blinded to the pharmacological intervention (STZ or saline) and intrathecal intervention (AS, MIS, or SAL).
Statistical analysis
PWLs were subjected to analysis of variance (ANOVA) containing one within-subject variable, test session (before the administration of STZ or vehicle versus each posttreatment day up to 10 days), and one between-subject variable, AS versus MIS versus SAL. Relevant pairwise comparisons were done and alpha levels were adjusted using the Bonferroni procedure when appropriate.
IV. Histological study
To test the spread of intrathecally injected oligonucleotides, we injected a group of anesthetized control rats with AS-Cav3.2 labeled with a fluorescent group (FITC) using the technique (intrathecal route and isoflurane anesthesia), volume (25 μl), and vehicle (neutral pH buffer) described earlier. For histological analysis, 10-μm sections of DRG and spinal cord were cut and examined by confocal microscopy to assess the presence of fluorescent oligonucleotides.
V. Electrophysiological studies
Because we were interested in functional indications of CaV3.2 T-current knock-down in diabetic rats, we performed voltage-clamp analysis of T-current density in two subpopulations of acutely dissociated rat DRG neurons after administering AS-CaV3.2 i.t. injections. Before the harvest of tissues, rats were deeply anesthetized with isoflurane and rapidly decapitated. For one experiment, we dissected 6–8 lumbar DRGs from both sides of rats. We prepared dissociated DRG cells and used them within 6–8 h for whole-cell recordings as previously described [18, 27, 28, 35]. We focused on both small-size cells (i.e., those with an average soma diameter of 20–30 μm), since functional studies have indicated that most of them are likely polymodal nociceptors capable of responding to noxious mechanical, chemical, and thermal stimuli in-vivo, and medium-size DRG cells (i.e., those with a 31–40 μm soma diameter), which, in-vivo, typically belong to thinly myelinated Aδ fibers that can subserve either nociceptive or non-nociceptive sensory functions [5, 25, 26, 39]. Our previous studies have confirmed that the majority of acutely dissociated small and medium DRG cells express T-currents [18, 27, 28, 35].
Recordings were made using standard whole-cell techniques. Series resistance (Rs) and capacitance (Cm) values were taken directly from readings of the amplifier after electronic subtraction of the capacitive transients. Series resistance was compensated to the maximum extent possible (usually ~60%–80%). In most experiments, we used a P/5 protocol for online leak subtractions. The percent reductions in peak current at various Ni2+ concentrations were used to generate a concentration-response curve. Mean values were fit to the following Hill function:
| (1) |
where PBmax is the maximal percent block of peak current, IC50 is the concentration that produces 50% inhibition, and n is the apparent Hill coefficient for blockade. The fitted value is reported with 95% linear confidence limits. The voltage dependencies of activation and steady-state inactivation were described with single Boltzmann distributions of the following forms:
| (2) |
| (3) |
In these forms, Imax is the maximal amplitude of current and Gmax is the maximal conductance, V50 is the voltage at which half of the current is activated or inactivated, and k represents the voltage dependence (slope) of the distribution. Drugs were prepared as 100 mM stock solutions of NiCl2 in H2O. The external solution used to isolate Ca2+ currents contained, in mM, 10 BaCl2 (or 2 CaCl2), 152 TEA-Cl, and 10 HEPES adjusted to pH 7.4 with TEA-OH. To minimize contamination of T-currents with even minimal HVA components, we used only fluoride (F−)-based internal solution to facilitate high voltage-activated (HVA) Ca2+ current rundown; this solution contained, in mM, 135 tetramethylammonium hydroxide (TMA-OH), 10 EGTA, 40 HEPES, and 2 MgCl2, adjusted to pH 7.2 with hydrofluoric acid (HF). This allowed studies of well-isolated and well-clamped T-currents in small DRG cells. All chemicals were obtained from Sigma (St. Louis, MO) unless otherwise noted. Statistical comparisons were made, where appropriate, using an unpaired Student t-test, Mann-Whitney sum test, and chi-square test. All quantitative data are expressed as means of multiple experiments ± standard error of the mean (SEM). The amplitude of T-current was measured from the peak, which was subtracted from the current at the end of the depolarizing test potential to avoid contamination with residual HVA currents that were present at more positive membrane potentials (typically −20 mV and higher). To record both T-type and HVA Ca2+ currents in the same cells, we used the same external solution but the internal solution contained, in mM: 110 Cs-methane sulfonate, 14 phosphocreatine, 10 HEPES, 9 EGTA, 5 Mg-ATP, and 0.3 tris-GTP, adjusted to pH 7.3 with CsOH.
Results
Body weight (BW) fluctuations were assessed daily and compared to the initial BW for each animal. The BWs of control animals injected with intravenous saline on day 0 remained within 3% over a 10-day period in all three groups (saline-SAL, antisense-AS and mismatch-MIS) where there was about 10 to 15% BW loss in STZ-injected animals in all three groups compared to the initial BW (data not shown).
To assess the severity of hyperglycemia we performed daily measurements of blood glucose (BG) levels in rats treated with STZ and compared BG levels with controls which received i.v. saline injection. Rats in all three groups were normoglycemic at day 0, with BG levels of about 80 mg/dl before STZ injection (Fig. 1). Whereas BG levels in the SAL group remained within normal range (from 70 to 100 mg/dl) throughout the experimental period (data not shown), STZ-injected animals in all three groups became severely hyperglycemic on day 3 and remained so for 10 days, with BG levels ranging from 400 to 540 mg/dl (*, p < 0.01 in all three groups compared to day 0).
Figure 1. Intravenously injected STZ induces rapid hyperglycemia in adult female rats.

Baseline BG levels (0 days) were determined before STZ injection (50 mg/kg, i.v.; marked with arrow) and found to be within normal limits. Daily BG measures were recorded in the three groups of rats that were given intrathecal injections of SAL, AS oligonucleotides or MIS oligonucleotides. STZ-injected animals in all three groups became severely hyperglycemic on day 3 and remained so for 10 days, with BG levels ranging from 400 to 540 mg/dl (*, p < 0.01 compared to day 0) (n = 5 to 20 animals per data point).
To study the development of PDN in-vivo, we examined thermal (Fig. 2) and mechanical sensitivity (Fig. 3) in STZ- (diabetic) rats (panels A and B) and SAL rats (control) (panels C and D). Baseline thermal nociception was assessed on day 0 before any treatment (Fig. 2). At that time point there were almost no fluctuations in the baseline PWLs from right and left paws in all animal groups. When the PWLs were assessed on day 3 after injection, STZ-injected rats (panel A, right paws; panel B, left paws) uniformly experienced thermal hyperalgesia as indicated by a significant decrease in PWLs in both paws (*, p < 0.05). None of the STZ-injected rats demonstrated thermal hypoalgesia. The thermal hyperalgesia coincided with severe hyperglycemia (Fig. 1). In contrast, saline-injected rats showed minimal changes in PWLs on day 3 (panel C, right paws) and (panel D, left paws) as compared to baseline PWLs, suggesting complete lack of thermal hyperalgesia.
Figure 2. Rapid onset STZ-induced PDN is manifested by thermal hypersensitivity, which is abolished by AS oligonucleotides designed to knock down the gene encoding the Cav3.2 isoform of T-channels.
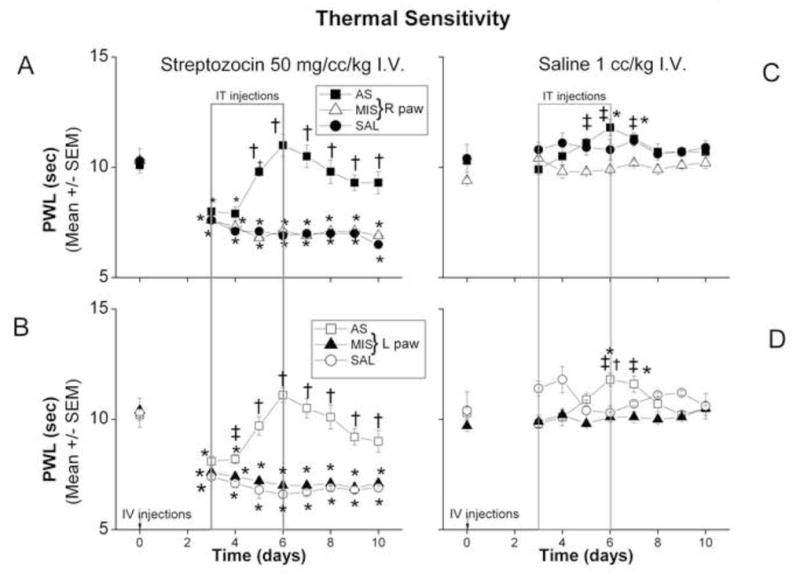
In STZ-injected animals (panel A, right paws; panel B, left paws), PWLs were obtained before i.v. STZ injection (arrow) and daily thereafter for up to 10 days. Compared to the baseline PWL, PWLs on day 3 after injection were significantly decreased in STZ-injected animals (*, p < 0.05). Intrathecal injection of AS (boxed area) starting on day 3 caused a significant increase in PWLs as compared to those of rats in the SAL and MIS treatment groups on day 5 and thereafter in both paws (†, p<0.01) (for the left paw on day 4 as well when compared to MIS group, ‡, p<0.05). In both SAL and MIS groups PWLs remain significantly decreased throughout the entire testing period in both right (A) and left (B) paws when compared to the baseline recordings (day 0) (*, p< 0.05) (n=5–10 animals per data point).
Saline-injected animals (panel C, right paws; panel D, left paws) showed no changes in PWLs on day 3 as compared to the baseline PWLs. Intrathecal injection of SAL or MIS on day 3 had no significant effect on PWLs throughout the testing period. The AS-injected group showed a significant increase in PWLs on the right side on days 5, 6 and 7 as compared to the MIS group (‡, p<0.05) but not SAL group. In left paw (panel D) PWLs recorded on days 6 and 7 were significantly increased compared to MIS group (‡, p<0.05) but only on day 6 when compared to SAL group (†, p< 0.05). The PWLs were also significantly higher in AS group on days 6 and 7 in both right (panel C) and left (panel D) paws as compared to their respective baseline PWLs (*, p< 0.05) (n = 5–9 animals per data point).
Figure 3. Rapid onset STZ-induced PDN is manifested by mechanical hypersensitivity, which is alleviated by AS oligonucleotides designed to knock down the gene encoding the Cav3.2 isoform of T-channels.
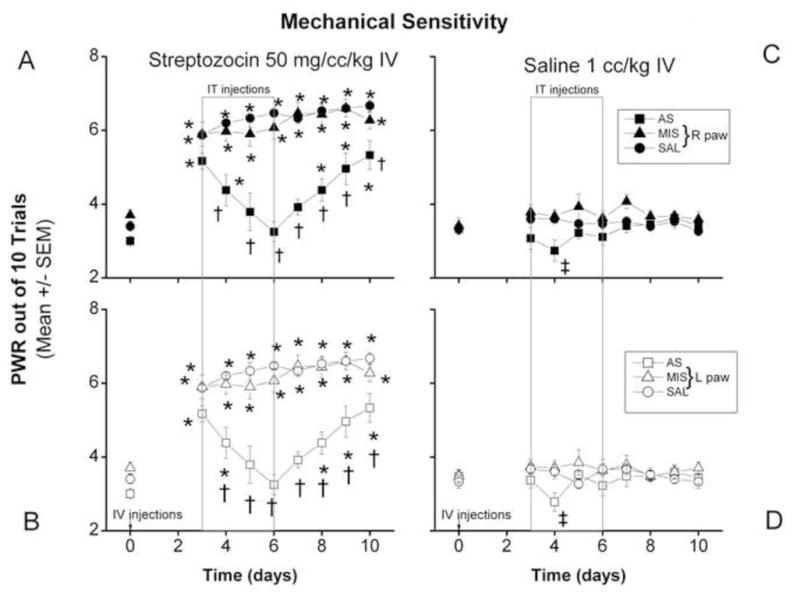
PWRs on day 3 after the injection of STZ (panel A, right paws; panel B, left paws) were significantly increased (*, p < 0.05) as compared to baseline recordings. Intrathecal injection of AS (boxed area) in i.v. STZ-treated rats caused a significant decrease in PWRs in both right paws as compared to PWRs of the SAL- and MIS- treated STZ rats and remained significantly decreased (†, p < 0.05) throughout the testing period. PWRs in both paws of the SAL and MIS groups remained significantly higher as compared to the baseline recording (day 0) throughout the testing period (*, p < 0.05) while PWRs in both paws of the AS group were no different on days 5 through 7 as compared to the baseline recording (n = 5–10 animals per data point).
Saline-injected animals showed no changes in PWRs of either paw (panel C, right; panel D, left) on day 3 as compared to baseline PWRs. Intrathecal injection of SAL or MIS had no significant effect on PWRs in mechanical sensitivity throughout the testing period compared to the baseline recordings. In the AS-injected group, there was small but significant increase in PWRs on day 4 in both paws (panel C, right; panel D, left) as compared to the MIS group (‡, p < 0.05) (n=5 to 9 animals per data point).
Immediately after pain testing, both STZ and control rats were randomly assigned to receive intrathecal injection of SAL, AS, or MIS every 12 h for the next 4 days (Fig. 2, outlined with dotted rectangle). As shown in panels A and B, AS injection caused, within two days, complete alleviation of thermal hyperalgesia. There was a significant increase in PWLs in AS group compared to SAL and MIS treatment groups on day 5 and thereafter in both paws (†, p< 0.05) with left paw showing significant PWL increase even sooner (on day 4 when compared to MIS group − ‡, p<0.05) resulting in normalization of thermal pain sensitivity in both right (A) and left (B) paws when compared to the baseline recordings (i.e. there was either no significant difference between PWLs from day 5 in AS treatment group compared to their respective baseline PWLs recorded on day 0). As predicted, neither SAL nor MIS injection had pain-alleviating effects. This was indicated by a significant decrease in PWLs throughout the testing period in both right (A) and left (B) paws as compared to the baseline recordings (day 0) (*, p < 0.05). When control rats (panels C and D) were subjected to intrathecal injection of SAL or MIS (panel C, right paw and panel D, left paw) there were small daily fluctuations in thermal sensitivity throughout the testing period which remained nonsignificant compared to the respective baseline recordings at day 0. AS injected group showed a small although significant increase in PWLs recorded on days 5, 6 and 7 in right paw (panel C) compared to MIS (‡, p<0.05) but not SAL group and on days 6 and 7 in left paw (panel D) compared to MIS group (‡, p<0.05) but only on day 6 when compared to SAL group (†, p< 0.05). The PWLs in AS group were also significantly higher on days 6 and 7 in both right (panel C) and left (panel D) paws compared to their respective baseline PWLs (*, p< 0.05).
To assess whether acutely induced hyperglycemia leads to mechanical hypersensitivity, we established the baseline number of paw withdrawals (PWRs) on day 0 before any treatment, finding that there was minimal fluctuation in the baseline PWRs from right and left paws in all groups. When PWRs were assessed on day 3 post-injection, we found that STZ-injected rats (panel A, right paws and panel B, left paws) uniformly experienced mechanical hypersensitivity as indicated by a significant increase in PWRs in both paws (*, p < 0.05). Again, no STZ-injected rats tested demonstrated mechanical hyposensitivity throughout the testing period. This hypersensitivity coincided with severe hyperglycemia, as indicated in Fig. 1. Saline-injected (control) rats showed no changes in PWRs on day 3 in either paw as compared to the baseline PWRs, indicating a lack of mechanical hypersensitivity (panel C, right paws, panel D, left paws). When AS was injected intrathecally, a significant decrease in PWRs occurred in STZ-injected rats the next day (day 4) in both right and left paws (panels A and B, respectively) as compared to SAL- and MIS-treated STZ rats. PWRs from AS-injected rats remained significantly decreased (†, p < 0.05) throughout the testing period. The alleviating effect of AS was significant: PWRs recorded from right paws (panel A) and left paws (panel B) on days 5 through 7 did not differ from the baseline recordings on day 0, indicating complete abolishment of mechanical hypersensitivity. The PWRs remained within the normal range until post-injection days 8 to 10, when a significant difference as compared to the baseline was again recorded (*, p < 0.05). Unlike AS injection, neither SAL nor MIS injection had any effect on mechanical hypersensitivity as indicated by significantly increased PWRs in both paws (A, right; B, left) throughout the testing period as compared to the baseline recordings (*, p < 0.05).
When saline-injected (control) rats (panels C and D) were given intrathecal injections of SAL or MIS (right paws, panel C; left paws, panel D) there were, despite small daily fluctuations, no significant changes in mechanical sensitivity throughout the testing period. However, in the AS-injected group there was a transient and small but significant decrease in PWRs on day 4 in both paws (panel C, right; panel D, left) compared to MIS group (‡, p < 0.05).
Since previous molecular studies indicated the existence of mRNA for all 3 isoforms of T-channels in DRG neurons [34], we tested the possible role of CaV3.1 and CaV3.3 in STZ-induced hyperalgesia in diabetic rats. Unlike AS-CaV3.2, i.t. injections of antisense oligonucleotides AS-CaV3.1 or AS-CaV3.3 had very little effects on STZ-induced thermal (Fig. 4A,B) and mechanical (Fig. 4C,D) hypersensitivity. The effects of i.t. AS-CaV3.1 and AS-CaV3.3 on thermal and mechanical hypersensitivity were not statistically different from the effects of i.t. injections of saline in diabetic rats (data not shown).
Figure 4. Rapid onset STZ-induced PDN manifested by thermal and mechanical hypersensitivity is not alleviated by AS oligonucleotides designed to knock down the genes encoding the Cav3.1 and CaV3.3 isoforms of T-channels.

Baseline thermal sensitivities measured with PWL in right paws (filled symbols panel A) and left paws (open symbols panel B) were significantly decreased 3 days following i.v. injections of STZ (arrows). Boxed area shows a lack of effects of i.t. injections of AS-CaV3.1 (circles) and AS-CaV3.3 (triangles) on new baseline achieved at day 3. In contrast, i.t. injections of AS-CaV3.2 (squares with dashed lines, same as Fig. 2A,B) completely reversed thermal hypersensitivity in diabetic rats.
Baseline mechanical sensitivities measured with PWR in right paws (filled symbols panel C) and left paws (open symbols panel D) were significantly increased 3 days following i.v. injections of STZ (arrows). Boxed area shows a lack of effects of i.t. injections of AS-CaV3.1 (circles) and AS-CaV3.3 (triangles) on new baseline achieved at day 3. In contrast, i.t. injections of AS-CaV3.2 (squares with dashed lines, same as Fig. 3A,B) completely reversed mechanical hypersensitivity in diabetic rats.
n=6 animals per data point; * p<0.05 AS-CaV3.1 vs. AS-CaV3.2; † p<0.05 AS-CaV3.3 vs. AS-CaV3.2.
Since in-vivo silencing of the Cav3.2 isoform of T-channels had a profound effect on STZ-induced PDN manifested as thermal and mechanical hypersensitivity, we examined the uptake of i.t. injected oligonucleotides using naïve rats. When the AS-Cav3.2 was labeled with a fluorescent group (FITC), we again gave rats i.t. injections every 12 h for 4 days, then dissected both L5-6 spinal cord sections and bilateral L5 and L6 DRGs. Examining these sections by confocal microscopy, we found that fluorescent AS-Cav3.2 had penetrated L5 and L6 DRGs. Many DRG cells had bright fluorescein epifluorescence, indicating effective uptake of oligonucleotides (Fig. 5A). In contrast, the corresponding transverse spinal cord sections showed that the epifluorescence was most intense in the periphery of the spinal cord around meninges, but was virtually absent from nerve tissue (Fig. 5B) (n=3 rats). These observations suggest that lumbar i.t. injection of oligonucleotides in adult rats leads to preferential uptake in corresponding DRGs making DRG neurons a likely target for gene silencing and the potential cause of the behavioral effects we identified.
Figure 5. Lumbar intrathecal injection of oligonucleotides led to preferential uptake in the corresponding DRG.

AS-Cav3.2 labeled with a fluorescent group, FITC, was intrathecally injected at the L5-6 level every 12 h for 4 days (same protocols as in Figs. 2 and 3). Four days after the first injection (three hours after the last injection) the rats were euthanized, perfused via the aorta with 4% paraformaldehyde and bilateral L5-6 DRGs and the corresponding L5-6 segment of the spinal cord harvested and kept in fixative solution until sectioning. Microphotograph on panel A (magnification 100x) indicates that many DRG cells had bright fluorescein epifluorescence, indicating effective uptake of oligonucleotides. The corresponding transverse spinal cord sections in the same rat shows that the epifluorescence was most intense around meninges, but was apparently absent from nerve tissue (panel B) (magnification 50x).
Based on these results, we studied the functional implications of AS-Cav3.2 knockdown in diabetic rats, using acutely dissociated lumbar DRG neurons from saline- and STZ-injected rats. We did voltage-clamp analysis of T-current density in two subpopulations of acutely dissociated rat DRG neurons after giving rats either AS-CaV3.2 or MIS-CaV3.2 i.t. injections. We recently described the up-regulation of T-current in acutely dissociated medium-size DRG neurons in STZ-induced neuropathy [17]. Here we tested the hypothesis that diabetes also alters T-current density in small DRG cells (< 30 μm soma diameter), most of which are likely classically described C-type nociceptors. To compare the expression of T-currents in small DRG cells after the induction of PDN, we used F−-containing internal solution, held cells at holding potentials (Vh) -90 mV, then imposed voltage commands of depolarizing pulses to test potentials (Vt) from −60 to 60 mV in 10-mV increments. Figures 6A and 6B show a representative family of inactivating inward currents in small DRG cells from control and diabetic animals, respectively. In both populations of cells, T-currents activated with small membrane depolarization and displayed fast, almost complete inactivation during 250 ms-long test potentials. The average current-voltage curves constructed from similar experiments which demonstrated significant enhancement of T-current amplitudes up to 1.9 fold (measured from peak to the end of the depolarizing pulse) in diabetic animals; these currents were enhanced over the broad range of membrane potentials and peaked at about Vt −20 mV (Fig. 6B).
Figure 6. Rapid onset STZ-induced PDN up-regulates T-current in acutely dissociated small DRG cells.
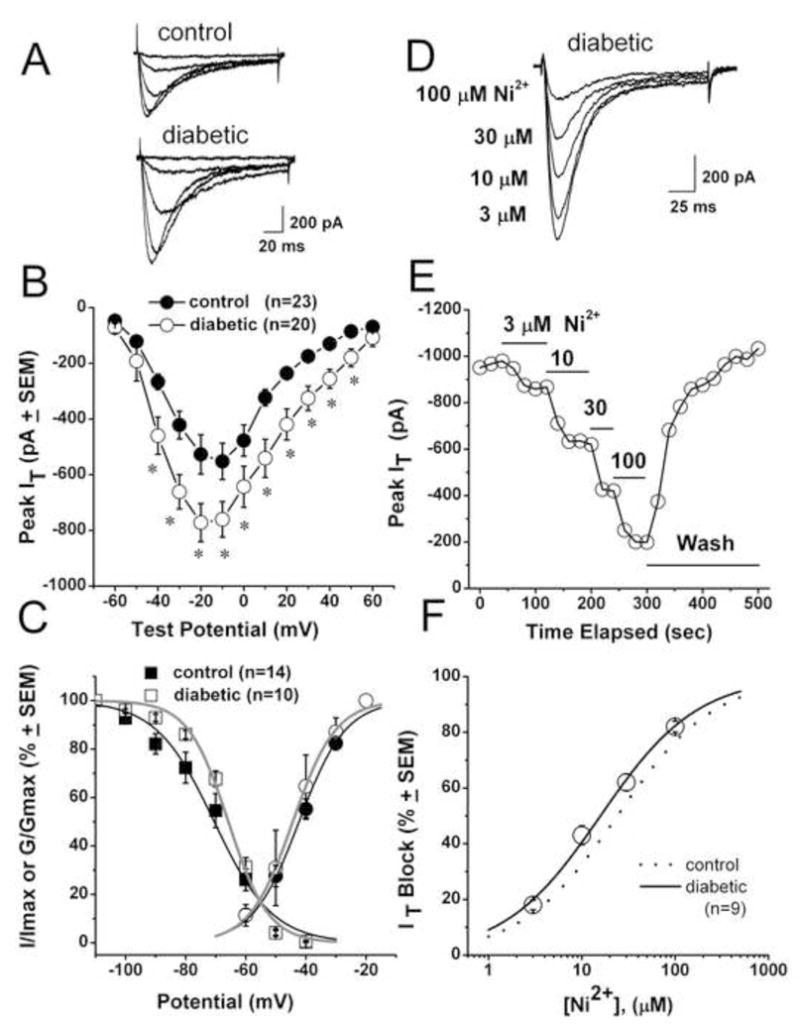
A. Traces represent families of Ca2+ currents evoked in representative small DRG cells from control (top panel: Cm 18.7 pF, Rs 1.0 MΩ) and STZ-treated diabetic rats (lower panel: Cm 22.2 pF, Rs 1.2 MΩ) by voltage steps from −90 mV (Vh) to Vt from −60 through −20 mV in 10-mV increments. Calibration bars pertain to both panels.
B. Average current-voltage curves represent control (black symbols) and diabetic (open symbols) DRG cells. Diabetes prominently increased the absolute average amplitude of Ca2+ current at most test potentials. Average capacitance was not different between the control group (21.56± 0.8 pF) and diabetic group (21.58±0.5 pF, p>0.05). Number of cells is indicated in parentheses.
C. Voltage-dependent kinetic properties of T-currents in STZ-treated rats. Square symbols represent steady-state inactivation curves using our standard double-pulse protocol [17, 18]: open symbols represent the diabetic group; filled black symbols represent the control group. Solid lines are fitted using equation #3 (see Methods), giving half-maximal availability (V50), which occurred at −70.5 ± 1.6 mV with a k of 9.7 ± 1.0 mV in the control group (black line); in the diabetic group (gray line), V50 was −65.8 ± 0.1 mV with a k of 6.7 ± 0.6 mV. Round symbols show apparent peak conductance values defined as Ipeak/(V–Er) plotted against command potentials in calculated from the same cells used for the current-voltage curve depicted in panel B. The extrapolated reversal potential (Er) was taken to be + 60 mV. Solid lines are fitted using equation #2, giving half-maximal conductance (V50), which occurred at −42.3 ± 0.7 mV with a k of 7.8 ± 0.6 mV in the control group (black line) and at −44.3 ± 0.7 mV with a k of 7.2 ± 0.6 mV in the diabetic group (gray line).
D. Representative T-current traces showing concentration-dependent block with escalating concentrations of Ni2+ in small DRG cell from the diabetic rat (Cm 20.3 pF; Rs 2.0 MΩ). Bars indicate calibration.
E. Time course of the effects of different concentrations of Ni2+ on the peak T-current from the same experiment presented in panel D. Horizontal bars indicate the time of drug applications. Note rapid onset and complete reversal of the effects of Ni2+ on the peak T-current after drug washout.
F. Open symbols show an average block of T-current by Ni2+ in control (dotted line) and diabetic (open symbols) small DRG cells. Lines are best fits obtained in controls (black dotted line) using equation #1, which gave IC50 of 29.4 ± 1.4 μM, and n of 0.85 ± 0.04 (from reference Jagodic et al., [18]); in diabetic cells (black solid line), there was an IC50 of 16.0 ± 0.8 μM and n of 0.8 ± 0.1. Both fits are constrained to 100% maximal block.
All recordings shown in this figure were done using an internal solution containing HF to isolate T-type from HVA Ca2+ current. * indicates statistical significance p<0.001.
We also measured time-dependent activation (10%–90% rise time) and time-dependent inactivation time constants (τ) (single exponential fit of decaying portion of the current waveforms) from current-voltage curves in these cells over the range of Vt from −50 mV to 0 mV. We found no significant difference between the two groups (data not shown). We also tested voltage-dependent (steady-state) inactivation, finding that diabetes caused no significant change in the midpoint (V50) of inactivation in these cells. For example, in Figure 6C the square symbols and fitted lines show that the inactivation, V50, was about −70 mV in control cells (n = 14) and −66 mV in DRG cells from STZ-induced neuropathic rats (n = 10). Similarly, there was little difference in the average V50 for T-current activation calculated from current-voltage curves in cells from control (−42 mV, n = 23) and diabetic rats (−44 mV, n = 20) groups (Fig. 6C, round symbols).
We examined the pharmacological properties of small DRG cells in an STZ model of PDN, testing their sensitivity to Ni2+, a CaV3.2-specific pharmacological tool that is a T-channel blocker in these cells [35]. Figure 6D shows representative traces; Figure 6E shows representative time courses from experiments in which Ni2+ reversibly blocked T-current in a concentration-dependent manner at 3, 10, 30 and 100 μM. However, when we compared IC50 for Ni2+ in control cells (about 29 μM, dashed line, from ref. [18]) with that in DRG cells from STZ-treated rats (about 16 μM, n = 9 cells, solid line), there was little difference between the two groups (Fig. 6F). In addition, we tested the sensitivity of T-currents in small DRG cells to 100 μM L-cysteine, a highly selective agonist for CaV3.2 currents [20]. L-cysteine increased the peak amplitude of T-currents in control cells and diabetic cells to similar degrees; i.e. 63 ±13% (n = 7) and 58 ± 9% (n = 8), respectively, with p > 0.05 (data not shown). The fact that pharmacological sensitivity to Ni2+ and L-cysteine was similar in both STZ-treated and control groups, strongly indicate that the molecular composition of CaV3.2 T-channel is little affected in small DRG cells from STZ-subjected rats.
We also tested the hypothesis that i.t. injections of AS-CaV3.2 reverse STZ-induced up-regulation of T-currents in small and medium-size DRG cells in parallel with the reversal of diabetic hyperalgesia in-vivo. For these recordings from small DRG cells, we used an internal solution that allowed us to assay both T-type and HVA Ca2+ currents. Figure 7 shows that STZ-induced PDN was associated with significantly increased T-current density in small DRG cells (from 21 ± 2 pA/pF in control rats, n = 20 cells, to 33 ± 3 pA/pF in diabetic rats, n = 21 cells; **, p < 0.01). After injection of diabetic rats with AS-CaV3.2, T-current density in small DRG cells decreased more than 50% (15 ± 2 pA/pF, n = 26 cells) as compared to that in both diabetic rats (***, p < 0.001) and control rats not treated with AS (p < 0.05,*). We then measured T-current density in small DRG cells in diabetic rats injected with mismatch ODNs (MIS-CaV3.2), finding that current density did not statistically differ from that in untreated diabetic rats (35 ± 5 pA/pF, n =13, p > 0.05, n.s.). Figure 7 summarizes these results. (All cells were recorded in at least 4 rats per group; the number of cells is indicated in columns). To test for the specificity of AS-CaV3.2, we also compared total HVA Ca2+ current density (Vh = −90 mV, Vt = 0 mV). In contrast to the robust effect on T-currents, we found no statistical difference between HVA current densities in small cells from diabetic rats treated with MIS-CaV3.2 (n = 14 cells) and those treated with AS-CaV3.2 ODNs (n = 33 cells, p > 0.4, data not shown).
Figure 7. In-vivo delivery of AS-CaV3.2 reverses up-regulation of T-current in small DRG cells.
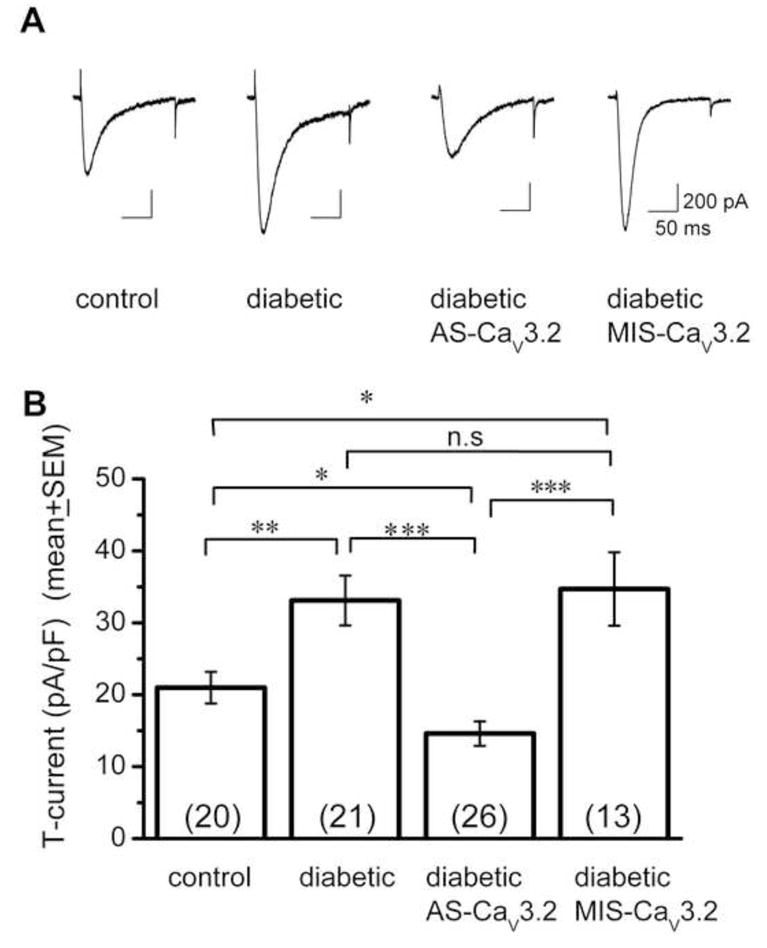
A. Traces of T-current (Vh −90 mV, Vt −30 mV) from representative small DRG cells from control, diabetic, diabetic AS-CaV3.2 treated and diabetic MIS-CaV3.2 treated rats are shown from left to right. Calibration pertains to all panels.
B. The amplitude of T-current was normalized to cell size and expressed as current density in pA/pF in four groups of animals, as indicated below each histogram bar. The number of cells in each group is indicated in parentheses. Note that AS-CaV3.2 treatment completely reversed up-regulation of T-current in diabetic cells, while MIS-CaV3.2 was ineffective. All recordings were done with internal solution that did not contain HF. n.s. not significant; * p < 0.05; ** p < 0.01, *** p < 0.001.
Furthermore, we also examined T-current density in medium-sized neurons (31–40 μm soma diameter), since we previously found [17] that T-current in these cells is up-regulated in STZ-injected animals. We used an internal solution with fluoride to isolate T-type from HVA currents [35]. Again, we found that diabetic rats not given AS had increased current density when measured at Vt of −30 mV (healthy rats 50 ± 6 pA/pF, n = 19 cells; diabetic rats 75 ± 6 pA/pF, n = 18 cells, p < 0.01). However, when AS-CaV3.2 was injected, there was a significant decrease in T-current density in diabetic rats under identical recording conditions (45 ± 12 pA/pF, n = 15 cells, p < 0.05), leading to normalization of current (i.e., T-current was similar in amplitude to that from control rats, data not shown). Thus, CaV3.2-AS effectively reversed STZ-induced up-regulation of T-current density in two sub-populations (small and medium) of acutely dissociated DRG neurons in parallel with its robust reversal of diabetic hyperalgesia in-vivo (Figs. 2 and 3).
Our results are consistent with the idea that up-regulated T-current density in DRG neurons may contribute to hyperalgesia in STZ-induced PDN. However, recent data indicate that STZ may induce alterations in vanilloid receptor family of ion channels in sensory neurons independent of its ability to induce hyperglycemia [29]. Thus, we were concerned about possible direct toxic effects of STZ on T-channels in sensory neurons in the setting of rapid onset of hyperglycemia. We conducted two sets of experiments to test this possibility. In the first experiment we administered insulin daily starting from the 3-rd day following i.v. STZ injections and reasoned that reversal of STZ-induced hyperglycemia should abolish or at least diminish hyperalgesia in-vivo and up-regulation of T-current density in DRG neurons in-vitro. Indeed, Figure 8 indicates that daily insulin treatments resulted in daily BG levels below 200 mg/dl (Fig. 8A) and almost complete gradual normalization of both thermal and mechanical hypersensitivity as indicated by measured PWLs (Fig. 8A,B) and PWRs (Fig. 8C,D), respectively. Importantly, in parallel with the reversal of hyperalgesia, insulin treatments also completely reversed up-regulation of T-current density in small DRG cells (Fig. 9).
Figure 8. Daily injections of insulin completely reverse thermal and mechanical hypersensitivity in diabetic rats after i.v. injections of STZ.
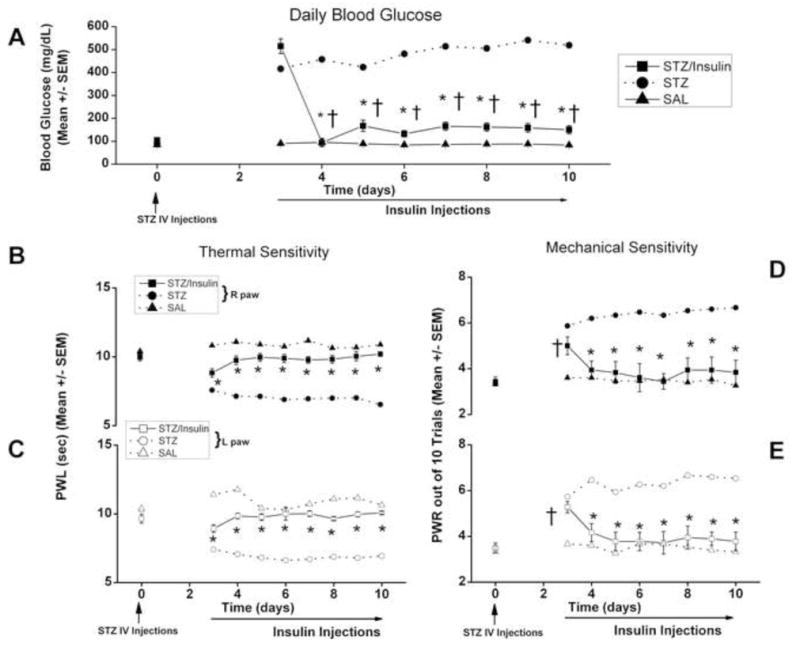
Panel A shows daily BG measures that were recorded in the three groups of rats given injections of saline i.v. (SAL, filled triangles), STZ i.v. and daily i.p. insulin injections starting from day 3 (STZ/Insulin, filled squares) and only i.v STZ (STZ, filled circles with dashed lines, same as Fig. 1). STZ-injected animals in both groups became severely hyperglycemic on day 3 to day 10, but BG decreased bellow 200 mg/dl in STZ/Insulin group from day 4 to day 10 (*, p < 0.01 STZ/Insulin vs. STZ; †, p<0.01 STZ/Insulin day 3 vs. day 4–10) (n = 5 to 10 animals per data point). Insulin NPH was given daily at the dose of 30 U i.p. on days 3 and 4; and 15 U i.p. from day 5 through day 10.
Panels B and C show complete reversal of thermal hypersensitivity in diabetic rats with daily insulin injections in the right paws (filled symbols, panel B) and in the left paws (open symbols, panel C).
*, p<0.05 STZ/insulin (square symbols) vs. STZ (circles); n = 5 to 10 animals per data point
Panels D and E show complete reversal of mechanical hypersensitivity measured with PWR in diabetic rats with daily insulin injections in the right paws (filled symbols, panel D) and left paws (open symbols, panel E).
*, p<0.05 STZ/insulin (square symbols) vs. STZ (circles); †, p<0.05 STZ/Insulin day 0 vs. day 4–10; n = 5 to 10 animals per data point.
Note that by day 10 there is very little difference in the average thermal and mechanical sensitivity between STZ/Insulin group (square symbols) and SAL (triangle symbols) group.
Figure 9. Daily injections of insulin completely reverse up-regulation T-current density in small DRG neurons in diabetic rats after i.v. injections of STZ.
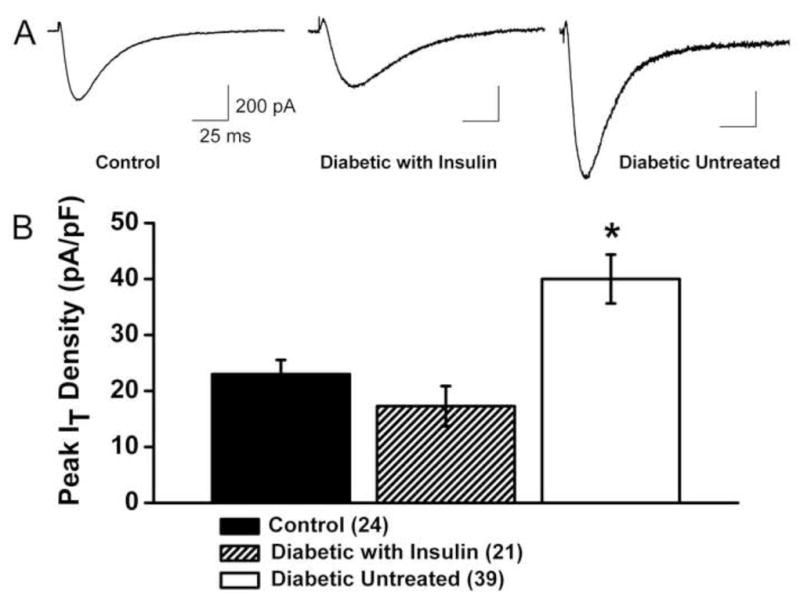
A. Traces of T-current (Vh −90 mV, Vt −30 mV) from representative small DRG cells from control (healthy), diabetic insulin-treated and diabetic untreated rats are shown from left to right. Calibration pertains to all panels.
B. The amplitude of T-current was normalized to cell size and expressed as current density in pA/pF in three groups of animals. The number of cells in each group is indicated in parentheses.
All recordings were done using an internal solution containing HF. All rats were subjected to behavioral pain testing, which confirmed the development of thermal hyperalgesia in diabetic untreated group in contrast to intact, healthy rats and diabetic, insulin-treated rats (n ≥ 4 rats per group, data not shown).
*, p < 0.01 for diabetic untreated group vs. control group (unpaired t-test).
In the second set of experiments, we injected rats with 50 mg/kg STZ i.p. and followed weekly the slower development of hyperglycemia and hyperalgesia in these animals compared with those injected with i.v. STZ. Stable high BG levels above 500 mg/dl and hyperalgesia were achieved during 4-th week following i.p. STZ injections (average 23 days, data not shown), at which point we began daily assessments of BG, thermal and mechanical hypersensitivities for 10 consecutive days (Figure 10). Next, we asked if i.p. injections of STZ and ensuing slower onset of hyperglycemia may lead to significant up-regulation of T-current density in small DRG cells. To address this issue, we used patch-clamp recordings from acutely dissociated DRG cells and assayed T-current density (Vh-90 mV, Vt −30 mV) with internal solution containing HF and in external solution containing 2 mM Ca2+. We found that diabetic animals had about 1.7-fold higher average T-current density (14.3±1.5 pA/pF; n=25 cells, 4 rats) than control animals (8.6±1.3 pA/pF, p<0.01; n=17 cells, 7 rats) (data not shown). Furthermore, i.t. injections of AS-CaV3.2, but not MIS-CaV3.2 for 4 consecutive days (from day 4–7 on the graphs 10B-E) completely reversed thermal (Fig. 10B,C) and mechanical (Fig. 10D,E) hypersensitivity in diabetic rats. Thus, it appears that T-currents in sensory neurons play an important role in the development of hyperalgesia of PDN in both models of STZ-induced hyperglycemia.
Figure 10. Slow onset STZ-induced PDN is manifested by thermal and mechanical hypersensitivity, which is abolished by AS oligonucleotides designed to knock down the gene encoding the Cav3.2 isoform of T-channels.
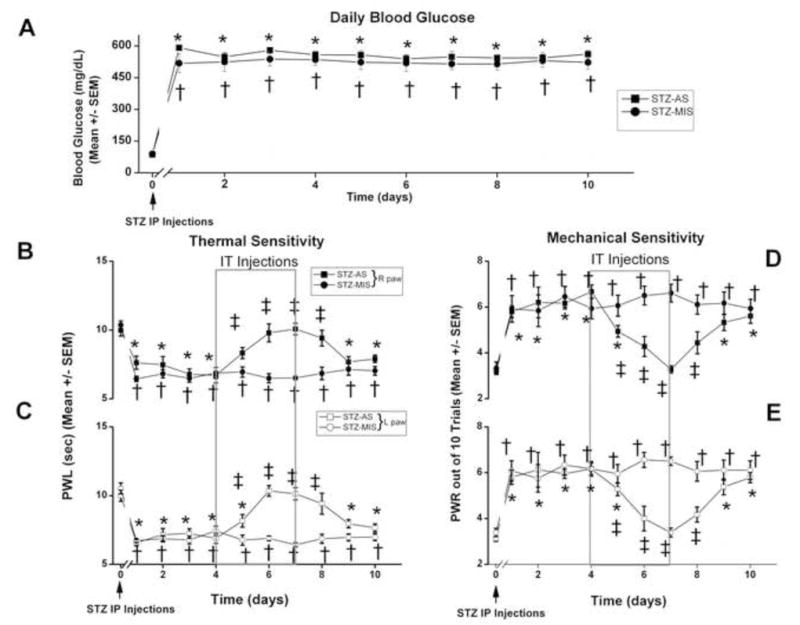
Panel A shows baseline BG levels (0 days) that were determined before STZ injection (50 mg/kg, i.p.; marked with arrow) and found to be within normal limits. Daily BG measures were recorded in the two groups of rats that were given either injections of STZ i.p and AS-CaV3.2 i.t. (STZ-AS, filled squares), or injections of STZ i.p. and MIS-CaV3.2 i.t. (STZ-MIS, filled circles). STZ-injected animals in both groups became severely hyperglycemic during 4-th week following injection (arrow) and remained so for at least 10 subsequent days, with BG levels ranging from 450 to 500 mg/dl.
Panels B and C show complete reversal of thermal hypersensitivity measured with PWL in diabetic rats with i.t injections of AS-CaV3.2 but not MIS-CaV3.2 (boxed area) in the right paws (filled symbols, panel B) and left paws (open symbols, panel C). Panels D and E show complete reversal of mechanical hypersensitivity measured with PWRs in diabetic rats with i.t injections of AS-CaV3.2 but not MIS-CaV3.2 (boxed area) in the right paws (filled symbols, panel D) and left paws (open symbols, panel E). n ≥ 6 animals per data point.
*, p<0.05 for baseline (day 0) vs. day 1–10 for STZ-AS;
†, p<0.05 for baseline (day 0) vs. day 1-10 for STZ-MIS;
‡, p<0.05 for day 0–10 for STZ-AS vs. STZ-MIS;
Discussion
Using gene-silencing technology, this study demonstrates that Cav3.2 molecular knockdown in diabetic rat sensory neurons with intrathecally injected antisense oligodeoxynucleotides alleviates painful PDN, as indicated by prolonged abolishment of thermal nociception and mechanical hypersensitivity. These beneficial in-vivo effects were accompanied by abolishment of STZ-induced up-regulation of T-current density in-vitro from small and medium-size acutely dissociated DRG neurons. These neurons likely belong to the classically described unmyelinated C-type nociceptors, and thinly myelinated Aδ sensory fibers, respectively.
Our findings and those of others [4, 22, 23, 33] suggest that i.t. injected oligodeoxynucleotides mainly localize in the meninges, but are preferentially uptaken by the corresponding DRGs. This, in addition to a robust decrease in T-current density in small and medium-size DRG neurons, suggests that DRGs are important and preferred targets for the pain-alleviating effects we describe here. Indeed, peripheral nociceptors in sensory ganglia can be sensitized by various mechanisms in pathological conditions. This can lead to their activation by stimuli that previously would not have been intense enough to cause activation, as well as by previously noxious stimuli that now produce an even greater sensation of pain, causing the symptoms observed in PDN. The electrophysiological correlates of this altered pain response include a lowered threshold for nociceptor activation, increased spontaneous activity, and increased frequency of firing in response to a suprathreshold stimulus [3, 7, 25, 26, 39]. However, in addition to the documented site of action at the level of DRGs, we can not completely rule out the possibility that i.t. injections of AS-CaV3.2 could affect the function of T-channels in the dorsal horn neurons.
Significant T-current up-regulation in DRG neurons from STZ-treated rats points at T-channels as a potential culprit for the development of painful PDN since they have an important function in the excitability of sensory neurons, decreasing the threshold for action potential firing [17, 18, 27, 28, 40]. However, remodeling of other ion channels has been reported in STZ-induced diabetic neuropathy in rats. For example, up-regulation of HVA Ca2+ channels [12], voltage-gated Na+ channels [14], and capsaicin-gated currents [15] in small DRG cells has been reported in rats with STZ-induced PDN. Our findings regarding the modulation of Cav3.2 T-channels in peripheral nociceptors of STZ-treated rats and the reversal of diabetic hyperalgesia of PDN with in-vivo silencing of this isoform in sensory neurons brings us a step closer to confirming the importance of T-channels for the development of diabetic neuropathy, a fact that will make them important targets in pain management. However, the function and relative contribution of other nociceptive ion channels to the development of painful PDN remains to be validated.
Recent cloning of pore-forming α1 subunits of T-channels has shown the existence of at least three subtypes that are likely to contribute to the heterogeneity of T-currents in native cells [13, 35]. These subtypes are G, or Cav3.1 [31], H, or Cav3.2 [8], and I, or Cav3.3 [24]. This is important since it suggests that isoform-specific targeting of T-channels should be considered when searching for effective therapies. The most recent evidence using gene silencing therapy and the transgenic mouse technology validates this notion. For example, Bourinet et al. [4] have used antisense oligodeoxynucleotides specifically engineered to knock down isoforms of T-channels in rat sensory neurons in which CaV3.2 isoforms, but not CaV3.1 or CaV3.3, isoforms, have a major function in boosting peripheral nociception in both acute and chronic animal pain paradigms of mononeuropathic pain. Their work was bolstered by that of Choi and et al. [6], who reported that mice lacking the CaV3.2 gene showed profound attenuation of acute thermal, chemical, and mechanical pain responses, as well as diminished responses to visceral pain and inflammatory pain induced by injections of formalin or capsaicin in receptive fields. Our findings add to the list of pathological pain states associated with PDN that might be effectively treated with CaV3.2 isoform-specific T-channel silencing.
Our finding led us to focus on the effectiveness of molecular knockdown of CaV3.2 but not Cav3.1 and Cav3.3, in alleviating PDN and was based on several factors. Transcripts of the CaV3.2 isoform are most prevalent in sensory ganglia, whereas Cav3.1 and CaV3.3 are 5–10-fold less abundantly expressed [2, 17, 34]. Consequently, the pain phenotype described in CaV3.2 knockout mice [6] is not evident in CaV3.1 knockouts [21]. Moreover, AS-Cav3.1 and AS-Cav3.3 had no beneficial effect on alleviating the pain phenotype associated with mononeuropathy [4] or painful PDN (our present study). Furthermore, our recent molecular and electrophysiological studies indicate that CaV3.2-mediated T-currents are selectively and robustly up-regulated in medium-size DRG neurons from STZ-treated rats [17]. Although mRNA for the CaV3.3 isoform is present in DRG neurons, functional studies of recombinant and native CaV3.3 currents display characteristic slow inactivation of macroscopic kinetics [24, 19] and little or no similarity to the functional properties of native DRG T-currents [17,18, 27, 28], thus raising questions about their physiological importance.
Although systemic in nature, somatic PDN tends to be most symptomatic in the extremities, making sensory ganglia important therapeutic targets. The fact that thermal hypersensitivity in the lower extremities of rats was alleviated by AS-CaV3.2 treatment suggests that CaV3.2 channels modulate somatic nociception. This is corroborated by findings that CaV3.2 channels in normal DRGs are not only dense in polymodal primary afferent neurons [27, 28, 34], but also significantly up-regulated in nociceptive DRG cells from STZ-treated rats. Our findings regarding the alleviation of mechanical hypersensitivity by AS-CaV3.2 treatment also implies that mechanoreceptors in DRG neurons, which are crucial in fine-touch perception and are dense in CaV3.2 channels [10, 38], could also contribute to the development of mechanical hypersensitivity in PDN. With currently available methods, it is difficult to test the function of low-threshold mechanosensory neurons in-vivo. However, future studies of their functional modulations after STZ treatment might examine this possibility. It is noteworthy that the pain-alleviating effects of repetitious AS-CaV3.2 treatment and the robust down-regulation of the STZ-induced increase in T-current density reached a peak within a few days after treatment was initiated, leading to normalization in pain responses (i.e., a complete lack of pain hypersensitivity) and significant decreases in T-current density as compared to controls. This correlates well with the estimated turn-over of T-channel protein synthesis, which undergoes a 2- to 3-day cycle [4].
Recently, we demonstrated that, in STZ-induced PDN, T-channels significantly contribute to an increase in cellular excitability in medium-size DRG neurons (17). However, in contrast to this study with small DRG cells, we found that increase in T-current density in medium-size DRG neurons from STZ-induced diabetic rats was accompanied by a depolarizing shift in steady-state inactivation (17). Here, we observed that the up-regulation of T-currents in small DRG cells from STZ-treated diabetic rats occurred without apparent kinetic alterations. This is similar to our findings in recent study using small DRG cells in a rat model of neuropathic pain resulting from chronic constrictive injury (CCI) of the sciatic nerve (18). The obvious reasons for this discrepancy are not known but it is likely that regulation of T-channel function is different among heterogenous subpopulations of sensory neurons in pathological conditions accompanied with abnormal pain perception.
Our findings with AS-CaV3.2-treated healthy rats show a transient anti-nociceptive effect that was much less impressive than the one observed in STZ-injected rats, suggesting higher anti-nociceptive efficacy of T-channel modulation in pathological conditions. Similarly selective effects have been reported with the pharmacological modulation of T-channels using mibefredil, a preferential T-channel blocker [9, 37] and the neuroactive steroid ECN, a selective T-channel blocker [30] in pain models of mononeuropathy.
In summary, our study validates CaV3.2 T-channels as important targets in the treatment of hyperalgesia associated with diabetes. T-channels are signal amplifiers in the peripheral pain pathways. Their up-regulation in sensory neurons may contribute to the hyperexcitability that ultimately leads to the development of painful PDN. Furthermore, our results suggest an unexplored avenue for the pharmacological development of specific ion channel therapies for pain control in patients with PDN.
Acknowledgments
Our research is supported by Dr. Harold Carron’s endowment (to V.J-T.), NIH R0-1 grant GM075229 (to S.M.T), funds from the Department of Anesthesiology at UVA (to V.J-T. and S.M.T.), funds from InJe University (W.J.C, W.Y.L) and generous gift from Mr. Joseph C. Palumbo and Mrs. Sandra C. Palumbo. We thank Lisa Carter, Bradley Bradenham and Daniel Greene for technical assistance. The authors have no conflicts of interest.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Aley KO, Levine JD. Rapid onset pain induced by intravenous streptozotocin in the rat. J Pain. 2001;2:146–50. doi: 10.1054/jpai.2001.21592. [DOI] [PubMed] [Google Scholar]
- 2.Beedle AM, McRory JE, Poirot O, Doering CJ, Altier C, Barrere C, Hamid J, Nargeot J, Bourinet E, Zamponi GW. Agonist-independent modulation of N-type calcium channels by ORL1 receptors. Nat Neurosci. 2004;7:118–125. doi: 10.1038/nn1180. [DOI] [PubMed] [Google Scholar]
- 3.Bhave G, Gereau RW. Posttranslational mechanisms of peripheral sensitization. J Neurobiol. 2004;61:88–106. doi: 10.1002/neu.20083. [DOI] [PubMed] [Google Scholar]
- 4.Bourinet E, Alloui A, Monteiol A, Barrere C, Couette B, Poirot O, Pages A, McRory J, Snutch TP, Eschalier A, Nargeot J. Silencing of the Cav3.2 T-type calcium channel gene in sensory neurons demonstrates its major role in nociception. EMBO J. 2005;24:315–324. doi: 10.1038/sj.emboj.7600515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Campbell JN, Meyer RA. Mechanisms of neuropathic pain. Neuron. 2006;52:77–92. doi: 10.1016/j.neuron.2006.09.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Choi S, Na HS, Kim J, Lee J, Lee S, Kim D, Park J, Chen CC, Campbell KP, Shin HS. Attenuated pain responses in mice lacking CaV3.2 T-type channels. Genes Brain Behav. 2007;6:425–431. doi: 10.1111/j.1601-183X.2006.00268.x. [DOI] [PubMed] [Google Scholar]
- 7.Coderre TJ, Katz J, Vaccarino AL, Melzack R. Contribution of central neuroplasticity to pathological pain: review of clinical and experimental evidence. Pain. 1993;52:259–285. doi: 10.1016/0304-3959(93)90161-H. [DOI] [PubMed] [Google Scholar]
- 8.Cribbs LL, Lee J, Yang J, Satin J, Zhang Y, Daud A, Barcley J, Williamson MP, Fox M, Rees M, Perez-Reyes E. Cloning and characterization of α1H from human heart, a member of the T-type Ca2+ channel gene family. Circ Res. 1998;83:103–109. doi: 10.1161/01.res.83.1.103. [DOI] [PubMed] [Google Scholar]
- 9.Dogrul A, Gardell LR, Ossipov MH, Tulunay FC, Lai J, Porecca F. Reversal of experimental neuropathic pain by T-type calcium channel blockers. Pain. 2003;105:159–168. doi: 10.1016/s0304-3959(03)00177-5. [DOI] [PubMed] [Google Scholar]
- 10.Dubreuil AS, Boukhaddaoui H, Desmadryl G, Martinez-Salgado C, Moshourab R, Lewin GR, Carroll P, Valmier J, Scamps F. Role of T-type calcium current in identified d-hair mechanoreceptor neurons studied in vitro. J Neurosci. 2004;24:8480–8484. doi: 10.1523/JNEUROSCI.1598-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Gooch C, Podwall D. The diabetic neuropathies. The Neurologist. 2004;10:311–322. doi: 10.1097/01.nrl.0000144733.61110.25. [DOI] [PubMed] [Google Scholar]
- 12.Hall KE, Liu J, Sima AA, Wiley JW. Impaired inhibitory G-protein function contributes to increased calcium currents in rats with diabetic neuropathy. J Neurophysiol. 2001;86:760–70. doi: 10.1152/jn.2001.86.2.760. Erratum in: J Neurophysiol 2001; 86: 4 pages inside back cover. [DOI] [PubMed] [Google Scholar]
- 13.Herrington J, Lingle CJ. Kinetic and pharmacological properties of low voltage-activated Ca2+ current in rat clonal (GH3) pituitary cells. J Neurophysiol. 1992;68:213–232. doi: 10.1152/jn.1992.68.1.213. [DOI] [PubMed] [Google Scholar]
- 14.Hong S, Morrow TJ, Paulson PE, Isom LL, Wiley JW. Early painful diabetic neuropathy is associated with differential changes in tetrodotoxin-sensitive and -resistant sodium channels in dorsal root ganglion neurons in the rat. J Biol Chem. 2004;279:29341–50. doi: 10.1074/jbc.M404167200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Hong S, Wiley JW. Early painful diabetic neuropathy is associated with differential changes in the expression and function of vanilloid receptor 1. J Biol Chem. 2005;280:618–27. doi: 10.1074/jbc.M408500200. [DOI] [PubMed] [Google Scholar]
- 16.Huguenard JR. Low-threshold calcium currents in central nervous system neurons. Annu Rev Physiol. 1996;58:329–358. doi: 10.1146/annurev.ph.58.030196.001553. [DOI] [PubMed] [Google Scholar]
- 17.Jagodic MM, Pathirathna S, Nelson MT, Mancuso S, Joksovic PM, Rosenberg ER, Bayliss DA, Jevtovic-Todorovic V, Todorovic SM. Cell-specific alterations of T-type calcium current in painful diabetic neuropathy enhance excitability of sensory neurons. J Neurosci. 2007;27:3305–3316. doi: 10.1523/JNEUROSCI.4866-06.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Jagodic MM, Pathirathna S, Joksovic PM, Lee W, Nelson MT, Naik AK, Su P, Jevtovic-Todorovic V, Todorovic SM. Up-regulation of the T-type calcium current in small rat sensory neurons after chronic constrictive injury of the sciatic nerve. J Neurophysiol. 2008;99:3151–3156. doi: 10.1152/jn.01031.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Joksovic PM, Brimelow BC, Murbartian J, Perez-Reyes E, Todorovic SM. Contrasting anesthetic sensitivities of slow T-type calcium channels of reticular thalamic neurons and recombinant Cav3.3 channels. Br J Pharmacol. 2005;144:59–70. doi: 10.1038/sj.bjp.0706020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Joksovic PM, Nelson MT, Jevtovic-Todorovic V, Patel MK, Perez-Reyes E, Campbell KP, Chen CC, Todorovic SM. CaV3.2 is the major molecular substrate for redox regulation of T-type Ca2+ channels in the rat and mouse thalamus. J Physiol. 2006;574:415–30. doi: 10.1113/jphysiol.2006.110395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Kim D, Park D, Choi S, Lee S, Sun M, Kim C, Shin HS. Thalamic control of visceral nociception mediated by T-type Ca2+ channels. Science. 2003;302:117–119. doi: 10.1126/science.1088886. [DOI] [PubMed] [Google Scholar]
- 22.Lai J, Gold MS, Kim CS, Bian D, Ossipov MH, Hunter JC, Porreca F. Inhibition of neuropathic pain by decreased expression of the tetrodotoxin-resistant sodium channel, NaV1.8. Pain. 2002;95:143–152. doi: 10.1016/s0304-3959(01)00391-8. [DOI] [PubMed] [Google Scholar]
- 23.Lai J, Hunter JC, Ossipov MH, Porreca F. Blockade of neuropathic pain by antisense targeting of tetrodotoxin-resistant sodium channels in sensory neurons. Methods Enzymol. 2000;314:201–213. doi: 10.1016/s0076-6879(99)14104-1. [DOI] [PubMed] [Google Scholar]
- 24.Lee JH, Daud AN, Cribbs LL, Lacerda AE, Pereverzev A, Klockner U, Schneider T, Perez-Reyes E. Cloning and expression of a novel member of the low voltage-activated T-type calcium channel family. J Neurosci. 1999;19:1912–1921. doi: 10.1523/JNEUROSCI.19-06-01912.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Levine JD, Reichling DB. Peripheral mechanisms of inflammatory pain. In: Wall PD, Melzack R, editors. Textbook of Pain. 4. Churchill Livingstone; London: 1999. pp. 59–84. [Google Scholar]
- 26.Levine JD, Fields HL, Basbaum AI. Peptides and the primary afferent nociceptor. J Neurosci. 1993;13:2273–2286. doi: 10.1523/JNEUROSCI.13-06-02273.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Nelson MT, Joksovic PM, Perez-Reyes E, Todorovic SM. The endogenous redox agent L-cysteine induces T-type Ca2+ channel-dependent sensitization of a novel subpopulation of rat peripheral nociceptors. J Neurosci. 2005;25:8766–75. doi: 10.1523/JNEUROSCI.2527-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Nelson MT, Woo J, Kang H-W, Barrett PQ, Vitko J, Perez-Reyes E, Lee J-H, Shin H-S, Todorovic SM. Reducing agents sensitize C-type nociceptors by relieving high-affinity zinc inhibition of T-type calcium channels. J Neurosci. 2007;27:8250–8260. doi: 10.1523/JNEUROSCI.1800-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Pabbidi RM, Cao DS, Parihar A, Pauza ME, Premkumar LS. Direct role of streptozotocin in inducing thermal hyperalgesia by enhanced expression of transient receptor potential vanilloid 1 in sensory neurons. Mol Pharmacol. 2008;73(3):995–1004. doi: 10.1124/mol.107.041707. [DOI] [PubMed] [Google Scholar]
- 30.Pathirathna S, Todorovic SM, Covey DF, Jevtovic-Todorovic V. 5alpha-reduced neuroactive steroids alleviate thermal and mechanical hyperalgesia in rats with neuropathic pain. Pain. 2005;117:326–39. doi: 10.1016/j.pain.2005.06.019. [DOI] [PubMed] [Google Scholar]
- 31.Perez-Reyes E, Cribbs LL, Daud A, Lacerda AE, Barclay J, Wiliamson MP, Fox M, Rees M, Lee J. Molecular characterization of a neuronal low-voltage-activated T-type calcium channel. Nature. 1998;391:896–900. doi: 10.1038/36110. [DOI] [PubMed] [Google Scholar]
- 32.Perez-Reyes E. Molecular physiology of low-voltage-activated T-type calcium channels. Physiol Rev. 2003;83:117–161. doi: 10.1152/physrev.00018.2002. [DOI] [PubMed] [Google Scholar]
- 33.Rydh-Rinder M, Berge OG, Hokfelt T. Antinociceptive effects after intrathecal administration of phosphodiester-, 2′-O-allyl-, and C-5-propyne-modified antisense oligodeoxynucleotides targeting the NMDAR1 subunit in mouse. Brain Res Mol Brain Res. 2001;86:23–33. doi: 10.1016/s0169-328x(00)00248-5. [DOI] [PubMed] [Google Scholar]
- 34.Talley EM, Cribbs LL, Lee JH, Daud A, Perez-Reyes E, Bayliss DA. Differential distribution of three members of a gene family encoding low voltage-activated (T-type) calcium channels. J Neurosci. 1999;19:1895–1911. doi: 10.1523/JNEUROSCI.19-06-01895.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Todorovic SM, Lingle CJ. Pharmacological properties of T-type Ca2+ current in adult rat sensory neurons: effects of anticonvulsant and anesthetic agents. J Neurophysiol. 1998;79:240–52. doi: 10.1152/jn.1998.79.1.240. [DOI] [PubMed] [Google Scholar]
- 36.Todorovic SM, Jevtovic-Todorovic V, Meyenburg A, Mennerick S, Perez-Reyes E, Romano C, Olney JW, Zorumski CF. Redox modulation of T-type calcium channels in rat peripheral nociceptors. Neuron. 2001;31:75–85. doi: 10.1016/s0896-6273(01)00338-5. [DOI] [PubMed] [Google Scholar]
- 37.Todorovic SM, Meyenburg A, Jevtovic-Todorovic V. Redox modulation of peripheral T-type Ca2+ channels in vivo: alteration of nerve injury-induced thermal hyperalgesia. Pain. 2004;109:328–339. doi: 10.1016/j.pain.2004.01.026. [DOI] [PubMed] [Google Scholar]
- 38.Shin JB, Martinez-Salgado C, Heppenstall PA, Lewin GR. A T-type calcium channel required for normal function of a mammalian mechanoreceptor. Nat Neurosci. 2003;6:724–730. doi: 10.1038/nn1076. [DOI] [PubMed] [Google Scholar]
- 39.Woolf CJ. Dissecting out mechanisms responsible for peripheral neuropathic pain: implications for diagnosis and therapy. Life Sci. 2004;74:2605–2610. doi: 10.1016/j.lfs.2004.01.003. [DOI] [PubMed] [Google Scholar]
- 40.White G, Lovinger DM, Weight FF. Transient low-threshold Ca2+ current triggers burst firing through an afterdepolarizing potential in an adult mammalian neuron. PNAS USA. 1989;86:6802–6806. doi: 10.1073/pnas.86.17.6802. [DOI] [PMC free article] [PubMed] [Google Scholar]