

Summary
Prolonged accumulation of misfolded proteins in the endoplasmic reticulum (ER) results in ER stress-mediated apoptosis. Cyclophilins are protein chaperones that accelerate the rate of protein folding through their peptidyl-prolyl cis-trans isomerase (PPIase) activity. In this study, we demonstrated that ER stress activates the expression of the ER-localized cyclophilin B (CypB) gene through a novel ER stress response element. Overexpression of wild-type CypB attenuated ER stress-induced cell death, whereas overexpression of an isomerase activity-defective mutant, CypB/R62A, not only increased Ca2+ leakage from the ER and ROS generation, but also decreased mitochondrial membrane potential, resulting in cell death following exposure to ER stress-inducing agents. siRNA-mediated inhibition of CypB expression rendered cells more vulnerable to ER stress. Finally, CypB interacted with the ER stress-related chaperones, Bip and Grp94. Taken together, we concluded that CypB performs a crucial function in protecting cells against ER stress via its PPIase activity.
Keywords: Cyclophilin B, Endoplasmic reticulum stress, Peptidyl prolyl cis-trans isomerase (Pplase), Reactive oxygen species (ROS)
Introduction
The lumen of the endoplasmic reticulum (ER) is a specialized cellular environment for the post-translational modification and folding of proteins. Properly folded proteins exit the ER and are trafficked through the secretory pathway, whereas unfolded proteins are retained and directed to the ER-associated protein degradation machinery (ERAD). ER stress is defined as an excess in the ER protein-folding load beyond the protein folding capacity (Harding et al., 1999). ER-stressed cells respond in two ways. First, protein synthesis is attenuated as the result of the reduced activity of the eukaryotic translation initiation factor 2 (eIF2) complex, which normally recruits charged initiator methionyl tRNA to the 40S ribosomal subunit (Kaufman, 2004). In addition, to relieve the protein-folding load, two more mechanisms have been described recently: IRE1 (also known as ERN1)-mediated ER-localized mRNA degradation (Hollien and Weissman, 2006) and co-translational proteasomal degradation of proteins destined to ER by p58IPK (also known as DNAJC3) in ER stressed-cells (Oyadomari et al., 2006; Rutkowski et al., 2007). Second, the transcription of genes encoding for ER-localized chaperones, but not cytosolic chaperones, is activated through a signaling pathway known as the unfolded protein response (UPR). In mammals, IRE1, PERK and ATF6 are three ER-localized transmembrane proteins that sense the presence of unfolded proteins in the ER lumen. Under normal conditions, these signaling molecules are maintained in an inactive state via the interaction of their luminal domains with the protein chaperone, Bip (also known as HSPA5). Bip is an ER-localized member of the Hsp70 protein chaperone family, which interacts with unfolded polypeptide chains. Upon exposure of the ER to stress, Bip is released from the stress sensors, in favor of binding to unfolded proteins. The release of IRE1 from Bip activates its protein kinase and endoribonuclease activity to initiate the splicing of XBP1 mRNA, which generates a potent transcription factor for UPR gene expression. Upon release from Bip, PERK is activated, phosphorylating the alpha subunit of eIF2 and attenuating protein synthesis. Paradoxically, eIF2α phosphorylation simultaneously promotes the translation of selective mRNAs. For example, the elevated translational activity of the transcription factor ATF4 by eIF2α phosphorylation results in an approximate 33% induction of UPR genes. Finally, Bip release causes ATF6 transport to the Golgi apparatus, where it is cleaved to yield a cytosolic fragment that migrates to the nucleus, activating the transcription of UPR genes including ER-resident folding enzymes and molecular chaperones such as Bip and Grp94 (also known as HSP90B1).
The ER stress response element (ERSE), a consensus sequence of CCAAT-N9-CCACG, is both necessary and sufficient for the transcriptional induction of genes encoding the major ER chaperones, such as Bip, Grp94, PDI and calreticulin (Kokame et al., 2001; Yamamoto et al., 2004). The increased expression of these proteins alleviates ER stress and enhances cell survival. However, when the protein-folding burden cannot be resolved, cells induce the expression of the CHOP proapoptotic transcription factor, and activate procaspase 12 (in mouse) or procaspase 4 (in human) to promote cell death (Hitomi et al., 2004).
Understanding the molecular mechanisms of how cells overcome ER stress is important because an increasing body of evidence has implicated ER stress in the etiology of a variety of diseases, including Alzheimer disease, polyglutamine-induced aggregation diseases, Parkinson’s disease, and diabetes (Kudo et al., 2002; Ozcan et al., 2008; Ozcan et al., 2006; Mattson and Chan, 2003; Conn et al., 2004; Katayama et al., 2004; Pereira et al., 2004; Ryu et al., 2005; Schroder and Kaufman, 2005).
Cyclophilins have been identified as cellular binding proteins for the immunosuppressive drug cyclosporin A (CsA) (Hong et al., 2002; Choi et al., 2007), and are constitutively expressed in most tissues (Andreeva et al., 1999). Cyclophilins have peptidyl prolyl cis-trans isomerase (PPIase) activity, which catalyzes protein folding in cells. Several classical isoforms of cyclophilins including CypA, CypB, CypC and CypD (also known as PPIA, PPIB, PPIC and PPID, respectively) have been identified and found to reside in their distinctive cellular locales, seemingly providing the compartment-specific functions. CypD is an integral component of the mitochondrial permeability transition complex, and plays a crucial role in apoptosis (Lin and Lechleiter, 2002; Baines et al., 2005; Schinzel et al., 2005). CypA is the most abundant CsA binding cytosolic protein. CypC localizes both in the cytosol and ER lumen, whereas CypB is detected mainly in the ER lumen. Although there has been some speculation that CypB plays a role in protein folding in the ER, there is currently no direct information supporting the notion that it provides a significant role in response to ER stress. In the present study, we have demonstrated that CypB performs vital functions to protect cells against ER stress.
Results
Cyclophilin B is transcriptionally upregulated by ER stress
Most ER molecular chaperones are upregulated during ER stress (Gillece et al., 1999; Uccelletti et al., 2004). In our experiments, the incubation of H9C2 cells with ER stress-inducing agents such as 1 µM thapsigargin (Tg) and 10 µg/ml tunicamycin (Tm) also rapidly increased expression levels of the major ER chaperone proteins Bip, Grp94, and PDI as well as CypB (Fig. 1A). Their induction by ER stress was observed under both proliferation and differentiation conditions. Densitometric analysis showed expression of CypB was approximately twofold greater on Tg and Tm treatment (Fig. 1A). Also, concurrent cell death upon ER stress was seen under microscopy (Fig. 1B). ER stress-induced cell death under both proliferation and differentiation conditions was further characterized by western blot analysis of caspase 3, procaspase 12, cytosolic cytochrome c, and mitochondrial Bax (Fig. 1C) and by Hoechst 33342 staining (Fig. 1D), indicating apoptotic death occurs when cells are subjected to ER stress.
Fig. 1.

Cyclophilin B is transcriptionally upregulated by ER stress. H9C2 cells were treated with 1 µM Tg and 10 µg/ml Tm under proliferation conditions (PM) for 48 hours or under differentiation conditions (DM) for 24 hours. Controls (con) are untreated cells. (A)Western blot analysis with ER stress marker antibodies to Bip, Grp94 and PDI. CypB protein level was quantified by densitometry. Actin was utilized as a loading control. Numbers under each band of CypB are representative of at least three different experiments and are expressed as the means ± s.d. *P<0.05 versus untreated cell in PM; #P<0.05, versus untreated cell in DM. (B) Micrographs of ER stress-induced cell death. (C)Western blot with antibodies to apoptosis markers: caspase 3 (cleaved form), Bax (mitochondrial), procaspase 12 and cytochrome c (cytosolic). When exposed to Tg or Tm, cleavage of caspase 3, reduction of procaspase 12, cytochrome c release and Bax translocation were observed. β-tubulin and HSP60 were used as a loading control for the cytosolic and mitochondrial fractions, respectively. Numbers under each band of procaspase 12 are representative of at least three different experiments and are expressed as the mean ± s.d. *P<0.05 versus untreated cell in PM; #P<0.05, versus untreated cell in DM. (D) Measurement of apoptosis-induced chromatin condensation by Hoechst 33342 staining. Yellow arrows indicate apoptosis-induced chromatin condensation and fragmentation. In the bar graph the data are expressed as the means ± s.d. obtained from five independent experiments. *P<0.05 versus untreated cells in PM condition; #P<0.05 versus untreated cell in DM condition. (E,F) Semi-quantitative RT-PCR analysis of CypB transcripts. GAPDH mRNA amplification was used as a control for RT-PCR. Transcript levels of CypB in untreated cells were used as a reference. Numbers under the each band of CypB are representative of at least three different experiments and are expressed as the means ± s.d. *P<0.05 versus untreated cell in PM; #P<0.05, versus untreated cell in DM (E). Actinomycin D (AD) was employed to inhibit mRNA synthesis. *P<0.05 actinomycin D-treated cell versus untreated cell (F).
To investigate if the elevated CypB is caused by transcriptional induction of CypB mRNA, we conducted semi-quantitative RT-PCR on the cells treated for 24 hours with ER stress-inducing agents. Upregulation of CypB mRNA on Tg and Tm treatment was clearly shown (Fig. 1E) and actinomycin D treatment abrogated the CypB mRNA induction (Fig. 1F), indicating that CypB mRNA was transcriptionally induced by ER stress, regardless of its mRNA stability.
Novel ER stress response element in cyclophilin B promoter
The transcriptional induction prompted our search for ER stress response elements on the CypB promoter; two putative ERSEs were found (Fig. 2A). In order to map the cis-acting element(s), serially deleted promoter regions were fused to the luciferase gene, the resultant reporter plasmids were transiently introduced into H9C2 cells and the activity of the CypB promoter was monitored by luciferase activity upon ER stress. More than a threefold increase in luciferase reporter activity in constructs harboring the full-length CypB upstream elements was observed on treatment of Tg and Tm. Deletion analysis showed that the region located between −400 bp and −1000 bp is dispensable for induction, but the region located between −200 bp and −400 bp is critical for the induction, as shown in Fig. 2B. The DNA sequence in this region harbors a cis-acting element similar to the ERSE consensus sequence (CCAAT-N9-CCACG, ERSE-I). To examine this further, we conducted mutagenesis studies using the pCypB/400 luciferase reporter plasmid with one putative ERSE from the CypB promoter. In the consensus-like sequence of CypB, the orientation of the CCACG element is inverted and the spacer sequence between the CCAAT and CCACG elements is 13 bases, rather than the consensus nine bases (Fig. 2A). Nucleotide replacements within each of these elements demonstrated that both elements are essential for ER stress induction from the CypB promoter (Fig. 2C). Indeed, it was reported that the transcription factors NF-Y and ATF6 bind simultaneously to the CCAAT and CCACG portions of the ERSE, respectively (Yoshida et al., 2001). Collectively, we concluded that the new ERSE found in the CypB promoter functions as the cis-acting element required for induction of CypB by ER stress, although the transcriptional activity of this ERSE is slightly weaker than that of the ERSE consensus sequence, ERSE-I (Fig. 2C, line 7).
Fig. 2.

Promoter analysis of cyclophilin B. (A) Sequence analysis of the CypB promoter with the Genomatix MatInspector program. Two putative ERSE elements of the CypB promoter are underlined. (B) Luciferase reporter assay of CypB promoter. Cells were incubated with 1 µM Tg or 10 µg/ml Tm for 24 hours in proliferation condition. Control were the cells without Tg or Tm treatment. The data are expressed as means ± s.d. from five independent experiments. §P<0.05 versus untreated pGL3 basic; *P<0.05 versus untreated pCypB/400; #P<0.05 versus untreated pCypB/400. (C) Mutation analysis of the ERSE consensus-like sequence. Luciferase assay was conducted using pCypB/400-based luciferase reporter constructs containing an ERSE mutated promoter. 1, pGL3 basic vector; 2, pCypB/400; 3, CCAAg mutant construct; 4, aaccT mutant construct; 5, CtTGG mutant construct; 6, aGTGG mutant construct; 7, pCypB/400 containing ERSE consensus sequence. The data are expressed as the means ± s.d. obtained from five independent experiments. *P<0.05 versus Tg-treated pCypB/400; #P<0.05 versus Tm-treated pCypB/400. (D) Activation of the CypB promoter by ATF6 or XBP1. Luciferase assay was conducted with pCypB/400 and one of the following constructs: pcDNA, inactive ATF6, active ATF6, unspliced XBP1, spliced XBP1, or ATF6-siRNA. The data are expressed as the means ± s.d. obtained from five independent experiments. *P<0.05 versus inactive ATF6 transfection; #P<0.05 versus unspliced XBP1 transfection; **P<0.05 versus active ATF6 transfection. pcDNA was employed as a negative control. All luciferase data are shown with the means ± s.d. relative to the basal activity of pCypB/400 (B–D). (E) EMSA and supershift EMSA of the CypB ERSE motif. Nuclear extracts prepared from H9C2 cells exposed to Tm for 24 hours or left unexposed, were incubated with the CypB-ERSE probe or a mutated probe (CGTtt) after preincubation with no antibody or with antiserum specific to ATF6. (F) ChIP assays with antibodies against ATF6, XBP1 or NF-Y along with extracts of H9C2 cells exposed to Tg or Tm for 24 hours or left unexposed. Input, sample representing amplification from a 1:100 dilution of total input chromatin. IgG: pre-immune serum. (G)Western blot with antibodies against ATF6, NF-Y or Lamin B using nuclear extracts. When exposed Tg and Tm, active ATF6 was increased whereas NF-Y remained unchanged. Lamin B was used as a loading control for nuclear extracts.
We next investigated binding of the ER stress-related transcription factors ATF6 and XBP1 to the CypB promoter through the novel CypB ERSE using a modified luciferase reporter assay and EMSA. A constitutively active form of the ATF6 gene or a spliced form of the XBP1 gene was transfected along with the luciferase reporter plasmid pCypB/400 to determine whether either active ATF6 or spliced XBP1 protein is alone sufficient to induce the luciferase reporter. Co-transfection with active ATF6 or spliced XBP1 increased the luciferase activity approximately twofold. Neither transfection with inactive ATF6 or unspliced XBP1 nor silencing ATF6 by ATF6-siRNA treatment, after evaluating the efficacy of siRNA treatment by western blot analysis (data not shown), could increase the luciferase activity (Fig. 2D).
EMSA was immediately followed to measure the direct binding of ATF6 or XBP1 to the CypB ERSE sequences. Nuclear extracts were prepared from H9C2 cells treated with or without Tm and then subjected to EMSA, using a 32P-labeled CypB-ERSE oligonucleotide probe, or a mutated version (CGTtt). The wild type formed two complexes with Tm-treated nuclear extracts: the lower band putatively with NF-Y protein, and the upper one with NF-Y plus ATF6. Supershift EMSA with anti-ATF6 antibody clearly showed ATF6 binding activity to the CypB-ERSE (Fig. 2E). By contrast, the mutated version was only minimally complexed with NF-Y and ATF6, suggesting the specificity of the novel ERSE of the CypB promoter. These bindings were abolished by incubation with a 100-fold excess of unlabeled competitor oligonucleotides. Moreover, ChIP assays also showed that CypB ERSE readily associates with ATF6 and NF-Y, but not XBP1 (Fig. 2F). Western blot analysis also demonstrated that active ATF6 is hardly detectable in control cells, but elevated upon ER stress whereas NF-Y remains unchanged (Fig. 2G).
Cyclophilin B overexpression suppresses thapsigargin or tunicamycin-induced cell death
In order to determine the protective role of CypB during the ER stress response, we overexpressed myc-tagged CypB/wt or myc-tagged CypB/R62A, which is defective in PPIase activity, in H9C2 cells. After establishing both CypB/wt and CypB/R62A stable transfectants, the expression level of CypB and other ER stress response proteins such as Bip and Grp94 were analyzed by immunoblotting (Fig. 3A). Among the three individual clones that we used for the study, overexpressed CypB did not appear to disturb expression level of the other ER stress proteins, Bip and Grp94.
Fig. 3.

Overexpression of cyclophilin B attenuates ER stress-induced cell death. (A) Expression level of CypB/wt or CypB/R62A in the indicated transfectants was analyzed by western blotting. Three different clones in each case were analyzed to exclude the possibility of clonal specificity. CypB was tagged with myc. ER chaperon proteins such as Bip and Grp94 were detected as a reference to monitor the effect of overexpressed CypB on other ER chaperon proteins. Actin was used as a loading control. (B) Localization of overexpressed CypB. After 24 hours of treatment with Tg or Tm under proliferation condition, localization of GFP-CypB/wt or GFP-CypB/R62A in the cells was examined by microscopy. ER tracker was used to identify of the ER. Upper panels shows the localization of GFP alone. Middle panels shows the localization of GFP-CypB/wt, and the lower panels shows the location of GFP-CypB/R62A. ER tracker, blue; GFP, green. Bars, 20 µm. (C) Attenuation of ER stress-induced cell death by overexpressed CypB/wt in proliferation (PM) or differentiation condition (DM) was determined by MTT assay. Note that CypB/R62A overexpression further reduces cell viability. The data are expressed as the means ± s.d. obtained from five independent experiments. *P<0.05 versus Tg-treated pcDNA transfected cells; #P<0.05 versus Tm-treated pcDNA transfected cells in PM and DM, respectively. (D)Western blot with apoptosis marker antibodies: caspase 3 (cleaved form), cytochrome c (cytosolic), procaspase 12 and Bax (mitochondrial). Numbers under each band are representative of at least five different experiments and are expressed as the means ± s.d. *P<0.05 versus Tg-treated pcDNA. (E) Changes in induction of ER resident proteins in CypB/wt and CypB/R62A transfectants. Total cellular extracts from the cells were analyzed for Bip, Grp94 and PDI by western blot. Numbers under each band are representative of at least five different experiments and are expressed as the means ± s.d. *P<0.05 versus Tg-treated pcDNA. (F) The defensive role of CypB is not limited to cell type. Chang and DU145 cells were infected with no adenovirus, Ad-GFP, Ad-CypB/wt, or Ad-CypB/R62A. The expression levels of CypB were detected by western blotting. Their transduction frequency (~60%) was monitored by GFP positivity. Numbers under each band are representative of at least five different experiments and are expressed as the means ± s.d. *P<0.05 versus non-infected Chang and DU145 cells, respectively. The survival rates of each infectant after ER stress were quantified by MTT assay. Ad-GFP, green fluorescence protein adenovirus; Ad-CypB/wt, CypB adenovirus; Ad-CypB/R62A, CypB/R62A adenovirus. The data represent the means ± s.d. obtained from five independent experiments. *P<0.05 versus Tg-treated non-infected cells; #P<0.05 versus Tm-treated non-infected Chang and DU145 cells.
Next, the location of the overexpressed CypB/wt and CypB/R62A was monitored via confocal microscopic analysis in order to rule out overexpression artifacts and to determine whether CypB/wt and CypB/R62A transfectants were correctly targeted to the ER. An ER tracker was utilized in order to locate the ER in the cells. Fig. 3B shows that overexpressed CypB/wt or CypB/R62A colocalizes with the ER tracker, thus suggesting that the overexpressed CypB/wt and CypB/R62A are targeted correctly to the ER and do not give rise to artifacts. Even when the cells were subjected to Tg and Tm, the location of the overexpressed proteins remained unchanged (Fig. 3B). Therefore, we concluded that the overexpressed CypB/wt is localized mainly in the ER, although we are currently unable to exclude the possibility that a small portion of CypB may be distributed elsewhere.
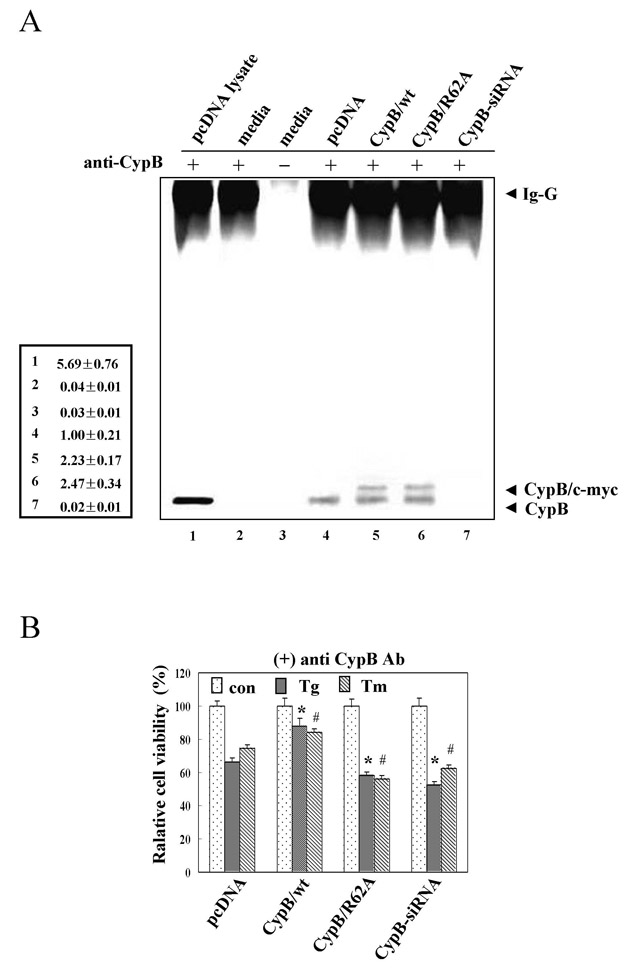
Effects of CypB/wt or CypB/R62A on cellular apoptosis under ER stress by Tg and Tm were determined by MTT conversion assay and western blot analysis of apoptotic molecules including cleaved caspase 3, cytosolic cytochrome c, procaspase 12 and mitochondrial Bax. As compared with pcDNA only, CypB/wt-overexpressing cells were significantly more resistant to Tg- and Tm-induced apoptosis, and CypB/R62A transfectants were even more sensitive (Fig. 3C,D). This acquired sensitivity from the PPIase-defective mutation indicates that PPIase activity might be involved in the protective role. On the premise that ER stress raises expression of the target molecules such as Bip, Grp94 and PDI, their diminution in CypB/wt-overexpressed cells indicated that overexpression of CypB/wt desensitized cells to ER stress; however, by contrast, overexpression of CypB/R62A sensitized them, as seen in Fig. 3E. Alleviation of ER stress by CypB overexpression, via its PPIase activity, suggests an anti-apoptotic role(s) of CypB against ER stress-induced cell death. Also, CypB is known to be secreted out of cells. To test the effect of secreted CypB, we performed immunoprecipitation of CypB from culture media (supplementary material Fig. S1A) and antibody neutralization using CypB antibody (supplementary material Fig. S1B). The results show that secreted CypB does not affect ER stress-induced cell death in cells transfected with pcDNA only, CypB/wt or CypB/R62A.
We next evaluated the anti-apoptotic effects of overexpressed CypB/wt against ER stress on Chang cells and DU145 prostate cancer cells to rule out the cell specificity. First, an adenovirus expressing GFP alone, CypB/wt or CypB/R62A was constructed and utilized in the transduction of Chang and DU145 cells (Fig. 3F). Immunoblotting analysis showed that exogenous CypB/wt or CypB/R62A was expressed at a similar level as endogenous CypB. In both cell types, CypB/wt infectants showed reduced cell death as compared with non-infected cells or Ad-GFP infectants under ER stress conditions (Fig. 3F). By contrast, CypB/R62A infectants showed significantly increased cell death. Collectively, these results suggest that the ability of overexpressed CypB/wt to suppress ER stress-induced apoptosis via PPIase activity is not limited to H9C2 cells.
Overexpression of cyclophilin B suppresses Ca2+ leakage from the ER, ROS generation, and mitochondrial membrane potential depolarization
Ca2+ in the ER has been reported to play an important role in apoptosis (Mattson and Chan, 2003; Benali-Furet et al., 2005). Therefore, we assessed the effects of overexpressed CypB/wt or CypB/R62A on ER Ca2+ homeostasis. After culturing in either Ca2+-containing (Fig. 4A) or Ca2+-free medium (Fig. 4B), cells were subjected to Tg. Cytosolic free Ca2+ was visualized as described in Materials and Methods. In both the Ca2+-containing and Ca2+-free media, far less Ca2+ was released into the cytosol in the CypB/wt transfectants after Tg treatment, as compared to pcDNA- or CypB/R62A-transfected cells. Interestingly, the overexpression of CypB/R62A caused a larger Ca2+ release from the ER than was observed in the pcDNA-transfected cells. These observations indicate that overexpressed CypB/wt abates, whereas overexpressed CypB/R62A exacerbates, Ca2+ leakage from the ER during ER stress.
Fig. 4.

Overexpressed cyclophilin B suppresses Ca2+ release from the ER in response to ER stress. Relative changes in cytosolic Ca2+ level in the indicated transfectants (n=20) are shown in the left panels and are plotted in the right panels. (A) Green color represents intracellular Ca2+. Red arrow (left panel) and dotted line (right panel). (B) Each transfectant was treated with 4 mM EGTA to reduce extracellular background Ca2+ prior to treatment with 1 µM Tg. In A and B the red arrowheads (left panel) and dotted line (right panel) indicate the Tg treatment point. In B the blue arrow (left panel) and dotted line (right panel) indicate the EGTA treatment point.
Reactive oxygen species (ROS) accumulation from unfolded protein response (UPR) under prolonged ER stress contributes to cell death (Haynes et al., 2004; Xue et al., 2005). We determined ROS levels, using DCF-DA (Fig. 5A), which detects intracellular ROS through the fluorometric measurement of the DCF oxidation. The loading of DCF-DA onto pcDNA-transfected cells showed that ROS accumulate in the presence of Tg. ROS accumulated by ER stress from Tg was attenuated by the overexpression of CypB/wt or by catalase treatment. By contrast, larger quantities of ROS were observed in the CypB/R62A transfectants using the same Tg concentration, indicating that CypB alleviates ROS generation at least partly through PPIase activity.
Fig. 5.

Cyclophilin B overexpression suppresses ROS generation, mitochondrial membrane damage and chromosomal DNA fragmentation in response to ER stress. (A) ROS measurement and its quantitative analysis. The dotted line represents the basal level of ROS in cells. Values are means ± s.d. obtained from five independent experiments. (B)MMP measurement and its quantitative analysis. Arrowhead indicates cells with mitochondrial damage. M1 and M2 are cells of damaged and undamaged mitochondria, respectively. (C) Cell viability by MTT assay. Con, untreated; Tg, 1 µM Tg treatment; Tg+catalase, pretreated with 2000 U/ml catalase prior to Tg treatment. The data are means ± s.d. obtained from five independent experiments. *P<0.05 versus Tg-treated pcDNA; #P<0.05 versus Tg-treated CypB/R62A. (D) TUNEL assay and its quantification. The green arrowheads in the top panels indicate TUNEL-positive cells. The data are expressed as the means ± s.d. obtained from five independent experiments. *P<0.05 versus Tg-treated pcDNA; #P<0.05 versus Tg-treated of CypB/R62A.
Mitochondrial membrane potential (MMP), which is associated with ER stress-induced cell death (Chae et al., 2004), was reduced in pcDNA transfected cells after 24 hours exposure to Tg (Fig. 5B). The MMP reduction by Tg, measured from the M2 region, was suppressed almost completely in the CypB/wt or by catalase treatment, but was exacerbated in CypB/R62A transfectants. Consistently, both cell viability as measured by MTT (Fig. 5C) and DNA fragmentation detected by the TUNEL assay (Fig. 5D) agreed with the results of the ROS and MMP measurement. Collectively, these results strongly indicate that CypB/wt overexpression significantly increased the resistance of the cells to ER stress-induced apoptotic cell death in a PPIase-dependent manner via the suppression of Ca2+ depletion in the ER, and attenuating ROS generation and mitochondrial damage.
Increased sensitivity of cyclophilin B deficient cells to ER stress-induced apoptosis
In order to assess in more detail the physiological roles of CypB in response to ER stress, we conducted RNA interference experiments using small interfering RNA (siRNA) to knockdown CypB. When evaluated by western blot analysis, the expression of CypB was suppressed almost completely by specific siRNA interference and the effects of CypB-siRNA on expression of other proteins such as Grp94 and Bip was not observed (Fig. 6A). The MTT assay, western blot analysis and TUNEL assay indicated that ER stress-induced apoptosis was significantly increased as a result of CypB knockdown (Fig. 6B,C). We also detected higher ROS levels and lower MMPs in CypB knockdown cells treated with Tg and Tm (data not shown), as compared with the untreated or control-siRNA-transfected cells. When z-VAD-fmk, a general caspase inhibitor, was added in order to inhibit apoptosis, ER stress-induced apoptosis was partially rescued under CypB knockdown conditions, thereby suggesting that CypB-mediated protection from ER stress may be partly caspase dependent (Fig. 6D).
Fig. 6.

siRNA knockdown of cyclophilin B enhances ER stress-induced cell death. (A) Knockdown efficiency of CypB-siRNA was determined by western blotting. ER chaperon proteins such as Grp94, Bip and PDI were detected as a reference to monitor the effect of CypB-siRNA on other ER chaperone proteins. (B) MTT assay. Relative cell viability of each cell line is shown based on that of the con-siRNA transfection. The data are means ± s.d. obtained from five independent experiments. *P<0.05 versus Tg-treated con-siRNA; #P<0.05 versus Tm-treated con-siRNA. (C)Western blotting with apoptosis marker antibodies: caspase 3 (cleaved form) and Bax. TUNEL assay performed after Tg treatment for 24 hours in con-siRNA and CypB-siRNA transfection. Green arrows indicate TUNELpositive cells. Numbers under each band are representative of at least five different experiments and are expressed as the means ± s.d. *P<0.05 versus Tg-treated con-siRNA transfection. (D) The defensive role of CypB is partly caspase dependent. PcDNA-, CypB/wt- and CypB-siRNA-transfected cells were cultured with 50 µM z-VAD-fmk prior to Tg treatment. The percentage of viable cells was determined using the MTT assay. The data are expressed as the means ± s.d. obtained from five independent experiments. *P<0.05 versus Tm-treated pcDNA transfectant; #P<0.05 versus Tm-treated CypB-siRNA transfectant.
Cyclophilin B physically interacts with Bip and Grp94
As ER-localized chaperones were reported to exist as a large complex, we attempted to determine whether CypB interacts with either Bip or Grp94 by GST pull-down (Fig. 7A) and co-immunoprecipitation assays (Fig. 7B). The results show that CypB interacts with Bip and Grp94 regardless of ER stress, and the apparent increases in the association after the ER stress may be a result of the increased chaperone expression. The interaction between CypB and Bip or Grp94 was finally verified by a yeast two-hybrid assay (Fig. 7C). In agreement with the GST pull-down assay (Fig. 7A), yeast two-hybrid assay showed an interaction of not only CypB/wt but also CypB/R62A with Bip, as well as Grp94, thus indicating that the PPIase activity of CypB does not influence binding to Bip or Grp94. Taken together, our results indicated that CypB interacts physically with Bip and Grp94, and that this interaction does not require the PPIase activity of CypB.
Fig. 7.

Cyclophilin B interacts with Bip and Grp94. (A) GST pull-down assay. A GST pull-down assay was conducted using bacterially expressed recombinant GST-CypB/wt and GST-CypB/R62A fusion proteins and H2C2 cell extracts treated with or without Tg and Tm. Western blotting was conducted using Bip, Grp94, CypB or GST antibodies. GST alone was used as a negative control. (B) Co-immunoprecipitation of CypB with Bip and Grp94. Immunoprecipitation was conducted with non-immune serum (Non-IS) or anti-CypB antibody (CypB-Ab). (C)Yeast two-hybrid binding assays. pGBKT7, pGADT7, pGBKT7/CypB/wt, pGBKT7/CypB /R62A, pGADT7/Grp78 and pGADT7/Grp94 were used in the yeast two-hybrid assay with AH109. In the left panel, CypB/wt interacts with Bip or Grp94. In the right panel, CypB/R62A also interacts with Bip or Grp94.
Discussion
CypB has been reported previously to exist in a complex with other molecular chaperones and folding enzymes (Menier et al., 2002), thereby suggesting a possible role in protecting cells against ER stress. In this study, we have demonstrated that CypB is induced in response to ER stress. In addition, the overexpression of wild-type CypB, but not of the catalytically inactive mutant CypB, protected cells against ER stress. Finally, CypB knockdown increased the sensitivity of cells to ER stress. All these findings suggest a crucial role(s) of CypB in protecting cells against ER stress.
The expression of genes, including those for Bip, Grp94 and PDI, is induced in response to ER stress (Ito et al., 2004). Analysis via RT-PCR and luciferase reporter assays showed that CypB expression is significantly upregulated in response to the ER stress-inducing agents, Tg [an inhibitor of Sarco-ER Ca2+ ATPase (SERCA) pump] and Tm (an inhibitor of N-linked glycosylation) (Patil and Walter, 2001). Consistent with the finding that the CypB gene is transcriptionally inducible by ER stress, we also discovered a novel ER stress response element (CCAAT-N9-gtaaCGTGG; herein referred to as ERSE-III) within the CypB promoter region, which is similar to the conventional ERSE-I motif (CCAAT-N9-CCACG). These sequences differ in two ways; the underlined CCACG in ERSE-I is oriented oppositely to that of ERSE-III, and the spacer between the CCAAT and CCACG motif is 4 bp longer. Several previous reports have described the existence of modified ERSE sequences, including ERSE-II (ATTGG-N-CCACG) (Kokame et al., 2001; Yamamoto et al., 2004). The lower fold induction of CypB may be the result of differences between ERSE-I and ERSE-III sequences. The mechanism by which ERSE-I mediates transcriptional activation has been extensively investigated. ATF6 has been shown to bind to the CCACG motif in the presence of NF-Y, a CCAAT binding factor. ATF6 has also been reported to bind independently to the sequence gtgaCGTGG, thereby indicating that the CGTGG element in the ERSE-III sequence may function as a direct binding site for ATF6 (Wang et al., 2000). Furthermore, the ACGT sequence of gtaaCGTGG is a proposed binding site for XBP1, another transcription factor that mediates ER stress-induced gene expression. However, EMSA and ChIP assays (Kanemoto et al., 2005) showed that NF-Y and ATF6, but not XBP1, are involved in CypB transcriptional regulation upon ER stress.
Our overexpression and siRNA-based knockdown studies indicate that CypB function is required for cell survival in response to ER stress. Previous studies using chemical cross-linking and immunoprecipitation have suggested that chaperones function in a large complex within the cells (Menier et al., 2002). This complex comprises the molecular chaperones including Bip, Grp94, PDI, ERp72, CypB, Grp170 and UDP-glucose:glycoprotein glucosyltransferase. Taking this a step further, we have demonstrated that CypB physically associates with ER stress-related proteins, including Bip and Grp94, thereby suggesting that CypB is one additional component of the ER stress-related complex. Also, our results showed that the CypB protective mechanism is characterized by reduction of Ca2+ release from the ER, ROS production, Bax translocation to the mitochondria, mitochondrial membrane depolarization, and also by suppression of activation of caspase 3 and caspase 12. In particular, the overexpression of CypB/wt inhibited Ca2+ leakage from the ER, whereas the overexpression of the PPIase-defective CypB/R62A mutant or siRNA-mediated knockdown of CypB (data not shown) increased Ca+2 leakage. In accordance with our data, CypB has been demonstrated to interact with a number of proteins that regulate Ca+2 homeostasis, including calreticulin, Bip, Grp94 and the Ca2+ signal modulating the cyclophilin ligand (Chae et al., 2004; Castilho et al., 1999; Chacon et al., 1991). Therefore, CypB PPIase activity may be directly associated with Ca2+ homeostasis during ER stress, or may indirectly influence the folding of Ca+2 regulatory proteins, although more investigations will be required to evaluate this hypothesis. Interestingly, the blockage of Ca2+ leakage from the ER, as a result of CypB overexpression in the ER, inhibited both mitochondrial damage and ROS production. Ca2+ concentrations in the mitochondria are dependent on those in the cytoplasm, which depend on Ca2+ influx through channels of the ER and the plasma membrane. A rise in Ca2+ concentrations in the cytoplasm accompanies their rise in mitochondria and results in impaired mitochondrial membrane potential (Castilho et al., 1999; Chacon et al., 1991). Thus, our data show that increases in cytosolic Ca2+ concentration resulting from Ca2+ leakage from the ER accelerate electron transport and stimulate ROS production. Our observations also indicate that the overexpressed CypB affects Ca2+ handling by the ER, in a manner resembling the effects of Bcl-2 overexpression or the knockout of Bax and Bak (Demaurex and Distelhorst, 2003), thereby suggesting that ER stress can trigger several parallel apoptosis signals, including disturbances in Ca2+ homeostasis in the ER, increased ROS generation, and reduced MMP, as was demonstrated by our results.
Intriguingly, the cellular behavior of CypB/R62A transfectants appears similar to that of CypB knockdown. A point mutation proximal to the binding pocket of CypA, R55A, abolished PPIase activity without affecting substrate binding (Zydowsky et al., 1992). Considering the highly homologous structures of CypA and CypB, the R62A PPIase mutation of CypB corresponds to the R55A mutation of CypA (Mikol et al., 1994). Thus, the enzymatically inactive CypB/R62A presumably retains substrate binding ability, which may account for the dominant negative-like behavior of CypB/R62A. Interestingly, a similar phenotype was detected for PPIase-defective Pin1 (the human homologue of Saccharomyces cerevisiae ESS1) (Rippmann et al., 2000). Little is currently known regarding the functional interactions between molecular chaperones in the ER, even though we have demonstrated that physical interactions occur among CypB, Bip and Grp94. The proteins that cooperate in their chaperone activity may rescue more substrates from protein aggregation than individual proteins without forming a complex. The molecular mechanisms relevant to the complex containing CypB, Bip and Grp94 remain to be investigated in the future.
In summary, our findings support a role(s) for CypB in ER stress. The overexpression of CypB prevents ER stress-associated phenotypes, including abnormal Ca2+ leakage, ROS generation, and mitochondrial membrane depolarization. Also, we have demonstrated for the first time that ER stress induces CypB expression via a novel ERSE in the CypB promoter region, and that CypB expression suppresses ER stress-mediated apoptosis. Future studies concerning the manner in which CypB prevents ER stress may prove helpful to our understanding of the maintenance of optimal cell function in a variety of diseases associated with protein misfolding in the ER.
Materials and Methods
Cell culture and reagents
H9C2 rat heart myoblasts were grown under proliferation conditions in (PM; proliferation medium) Dulbecco’s modified Eagle’s medium-F12 (DMEM-F12) supplemented with 10% (v/v) fetal calf serum, or under differentiation conditions in (DM; differentiation medium) DMEM-F12 supplemented with 1% horse serum. DU145 and Chang cells were cultured in DMEM supplemented with 10% fetal bovine serum and antibiotics (100 units/ml penicillin and 100 µg/ml streptomycin sulfate). In order to induce ER stress, thapsigargin (Tg) and tunicamycin (Tm) (BIOMOL) were utilized at 1 µM and 10 µg/ml, respectively.
RT-PCR
H9C2 cells were co-treated with actinomycin D (2 µM) and Tg (1 µM) or Tm (10 µg/ml) for 48 hours. For RT-PCR, total RNA (2 µg) was isolated with TRIzol reagent according to manufacturer’s protocol. cDNAs were synthesized with reverse transcriptase. PCR was performed for 32 cycles. Primers for the amplification of CypB transcripts were as follows; forward, 5′-GCATGAAGGTG CCCCC C-TCTTCGCC-3′; reverse, 5′-GCCTCCTTGGCAATGGCAAAGG-3′. GAPDH was amplified using primers (5′-GAAGGTGAAGGTCGGAGTC-3′ and 5′-GAAGA-TGGTGATGGGATTTC-3′) as an internal control for RT-PCR.
Plasmids
The wild-type rat CypB gene (CypB/wt) was amplified from a total RNA preparation via RT-PCR using the forward primer, 5′-GCAAGCTTATGAAGGT GCCC-CCCTCTTCGCC-3′ (HindIII) and the reverse primer, 5′-GCGGATCCCTCCTT-GGCAATGGCAAAGG-3′ (BamHI). The PPIase-defective mutant gene (CypB/R62A) was prepared by mutation of the arginine residue (R62) into alanine (A) using the following primers: 5′-AAGTTCCATGCTGTCATCAAGGAC-3′ and 5′-GTCCTTGATGACAGCATGGAACTT-3′. The PCR products were then inserted into a pcDNA 3.0 mammalian expression vector (Invitrogen) to make pcDNA/CypB/wt or pcDNA/CypB/R62A. CypB/wt or CypB/R62A was expressed with a myc tag at the C terminus. For the luciferase assays, 1000 bp of the CypB promoter sequence was serially deleted by 200 bp through PCR. The deleted fragments were then cloned into pGL3 basic vector (Promega) with KpnI and XhoI. The putative NF-Y or ATF6 binding sites on the CypB promoter were mutated via PCR-based site-directed mutagenesis. pEGFP NI (Clontech) containing CypB/wt or CypB/R62A cDNA was utilized for the localization of CypB/wt or CypB/R62A in cells. For the GST pull-down experiments, pGEX-KG (GE Healthcare) containing CypB/wt or CypB/R62A was constructed via PCR-based cloning. For the yeast two hybrid experiment, CypB or CypB/R62A was cloned into the EcoRI and BamHI restriction sites of pGBKT7.0 (Clontech). Bip or Grp94 was cloned into pGADT7.0 (Clontech) with the appropriate enzymes. The ATF6 inactive form, ATF6 active form, XBP1 unspliced form, and XBP1 spliced form plasmids were utilized in the luciferase assay (Lee et al., 2002).
Western blot
Antibodies against CypB, Bip, Bax, cytochrome c, Grp94, PDI, caspase 3, procaspase 12, actin, β-tubulin and myc were obtained from Abcam, Santa Cruz Biotechnology, and StressGen Biotechnologies. Unless otherwise specified, actin protein was immunoblotted in order to standardize the quantity of sample proteins.
Promoter analysis and luciferase assay
The CypB promoter sequence was analyzed using Genomatrix MatInspector (http://www.genomatix.de). One ERSE candidate was located at −243 bp, approximately −225 bp from the CypB ORF and the other was at −693bp, approximately −673 bp. H9C2 cells were transfected with 0.2 µg of the pGL3 basic-derived plasmids together with the internal control plasmid, pCMV-Lac (Promega). Luciferase and β-gal activities (not shown) were measured using 50 µl of each cell lysate using a microplate reader (Bio-Rad), and the luciferase activity was normalized on the basis of β-gal activity.
Electrophoretic mobility shift assay (EMSA)
H9C2 cells were treated with Tm (10 µg/ml) for 24 hours. For isolation of the nuclear fraction in Tm-treated or non-treated cells (approximately 80% confluence), cells were trypsinized and centrifuged at 220 g for 10 minutes. After removal of supernatants, total cells were resuspended with hypotonic buffer (10 mM Hepes pH, 7.9, 1.5 mM MgCl2, 10 mM KCl, 0.2 mM PMSF, 0.5 mM DTT, 1×protease inhibitor cocktail) and incubated at 4°C for 5 minutes. The cells were centrifuged at 1550 g at 4°C for 20 minutes. In order to isolate nuclear extracts, precipitates were incubated with hypotonic buffer: high salt buffer (20 mM Hepes pH, 7.9, 25% glycerol, 0.4 M KCl, 1.5 mM MgCl, 0.2 mM EDTA, 0.2 mM PMSF, 0.5 mM DTT; 1:1, v/v) at 4°C for 15 minutes. For removal of debris, cells were centrifuged at 9300 g at 4°C for 20 minutes. Nuclear protein concentration was determined by Bradford assays. Oligonucleotides (CypB-ERSE, 5′-CTATCCAATGAGAGGGCTGTAACGTGGC-AGC-3′; CGTtt, 5′-CTATCCAATGAGAGGGCTGTAACGTttCAGCCACCCGCA-3′; underlined residues indicate the NF-Y1-binding motif, lowercase residues indicate the ATF6-binding motif) were labeled with [γ-32P]ATP (Amersham Biosciences) using T4 polynucleotide kinase (Promega). Nuclear extracts (15 µg) treated with or without 10 µg/ml Tm and radiolabeled probe (CypB-ERSE or CGTtt) were incubated in the presence of 5× binding buffer [25% glycerol, 50 mM Tris, pH 7.5, 250 mM NaCl, 5 mM DTT, 5 mM EDTA, 20 µg/ml poly(dI-dC)] for 1 hour. For the competition assay, each sample was treated with a 100-fold excess of unlabeled competitor oligonucleotides. Rabbit antibodies against rat ATF6 (Santa Cruz Biotechnology) were utilized for the supershift assay. Nuclear extracts (15 µg) were loaded onto non-denatured TBE-polyacrylamide gel (4%) to resolve protein and γ-32P-labeled CypB probe complexes. Non-denatured TBE-polyacrylamide gel was run using 120 V for 6 hours. Dried gel was exposed with X-ray film for 36 hours.
Chromatin immunoprecipitation (ChIP)
Conventional chromatin IP was conducted as described previously (Luo et al., 2003) except that cross-linked H9C2 chromatin was subjected to immunoprecipitation with antibodies against ATF6, XBP1 or NF-Y (Santa Cruz Biotechnology). CypB-ERSE containing the binding sites of ATF6 and NF-Y was amplified using primers (5′-ATCCCAACGCCGCCTTCCGC-3′ and 5′-AAGCAATGCGGAGTGGTAGG-3′). Non-ERSE was amplified using primers (5′-ACTACAGAGAAGAGGCCTTA-3′ and 5′-CCTTGCTTGGGGACCGGGAG-3′) as a negative control.
Location of cyclophilin B
In order to determine the location of CypB, GFP, GFP-CypB/wt or GFP-CypB/R62A constructs were transfected into H9C2 cells. The transfectants, treated or untreated with Tg or Tm on coverslips, were loaded with 1 µM of ER tracker White-Blue DPI (Molecular Probes) for 30 minutes, then fixed for 15 minutes in 4% formaldehyde at room temperature. The stained cells were monitored using a LSM510 confocal laser microscope (Carl Zeiss).
MTT conversion assay
Cell viability was evaluated using the MTT reduction conversion assay in 12-well plates. The optical density was assessed at 550 nm in a microplate reader (Bio-Rad). Cell survival was expressed as the percentage of absorbance relative to that of untreated cells.
Hoechst 33342 staining and TUNEL assay
After treatment with Tg and Tm, H9C2 cells were incubated for 30 minutes with Hoechst 33342 (Molecular Probes) loading dye and washed three times in ice cold 1×PBS. The stained cells were monitored using a LSM510 confocal laser microscope (Carl Zeiss).
The TUNEL assay was conducted using the ApopDIRECT™ DNA fragmentation kit (MBL). Positive apoptotic nuclei were assessed using confocal microscopy.
Recombinant adenovirus
CypB/wt or CypB/R62A was cloned into pCA14. For homologous recombination, pCA14-CypB/wt or pCA14-CypB/R62A was transformed into E. coli BJ 5183 along with vmRL-H5dl324Bst. Recombinant adenovirus was propagated in HEK293 cells. Ad-GFP or uninfected cells were employed as a negative control for CypB expression.
Measurement of intracellular Ca2+ level
CypB/wt and CypB/R62A transfectants were seeded onto 25 mm coverslips in DMEM-F12 containing 10% serum. After 40 minutes of loading with the Ca2+-sensitive dye 10 µM Fluo-4 (Molecular Probes), the cells were rinsed twice in serum-free medium and stimulated with DMSO or 1 µM Tg. In order to remove the intracellular Ca2+, the cells were preincubated with 4 µM EGTA (Sigma-Aldrich) prior to Tg treatment. After 30 seconds, the cytosolic Ca2+ levels were assessed (n=20) using a LSM510 confocal microscope (Carl Zeiss).
Reactive oxygen species (ROS) and mitochondrial membrane potential (MMP) analyses (Δψm)
After incubation with 10 µM DCF-DA (Molecular Probes), cells were analyzed for ROS using flow cytometry. For MMP analysis, flow cytometry was used after incubation with 40 nM DiOC6 (Molecular Probes).
RNA interference
siRNA target sequences were as follows: CypB-siRNA (sense, 5′-UCCCAGAUGAGACUUCAAdTdT-3′; antisense, 5′-UUGAAGUUCUCAUCUG-GGAdTdT-3′), ATF6-siRNA (sense: 5′-GGGUUCAGAUAUUGCCGUAdTdT-3′; antisense: 5′-UACGGCAAUAUCUGAACCCdGdT-3′), and control-siRNA (sense, 5′-UCCCAGAUAGAGACUUCAATT-3′; anti-sense, 5′-UUGAAGUCUCUAUCU-GGGATT-3′). siRNA interference of CypB or ATF6 was analyzed using western blotting.
GST pull-down assay
Fusion proteins of pGEX-KG/CypB (GST-CypB/wt) and pGEX-KG/CypB/R62A (GST-CypB/R62A) were prepared as previously described (Bourdoncle et al., 2005). The affinity beads were incubated with 500 µg of H9C2 total cell extracts treated for 24 hours with or without Tg and Tm. The proteins retained on the beads were detected using western blot analysis.
Co-immunoprecipitation
H9C2 cell extracts (500 µg) treated with or without 1 µM Tg and 10 µg/ml Tm were incubated with non-immune serum-coupled protein G or A Sepharose beads or anti-CypB antibody-coupled protein G or A Sepharose beads. The immunoprecipitates were subjected to western blot analysis.
Yeast two-hybrid assay
The CypB/wt or CypB/R62A was ligated into pGBKT7 encoding for the GAL4 DNA binding domain (BD). The Bip or Grp94 gene was cloned into pGADT7 encoding for the activation domain (AD). To assess binding between CypB and Bip or Grp94, the experiments were conducted as described previously (Chen et al., 2004).
Statistical analyses
The results were expressed as the means ± s.d. from at least three independent experiments. Statistical analyses were conducted using Student’s t-tests. Unless indicated otherwise, P<0.05 was deemed to be significant.
Supplementary Material
{kind=link}
Acknowledgments
This work was supported by grants from the Korea Science and Engineering Foundation (No. R13-2002-020-02001-0 (2007)) to S.S.K., a grant from the Seoul RNBD program (No.11062) to W.C., Republic of Korea, and by NIH grant DK42394 to R.J.K. R.J.K. is an Investigator of the Howard Hughes Medical Institute.
Footnotes
Supplementary material available online at http://jcs.biologists.org/cgi/content/full/121/21/3636/DC1
References
- Andreeva L, Heads R, Green CJ. Cyclophilins and their possible role in the stress response. Int. J. Exp. Pathol. 1999;80:305–315. doi: 10.1046/j.1365-2613.1999.00128.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baines CP, Kaiser RA, Purcell NH, Blair NS, Osinska H, Hambleton MA, Brunskill EW, Sayen MR, Gottlieb RA, Dorn GW, et al. Loss of cyclophilin D reveals a critical role for mitochondrial permeability transition in cell death. Nature. 2005;434:658–662. doi: 10.1038/nature03434. [DOI] [PubMed] [Google Scholar]
- Benali-Furet NL, Chami M, Houel L, De Giorgi F, Vernejoul F, Lagorce D, Buscail L, Bartenschlager R, Ichas F, Rizzuto R, et al. Hepatitis C virus core triggers apoptosis in liver cells by inducing ER stress and ER calcium depletion. Oncogene. 2005;24:4921–4933. doi: 10.1038/sj.onc.1208673. [DOI] [PubMed] [Google Scholar]
- Bourdoncle A, Labesse G, Margueron R, Castet A, Cavailles V, Royer CA. The nuclear receptor coactivator PGC-1alpha exhibits modes of interaction with the estrogen receptor distinct from those of SRC-1. J. Mol. Biol. 2005;347:921–934. doi: 10.1016/j.jmb.2005.01.048. [DOI] [PubMed] [Google Scholar]
- Castilho RF, Ward MW, Nicholls DG. Oxidative stress, mitochondrial function, and acute glutamate excitotoxicity in cultured cerebellar granule cells. J. Neurochem. 1999;72:1394–1401. doi: 10.1046/j.1471-4159.1999.721394.x. [DOI] [PubMed] [Google Scholar]
- Chacon E, Acosta D. Mitochondrial regulation of superoxide by Ca2+: an alternate mechanism for the cardiotoxicity of doxorubicin. Toxicol. Appl. Pharmacol. 1991;107:117–128. doi: 10.1016/0041-008x(91)90336-d. [DOI] [PubMed] [Google Scholar]
- Chae HJ, Kim HR, Xu C, Bailly-Maitre B, Krajewska M, Krajewski S, Banares S, Cui J, Digicaylioglu M, Ke N, et al. BI-1 regulates an apoptosis pathway linked to endoplasmic reticulum stress. Mol. Cell. 2004;15:355–366. doi: 10.1016/j.molcel.2004.06.038. [DOI] [PubMed] [Google Scholar]
- Chen Q, Chen J, Sun T, Shen J, Shen X, Jiang H. A yeast two-hybrid technology-based system for the discovery of PPARgamma agonist and antagonist. Anal.Biochem. 2004;335:253–259. doi: 10.1016/j.ab.2004.09.004. [DOI] [PubMed] [Google Scholar]
- Choi KJ, Piao YJ, Lim MJ, Kim JH, Ha J, Choe W, Kim SS. Overexpressed cyclophilin A in cancer cells renders resistance to hypoxia- and cisplatin-induced cell death. Cancer Res. 2007;67:3654–3662. doi: 10.1158/0008-5472.CAN-06-1759. [DOI] [PubMed] [Google Scholar]
- Conn KJ, Gao W, McKee A, Lan MS, Ullman MD, Eisenhauer PB, Fine RE, Wells JM. Identification of the protein disulfide isomerase family member PDIp in experimental Parkinson’s disease and Lewy body pathology. Brain Res. 2004;1022:164–172. doi: 10.1016/j.brainres.2004.07.026. [DOI] [PubMed] [Google Scholar]
- Demaurex N, Distelhorst C. Cell biology. Apoptosis - the calcium connection. Science. 2003;300:65–67. doi: 10.1126/science.1083628. [DOI] [PubMed] [Google Scholar]
- Gillece P, Luz JM, Lennarz WJ, de La Cruz FJ, Römisch K. Export of a cysteine-free misfolded secretory protein from the endoplasmic reticulum for degradation requires interaction with protein disulfide isomerase. J. Cell Biol. 1999;147:1443–1456. doi: 10.1083/jcb.147.7.1443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harding HP, Zhang Y, Ron D. Protein translation and folding are coupled by an endoplasmic-reticulum-resident kinase. Nature. 1999;397:271–274. doi: 10.1038/16729. [DOI] [PubMed] [Google Scholar]
- Haynes CM, Titus EA, Cooper AA. Degradation of misfolded proteins prevents ER-derived oxidative stress and cell death. Mol. Cell. 2004;15:767–776. doi: 10.1016/j.molcel.2004.08.025. [DOI] [PubMed] [Google Scholar]
- Hitomi J, Katayama T, Eguchi Y, Kudo T, Taniguchi M, Koyama Y, Manabe T, Yamagishi S, Bando Y, Imaizumi K, et al. Involvement of caspase-4 in endoplasmic reticulum stress-induced apoptosis and Abeta-induced cell death. J. Cell Biol. 2004;165:347–356. doi: 10.1083/jcb.200310015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hollien J, Weissman JS. Decay of endoplasmic reticulum-localized mRNAs during the unfolded protein response. Science. 2006;313:104–107. doi: 10.1126/science.1129631. [DOI] [PubMed] [Google Scholar]
- Hong F, Lee J, Song JW, Lee SJ, Ahn H, Cho JJ, Ha J, Kim SS. Cyclosporin A blocks muscle differentiation by inducing oxidative stress and inhibiting the peptidyl-prolyl-cis-trans isomerase activity of cyclophilin A: cyclophilin A protects myoblasts from cyclosporin A-induced cytotoxicity. FASEB J. 2002;16:1633–1635. doi: 10.1096/fj.02-0060fje. [DOI] [PubMed] [Google Scholar]
- Ito D, Walker JR, Thompson CS, Moroz I, Lin W, Veselits ML, Hakim AM, Fienberg AA, Thinakaran G. Characterization of stanniocalcin 2, a novel target of the mammalian unfolded protein response with cytoprotective properties. Mol. Cell. Biol. 2004;24:9456–9469. doi: 10.1128/MCB.24.21.9456-9469.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kanemoto S, Kondo S, Ogata M, Murakami T, Urano F, Imaizumi K. XBP1 activates the transcription of its target genes via an ACGT core sequence under ER stress. Biochem. Biophys. Res. Commun. 2005;331:1146–1153. doi: 10.1016/j.bbrc.2005.04.039. [DOI] [PubMed] [Google Scholar]
- Katayama T, Imaizumi K, Manabe T, Hitomi J, Kudo T, Tohyama M. Induction of neuronal death by ER stress in Alzheimer’s disease. J. Chem. Neuroanat. 2004;28:67–78. doi: 10.1016/j.jchemneu.2003.12.004. [DOI] [PubMed] [Google Scholar]
- Kaufman RJ. Regulation of mRNA translation by protein folding in the endoplasmic reticulum. Trends Biochem. Sci. 2004;29:152–158. doi: 10.1016/j.tibs.2004.01.004. [DOI] [PubMed] [Google Scholar]
- Kokame K, Kato H, Miyata T. Identification of ERSE-II, a new cis-acting element responsible for the ATF6-dependent mammalian unfolded protein response. J. Biol. Chem. 2001;276:9199–9205. doi: 10.1074/jbc.M010486200. [DOI] [PubMed] [Google Scholar]
- Kudo T, Katayama T, Imaizumi K, Yasuda Y, Yatera M, Okochi M, Tohyama M, Takeda M. The unfolded protein response is involved in the pathology of Alzheimer’s disease. Ann. N. Y. Acad. Sci. 2002;977:349–355. doi: 10.1111/j.1749-6632.2002.tb04837.x. [DOI] [PubMed] [Google Scholar]
- Lee K, Tirasophon W, Shen X, Michalak M, Prywes R, Okada T, Yoshida H, Mori K, Kaufman RJ. IRE1-mediated unconventional mRNA splicing and S2P-mediated ATF6 cleavage merges to regulate XBP1 in signaling the unfolded protein response. Genes Dev. 2002;16:452–466. doi: 10.1101/gad.964702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin DT, Lechleiter JD. Mitochondrial targeted cyclophilin D protects cells from cell death by peptidyl prolyl isomerization. J. Biol. Chem. 2002;277:31134–31141. doi: 10.1074/jbc.M112035200. [DOI] [PubMed] [Google Scholar]
- Luo S, Baumeister P, Yang S, Abcouwer SF, Lee AS. Induction of Grp78/BiP by translation block: Activation of the Grp78 promoter by ATF4 through an upstream ATF/CRE site independent of the endoplasmic reticulum stress elements. J. Biol. Chem. 2003;278:37375–37385. doi: 10.1074/jbc.M303619200. [DOI] [PubMed] [Google Scholar]
- Mattson MP, Chan SL. Neuronal and glial calcium signaling in Alzheimer’s disease. Cell Calcium. 2003;34:385–397. doi: 10.1016/s0143-4160(03)00128-3. [DOI] [PubMed] [Google Scholar]
- Menier L, Usherwood YK, Chung KT, Hendershot LM. A subset of chaperones and folding enzymes form multiprotein complexes in endoplasmic reticulum to bind nascent proteins. Mol. Biol. Cell. 2002;13:4456–4469. doi: 10.1091/mbc.E02-05-0311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mikol V, Kallen J, Walkinshaw MD. X-ray structure of a cyclophilin B/cyclosporin complex: comparison with cyclophilin A and delineation of its calcineurin-binding domain. Proc. Natl. Acad. Sci. USA. 1994;91:5183–5186. doi: 10.1073/pnas.91.11.5183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oyadomari S, Yun C, Fisher EA, Kreglinger N, Kreibich G, Oyadomari M, Harding HP, Goodman AG, Harant H, Garrison JL, et al. Cotranslocational degradation protects the stressed endoplasmic reticulum from protein overload. Cell. 2006;126:727–739. doi: 10.1016/j.cell.2006.06.051. [DOI] [PubMed] [Google Scholar]
- Ozcan U, Yilmaz E, Ozcan L, Furuhashi M, Vaillancourt E, Smith RO, Görgün CZ, Hotamisligil GS. Chemical chaperones reduce ER stress and restore glucose homeostasis in a mouse model of type 2 diabetes. Science. 2006;313:1137–1140. doi: 10.1126/science.1128294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ozcan U, Ozcan L, Yilmaz E, Düvel K, Sahin M, Manning BD, Hotamisligil GS. Loss of the tuberous sclerosis complex tumor suppressors triggers the unfolded protein response to regulate insulin signaling and apoptosis. Mol. Cell. 2008;29:541–551. doi: 10.1016/j.molcel.2007.12.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Patil C, Walter P. Intracellular signaling from the endoplasmic reticulum to the nucleus: the unfolded protein response in yeast and mammals. Curr. Opin. Cell Biol. 2001;13:349–355. doi: 10.1016/s0955-0674(00)00219-2. [DOI] [PubMed] [Google Scholar]
- Pereira C, Ferreiro E, Cardoso SM, de Oliveira CR. Cell degeneration induced by amyloid-beta peptides: implications for Alzheimer’s disease. J. Mol. Neurosci. 2004;23:97–104. doi: 10.1385/JMN:23:1-2:097. [DOI] [PubMed] [Google Scholar]
- Rippmann JF, Hobbie S, Daiber C, Guilliard B, Bauer M, Birk J, Nar H, Garin-Chesa P, Rettig WJ, Schnapp A. Phosphorylation-dependent proline isomerization catalyzed by Pin1 is essential for tumor cell survival and entry into mitosis. Cell Growth Differ. 2000;11:409–416. [PubMed] [Google Scholar]
- Rutkowski DT, Kang SW, Goodman AG, Garrison JL, Taunton J, Katze MG, Kaufman RJ, Hegde RS. The Role of p58IPK in protecting the Stressed Endoplasmic Reticulum. Mol Biol. Cell. 2007;18:3681–3691. doi: 10.1091/mbc.E07-03-0272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ryu EJ, Angelastro JM, Greene LA. Analysis of gene expression changes in a cellular model of Parkinson disease. Neurobiol. Dis. 2005;18:54–74. doi: 10.1016/j.nbd.2004.08.016. [DOI] [PubMed] [Google Scholar]
- Schinzel AC, Takeuchi O, Huang Z, Fisher JK, Zhou Z, Rubens J, Hetz C, Danial NN, Moskowitz MA, Korsmeyer SJ. Cyclophilin D is a component of mitochondrial permeability transition and mediates neuronal cell death after focal cerebral ischemia. Proc. Natl. Acad. Sci. USA. 2005;102:12005–12010. doi: 10.1073/pnas.0505294102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schröder M, Kaufman RJ. ER stress and the unfolded protein response. Mutat. Res. 2005;569:29–63. doi: 10.1016/j.mrfmmm.2004.06.056. [DOI] [PubMed] [Google Scholar]
- Uccelletti D, O’Callaghan C, Berninsone P, Zemtseva I, Abeijon C, Hirschberg CB. Ire-1-dependent transcriptional up-regulation of a luminal uridine diphosphatase from Caenorhabditis elegans. J. Biol. Chem. 2004;279:27390–27398. doi: 10.1074/jbc.M402624200. [DOI] [PubMed] [Google Scholar]
- Wang Y, Shen J, Arenzana N, Tirasophon W, Kaufman RJ, Prywes R. Activation of ATF6 and an ATF6 DNA binding site by the endoplasmic reticulum stress response. J. Biol. Chem. 2000;275:27013–27020. doi: 10.1074/jbc.M003322200. [DOI] [PubMed] [Google Scholar]
- Xue X, Piao JH, Nakajima A, Sakon-Komazawa S, Kojima Y, Mori K, Yagita H, Okumura K, Harding H, Nakano H. TNFalpha induces the unfolded protein response (UPR) in a ROS-dependent fashion, and the UPR counteracts ROS accumulation by TNF alpha. J. Biol. Chem. 2005;280:33917–33925. doi: 10.1074/jbc.M505818200. [DOI] [PubMed] [Google Scholar]
- Yamamoto K, Yoshida H, Kokame K, Kaufman RJ, Mori K. Differential contributions of ATF6 and XBP1 to the activation of endoplasmic reticulum stress-responsive cis-acting elements ERSE, UPRE and ERSE-II. J. Biochem. 2004;136:343–350. doi: 10.1093/jb/mvh122. [DOI] [PubMed] [Google Scholar]
- Yoshida H, Okada T, Haze K, Yanagi H, Yura T, Negishi M, Mori K. Endoplasmic reticulum stress-induced formation of transcription factor complex ERSF including NF-Y (CBF) and activating transcription factors 6alpha and 6beta that activates the mammalian unfolded protein response. Mol. Cell. Biol. 2001;21:1239–1248. doi: 10.1128/MCB.21.4.1239-1248.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zydowsky LD, Etzkorn FA, Chang HY, Ferguson SB, Stolz LA, Ho SI, Walsh CT. Active site mutants of human cyclophilin A separate peptidyl-prolyl isomerase activity from cyclosporin A binding and calcineurin inhibition. Protein Sci. 1992;1:1092–1099. doi: 10.1002/pro.5560010903. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.