

Abstract
Hepatic oxidative stress and lipid peroxidation are common features of several prevalent disease states, including alcoholic liver disease (ALD) and non-alcoholic fatty liver disease (NAFLD), a common component of the metabolic syndrome. These conditions are characterized in part by excessive accumulation of lipids within hepatocytes, which can lead to autocatalytic degradation of cellular lipids giving rise to electrophilic end products of lipid peroxidation. The pathobiology of reactive lipid aldehydes remains poorly understood. We therefore sought to investigate the effects of 4-hydroxynonenal (4-HNE) and 4-oxononenal (4-ONE) on the transport and secretion of very low-density lipoprotein using HepG2 cells as a model hepatocyte system. Physiologically relevant concentrations of 4-HNE and 4-ONE rapidly disrupted cellular microtubules in a concentration-dependent manner. Interestingly, 4-ONE reduced apolipoprotein B-100 (ApoB) secretion while 4-HNE did not significantly impair secretion. Both 4-HNE and 4-ONE formed adducts with ApoB protein, but 4-HNE adducts were detectable as mono-adducts, while 4-ONE adducts were present as protein-protein cross-links. These results demonstrate that reactive aldehydes generated by lipid peroxidation can differ in their biological effects, and that these differences can be mechanistically explained by the structures of the protein adducts formed.
Keywords: 4-hydroxynonenal, 4-oxononenal, apolipoprotein B-100, microtubules, lipid peroxidation
1. Introduction
Excessive accumulation of lipid droplets within the liver as a result of metabolic disturbances is a common feature of several important diseases, including alcoholic liver disease (ALD) and non-alcoholic fatty liver disease (NAFLD) [1–3]. Fatty liver is now recognized as an important condition that can lead to more severe pathology, and it has been estimated that nearly one third of individuals in the United States have steatotic livers [4, 5]. A consequence of cellular lipid accumulation and oxidative stress is the autocatalytic process of lipid peroxidation and the production of biogenic lipid aldehydes, which may be biomarkers as well as mediators of disease [6, 7]. Indeed, lipid peroxidation has been reported in ALD and in NAFLD, and may play a role in progression of these diseases [8–12]. The α,β-unsaturated aldehyde 4-hydroxynonenal (4-HNE) is a prototypical end product of lipid peroxidation and has been widely studied in the context of inflammatory diseases [6]. 4-oxononenal (4-ONE) is generated through an independent pathway and has more recently been recognized as an important lipid peroxidation product [13–15]. These related aldehydes can react with protein Cys, His, and Lys residues to form Michael adducts, or with Lys ε-amino groups to generate Schiff base products [6, 14]. In addition to simple adducts, protein-protein cross-links can also be formed, with 4-ONE showing a much greater cross-linking propensity than 4-HNE.
Because the liver actively secretes lipids as very low-density lipoprotein (VLDL), fatty liver may increase exposure of VLDL components to lipid peroxidation products within the liver. VLDL is an important secretory product of the liver responsible for supplying fuel in the form of lipids to peripheral tissues, and undergoes enzymatic delipidation to generate low-density lipoprotein (LDL). The major protein component of VLDL and LDL is apolipoprotein B-100 (ApoB).
Modification of protein and lipid components of VLDL and LDL by reactive aldehydes is of particular interest because of the association between oxidized LDL and cardiovascular disease (CVD) [10, 11, 16–20]. Despite this association, the structures and biological effects of specific lipid peroxidation products on hepatic VLDL secretion remain poorly understood [16, 20–22]. A recent study suggests that peroxidation of polyunsaturated fatty acids within the liver has a negative regulatory effect on VLDL secretion, but did not examine the effects of specific lipid peroxidation products [23]. It was therefore important to investigate the effects of the lipid aldehydes 4-HNE and 4-ONE on hepatic VLDL secretion and to elucidate the mechanisms involved. These aldehydes were selected as model compounds because they can be generated in significant quantities within the liver, and have been reported to form potentially atherogenic adducts with VLDL [22]. Additionally, because of their structural similarity and the absence of biological interconversion, differences in their biological effects can be directly correlated to specific differences in chemical reactivity [13, 24, 25].
Several studies have demonstrated that the microtubule system plays an important role in hepatic secretion of VLDL and the accompanying triglyceride [26, 27]. Tubulin protein is present within the cell as either free heterodimers comprised of α- and β-tubulin, or as large microtubule polymers formed by the association of many heterodimers. The lipid aldehyde 4-HNE has been reported to cause impairment of tubulin polymerization in neuronal cells [28, 29], and studies using purified tubulin protein have characterized the disruptive effects of 4-HNE and related aldehydes on tubulin [30–32]. It was therefore important to investigate the effects of 4-HNE and 4-ONE on hepatic microtubules, and to characterize any resulting effects on ApoB secretion.
In order to further elucidate the role reactive aldehydes play in altering hepatic lipoprotein synthesis, transport, and secretion, we sought to characterize the effects of hepatic exposure to subcytotoxic concentrations of 4-HNE and 4-ONE on microtubule structure and secretion of the major VLDL protein component ApoB. Interestingly, although the chemical structures of 4-HNE and 4-ONE are very similar and their effects on microtubules are comparable, their effects on secretion of ApoB were found to be different. These differences in the effects of 4-HNE and 4-ONE can be explained mechanistically based on the presence or absence of protein cross-linking adducts. The results presented here indicate that the chemical structures of 4-HNE- and 4-ONE-ApoB adducts may be critical in determining the hepatic fate of VLDL, and may thereby modulate liver and cardiovascular pathology.
2. Materials and methods
2.1 Materials
Unless otherwise specified, all chemicals were obtained from Sigma-Aldrich Chemical Co. (St. Louis, MO). Lactate dehydrogenase (LDH) release assays, α-tubulin antibody, β-actin antibody, colchicine, brefeldin A, Hoechst 33342, RPMI 1640 medium, fetal bovine serum, 2, 4-dinitrophenylhydrazine (DNPH), and streptavidin-coated plates were obtained from Sigma (St. Louis, MO). Synthesis of 4-HNE and 4-ONE was performed as previously described and was greater than 99% pure by TLC and UV/vis spectrophotometry [33]. Coverslip mounting medium containing DAPI was purchased from Vector Laboratories (Burlingame, CA). A CCK-8 cell counting kit was purchased from Dojindo Molecular Technologies (Rockville, MD). Fluorescent goat anti-mouse AlexaFluor 488 secondary antibody was obtained from Invitrogen (Eugene, OR). Microsomal triglyceride transfer protein (MTP) and MTP activity assay kits were obtained from Roar Biomedical (New York, NY). ApoB ELISA kits were purchased from AlerChek (Portland, ME), and human ApoB standard was purchased from Meridian Life Science, Inc. (Saco, ME). Protein desalting spin columns and biotin hydrazide were obtained from Pierce (Rockford, IL). HepG2 cells were obtained from ATCC (Manassas, VA).
Cell Proliferation, and Cytotoxicity Assays, and Cellular DNA Measurement
Cell proliferation assays were performed according to the manufacturer’s instructions using a CCK-8 kit from Dojindo Molecular Technologies (Rockville, MD), and lactate dehydrogenase (LDH) release assays were performed using a kit from Sigma. 4-HNE and 4-ONE solutions for cell treatment were made fresh in 50 mM sodium phosphate, pH 7.4. HepG2 cells were grown on 6-well plates to >90% confluence and were treated in serum-free RPMI 1640 for 1 hour with 4-HNE or 4-ONE to give final concentrations of ranging from 20 μM to 250 μM, followed by a 24 hour recovery at 37° C in serum-containing RPMI 1640 media. Hoechst 33342 was used to measure cellular DNA as previously described [34]. Briefly, cells were collected in 1 mL of phenol red-free, serum-free RPMI 1640, and 50 μL of cell suspension was incubated in sterile 96 well fluorescence plates for 1 hour at 37° C with 200 μL of serum-free, phenol red-free RPMI 1640 containing Hoechst 33342 at 10 μg/mL. Plates were shaken for 10 seconds prior to reading, and cellular DNA was measured by Hoechst 33342 staining with excitation at 355 nm and emission at 465 nm.
2.2 Soluble Tubulin Extraction and Soluble/Microtubule Ratios
HepG2 cells were plated in 6 well plates at a density of 500,000 cells per well, and grown to >90% confluence in RPMI 1640 media supplemented with 10% fetal calf serum and 1% penicillin/streptomycin. Cells were exposed to final concentrations of 20 μM, 50 μM, or 100 μM of 4-HNE or 4-ONE in 6 well plates with a 3 mL treatment per well in serum-free RPMI 1640 for 15 minutes, 30 minutes, or 1 hour time points. Following treatment, aldehyde-containing media was removed and soluble and insoluble microtubule fractions were extracted by a modification of the technique reported by Kannarkat et. al. [35]. Briefly, cells were washed once with warm PEM (80 mM Pipes, 2 mM EDTA, 1 mM MgCl2, pH 6.8) and extracted for 1 minute in 1 mL PEM buffer containing 0.15% Triton X-100 to extract soluble proteins. The soluble fraction was then subjected to centrifugation at 15,000 rpm at 37° C to pellet any cell debris, and the supernatant was collected and transferred to a fresh tube and boiled in 6X SDS loading buffer. The insoluble fractions were collected in 1 mL of PEM buffer, sonicated to shear DNA, and boiled in SDS loading buffer. Paired soluble and insoluble fractions were loaded at 25 μL on 10% SDS-PAGE gels, and separated by electrophoresis. Proteins were transferred to PVDF membranes, and immunoblotting experiments were performed using an α-tubulin antibody (Sigma, St. Louis, MO) at 1:5000, and densitometry was performed on the resulting bands to calculate ratios of soluble: polymerized α-tubulin from each treatment group. Results were expressed as percent control. For determination of total cellular tubulin, cells were grown and treated as in the extraction protocol, except that whole cell pellets were collected in 1 mL phosphate buffered saline (PBS), pH 7.4, sonicated, and boiled in 6X SDS loading buffer. Immunoblotting experiments were performed as above, but with the addition of 1:5000 β-actin antibody. Results were calculated as the ratio of a tubulin to β actin.
2.3 Immunocytochemistry and Confocal Microscopy
HepG2 cells were grown on glass coverslips and treated with 100 μM 4-HNE, 4-ONE, or colchicine in serum-free media and microtubules were stained after 30 minutes of treatment, or aldehyde-containing treatment media was removed and cells were allowed to recover in serum-containing media for 24 hours, after which microtubules were stained. For microtubule staining, cells were fixed with 10% neutral buffered formalin for 30 minutes at room temperature, and the coverslips were then extracted for 20 minutes at room temperature with PBS, pH 7.4, containing 2.5% bovine serum albumin (BSA) and 1% Triton X-100. Coverslips were then washed three times for 5 minutes with TBS and blocked with 2.5% BSA in PBS for 10 minutes, followed by incubation with 1:500 α tubulin antibody (Sigma) in PBS for 30 minutes. After three five minute washes in TBST, coverslips were then incubated with 1:1000 secondary antibody (Invitrogen, Eugene, OR) goat anti-mouse AlexaFluor 488 for 30 minutes. Coverslips were again washed three times for 5 minutes in TBST, rinsed in TBS, and coverslips were mounted on microscope slides using mounting medium containing DAPI (Vector Laboratories, Burlingame, CA). Confocal images were taken with a 100X oil immersion objective on a Nikon Eclipse TE2000-E instrument, with identical instrument laser and contrast settings used within each group. Images were acquired using EZ-C1 software (Nikon, Melville, NY).
2.4 Microsomal Triglyceride Transfer Protein Activity Assay
Microsomal triglyceride transfer protein (MTP) activity was measured using a kit from Roar Biomedical (New York, NY). For cell experiments, HepG2 cells were treated with 100 μM 4-HNE or 4-ONE for 30 minutes in serum-free media, and cells were immediately collected and sonicated, and the manufacturer’s instructions were followed to measure MTP activity using 100 μg of cellular protein per well. MTP activity was assessed as the transfer of a labeled lipid from a donor particle to an acceptor particle.
2.5 ApoB ELISA
Secreted and intracellular ApoB were measured using an ApoB ELISA kit (AlerChek, Portland, Maine). HepG2 cells were treated with 20 μM, 50 μM, or 100 μM 4-HNE or 4-ONE for a period of 1 hour in 6 well plates with 3 mL treatment per well. Brefeldin A was used at 5 mg/mL as a control for impairment of VLDL secretion, and 100 μM colchicine was used as a control for microtubule disruption. Treatments were then terminated, and RPMI 1640 media containing stripped serum was added for 24 hours. Media was collected and spun at 15,000 rpm for 5 minutes to pellet any detached cells, and transferred to a fresh tube. The ApoB ELISA was then performed using the media according to the manufacturer’s instructions, and cell DNA was measured using Hoechst 33342 staining as described above. Secreted ApoB was then normalized to cellular DNA.
2.6 Biotin Hydrazide Derivatization, 2,4-Dinitrophenyl Hydrazine Derivatization, and Fluorescent Cross-link Detection
For determination of ApoB adducts within cells, HepG2 cells grown in 60 mm plates to >90% confluence were treated for 30 minutes with 100 μM 4-HNE or 4-ONE in serum-free media, and were collected in 0.5 mL sodium phosphate buffer, pH 7.4. Cells were sonicated and lysates were derivatized for 2 hours with 0.5 mM biotin hydrazide at room temperature. Samples were desalted three times using spin columns (Pierce) to remove unreacted biotin hydrazide. Using streptavidin plates (Sigma), wells were pre-washed 3 times with TBST, after which 100 μg of derivatized HepG2 lysate was added per well and incubated for 1 hour at room temperature. Subsequent steps were performed as per the ApoB ELISA, but with plate binding selecting for adducts. For determination of total carbonyls incorporated in ApoB and fluorescent cross-links, human ApoB (Meridian Life Science, Inc., Saco, ME) was treated with concentrations of 4-HNE or 4-ONE ranging from 3 nM to 500 μM for 30 minutes at 37° C in either 50 mM sodium phosphate or TRIS. Aldehyde Lys-Lys cross-links were detected by measuring fluorescence as previously reported [36] using excitation at 360 nm and emission at 430 nm. Samples were subsequently derivatized with 2, 4-dinitrophenylhydrazine (DNPH) as previously [37] reported, and absorbance was measured at 370 nm to quantify total carbonyls.
2.7 Statistical Analysis
Statistical analyses were performed using GraphPad Prism (GraphPad Software, La Jolla, CA). For comparison of 4-HNE and 4-ONE adducts, Student’s paired t-tests were performed. For comparisons of multiple groups, one-way analysis of variance was used with Tukey’s post-test. In all experiments, aldehyde concentrations ranged from 3 nM to 500 μM, as indicated in each figure. Data are presented as mean ± the standard error of the mean, with n = 3 to 12 independent experiments.
3. Results
In order to investigate the potential of 4-HNE and 4-ONE to dysregulate selected aspects of hepatic VLDL synthesis and secretion, experiments were conducted using the human HepG2 cell line. Experiments using this cell line showed that a single, 1 hour exposure of sub-confluent cells to 4-HNE or 4-ONE in serum-free media, followed by a 24 hour recovery period in serum-containing media, inhibited cell proliferation in a concentration-dependent manner, as shown in Figure 1A (4-HNE) and Figure 1B (4-ONE). However, as presented in Figures 1C and 1D, when cells were more than 90% confluent, a single treatment followed by 24 hours recovery did not result in a significant loss of cell number as measured by cellular DNA. Lactate dehydrogenase (LDH) release assays were used to assess cell damage following 2 hours exposure of the cells to various concentrations of 4-HNE and 4-ONE (Figure 1E), showing that these aldehydes are not toxic to HepG2 cells at or below concentrations of 100 μM. Taken together, these results indicate that HepG2 cells are capable of tolerating relatively high concentrations of 4-HNE and 4-ONE, and that the cellular effects observed in subsequent experiments are not due to cell death. The aldehyde concentrations used in these experiments are within the range of previous reports using primary rat heptocytes, and are believed to be biologically relevant for the liver [6, 38–41].
Fig. 1. Proliferation inhibition and cytotoxicity characteristics of 4-HNE and 4-ONE in HepG2 cells.
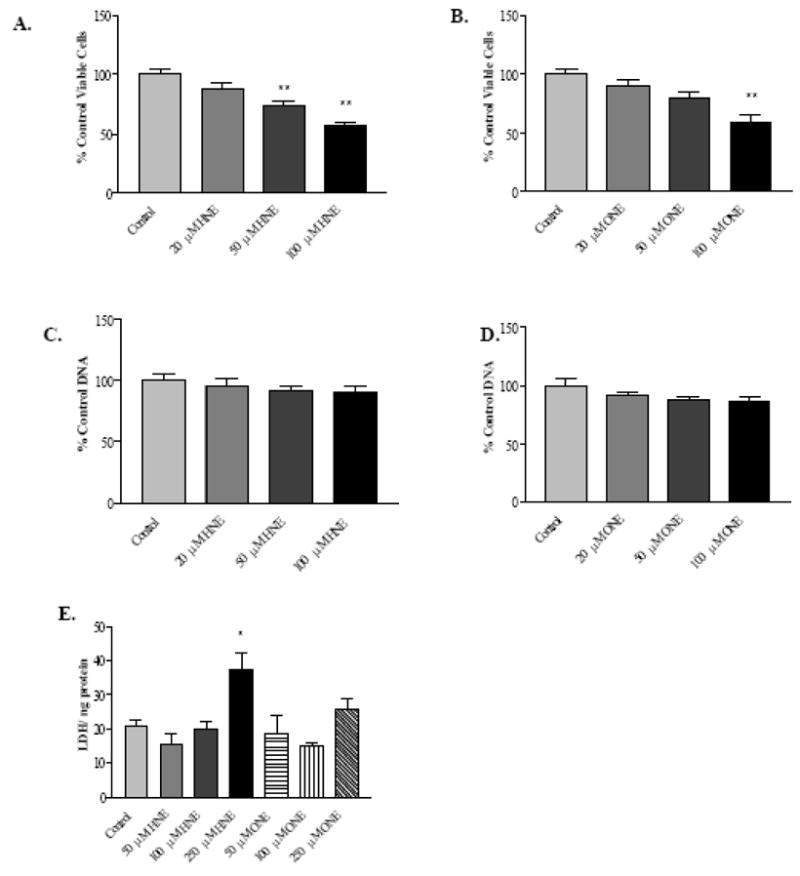
A, 4-HNE-mediated cell proliferation inhibition following 1 hour treatment and 24 hours recovery. B, 4-ONE-mediated cell proliferation inhibition following 1 hour treatment and 24 hours recovery. C, 4-HNE cytotoxicity measured as cellular DNA following 1 hour treatment and 24 hours recovery. D, 4-ONE cytotoxicity measured as cellular DNA following 1 hour treatment and 24 hours recovery. E, LDH release following 2 hours aldehyde treatment. N = 8 for proliferation inhibition experiments; n = 3 for cytotoxicity experiments, * P < 0.05, ** P < 0.001. Bars represent means +/− SEM.
As previously noted, microtubules are believed to play an important role in the transport of cellular cargo, including lipids destined to be secreted as VLDL. Cellular tubulin can be partitioned into two fractions using the extraction technique described previously, namely the soluble fraction (free dimer) and the polymerized fraction (microtubules). An increase in the ratio of soluble: polymerized tubulin indicates a loss of microtubule structures within the cell, and a corresponding decrease in transport along microtubule pathways. The data presented in Figure 2A and 2B demonstrate that 4-HNE and 4-ONE, respectively, significantly disrupt HepG2 microtubules in a concentration-dependent manner within 30 minutes of exposure. Based on the observed cellular microtubule disruption mediated by 4-HNE and 4-ONE with 30 minutes exposure, it was important to characterize the time course of this effect. Microtubule disruption occurs rapidly in that the aldehyde-mediated effects were observed as early as 15 minutes after exposure to 100μM 4-HNE or 4-ONE (Figure 2C), with an apparent maximal disruptive effect at 30 minutes exposure time (Figure 2D). HepG2 cells begin to recover from microtubule disruption by 1 hour post-exposure (Figure 2E), and cells treated with 4-HNE and 4-ONE, but not colchicine-treated cells, show partial (4-HNE) to full recovery (4-ONE) by 24 hours post-treatment (Figure 2F).
Fig. 2. 4-HNE and 4-ONE cause concentration- and time-dependent loss of microtubule polymer in HepG2 cells.
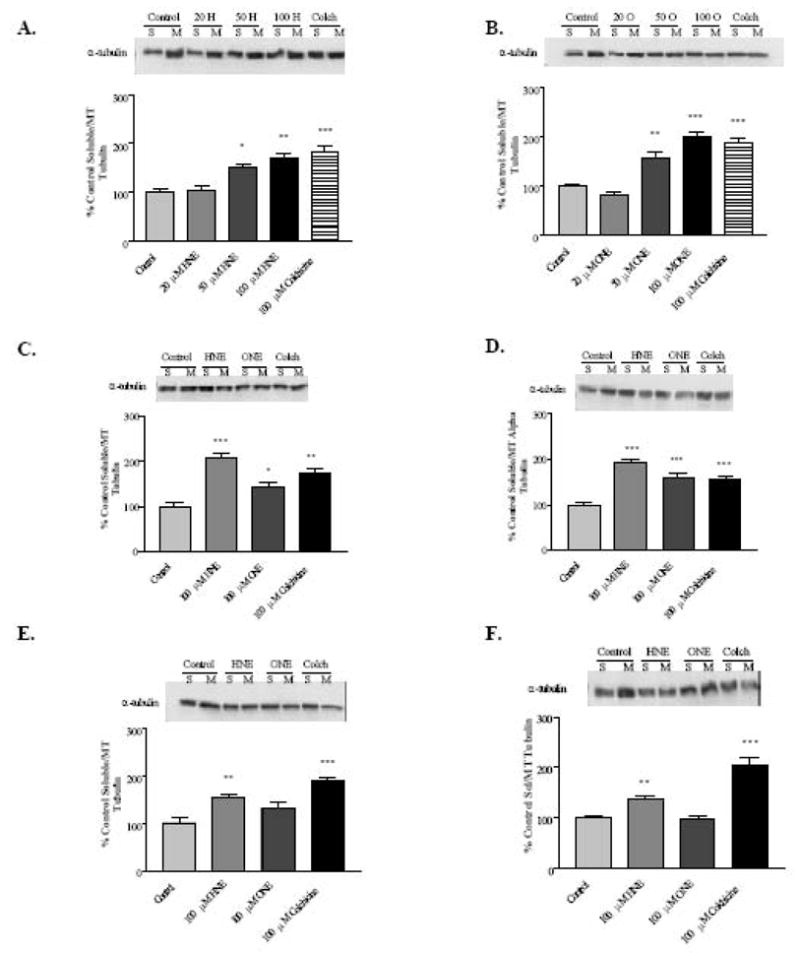
A, 4-HNE concentration-dependent loss of microtubule polymer at 30 minutes treatment. B, 4-ONE concentration-dependent loss of microtubule polymer at 30 minutes treatment. C-F, Lipid aldehyde-mediated microtubule disruption time course in HepG2 cells. C, 15 minutes exposure. D, 30 minutes exposure. E, 1 hour exposure. F, 30 minute exposure with 24 hour recovery. N = 4–12; * P < 0.05, ** P < 0.01, *** P < 0.001. H = 4-HNE, O = 4-ONE, Colch = Colchicine, S = Soluble fraction, M = Microtubule fraction. Bars represent means +/− SEM.
Confocal microscopy experiments confirm microtubule disruption by 4-HNE, 4-ONE, and colchicine at 30 minutes exposure (Figure 3A), as indicated by a loss of filamentous microtubule structures from the cytosol and the restriction of staining to bands surrounding the nuclei. A high degree of microtubule recovery was observed at 24 hours post-treatment with 4-HNE and 4-ONE, but not in cells treated with colchicine, a specific microtubule-disrupting agent. Potential interactions of these α,β-unsaturated agents with tubulin include inter-and intrapeptide cross-linking. As shown in Figure 3B, cross-linking of tubulin in HepG2 cells was observed with 100 μM 4-ONE treatment at 30 minutes, and cross-links were also detected at later time points with 4-ONE treatment, but not with 4-HNE treatment (data not shown). Treatment of cells with 4-HNE or 4-ONE did not significantly alter the total amount of tubulin protein present in the cells, while colchicine treatment reduced total cellular tubulin (Figure 3C). These results show that microtubule disruption by 4-HNE and 4-ONE occurs very rapidly following exposure, that cells recover from this disruption, and that the tubulin polymerization state, but not the total amount of tubulin present in the cell, is altered by 4-HNE or 4-ONE.
Fig. 3. HepG2 microtubule staining, microtubule cross-links, and total cellular tubulin.
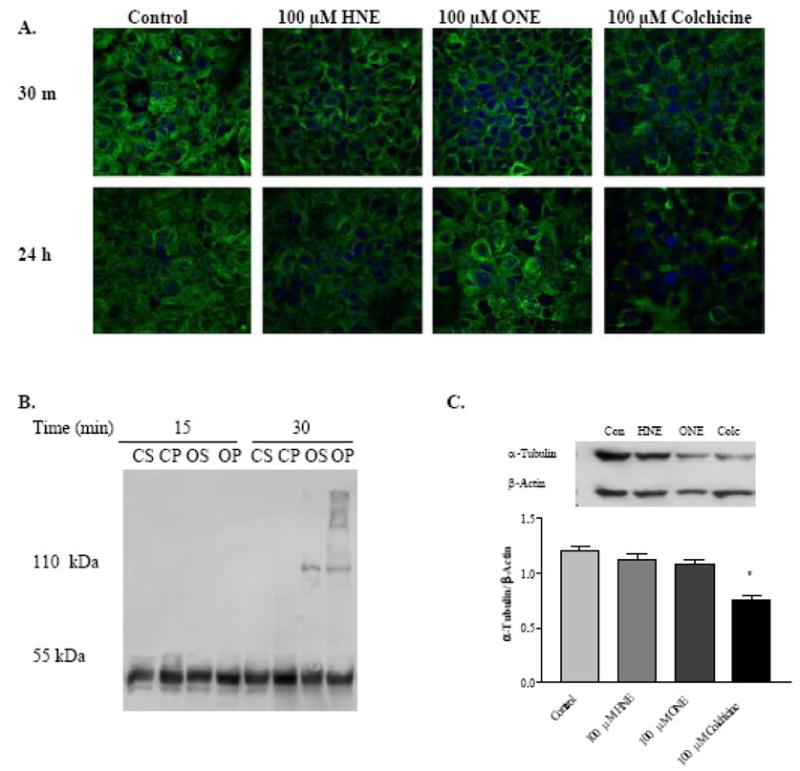
A, Confocal microscopy of microtubule structures at 30 minutes exposure or 30 minute treatment followed by 24 hours recovery. Blue = DAPI nuclear staining, Green = alpha tubulin staining. B, 4-ONE Cross-linking of tubulin in HepG2 cells exposed to 100 μM 4-ONE for 15 or 30 minutes. CS = Control soluble fraction, CP = Control polymerized fraction, OS = 4-ONE soluble fraction, OP = 4-ONE polymerized fraction. C, Total tubulin content following 30 minute aldehyde exposure and 24 hours recovery. N = 12, * P < 0.001. Bars represent means +/− SEM.
ApoB is an important protein component of VLDL and LDL, and alterations in VLDL secretion or oxidative modification of ApoB may play a role in progression of ALD, the metabolic syndrome, as well as CVD [10, 42–44] HepG2 cells exposed to 4-HNE did not show significant changes in ApoB secretion, while colchicine treatment caused a modest decrease in secretion (Figure 4A). In contrast, 4-ONE treatment was observed to strongly inhibit ApoB secretion in a concentration-dependent manner (Figure 4B). Brefeldin A, an agent that disrupts ER to Golgi transport [45], was used as a positive control for inhibition of VLDL secretion (Figure 4B). Thus, 4-HNE and 4-ONE had differing effects on ApoB secretion from HepG2 cells, suggesting that temporary microtubule disruption mediated by lipid aldehydes was not primarily responsible for alterations in ApoB secretion. Binding of the ELISA ApoB antibody was not affected by ApoB aldehyde modification, and therefore a decrease in signal from the ELISA indicates a lower level of ApoB protein rather than a loss of antibody recognition of the modified protein.
Figure 4. Divergent effects of 4-HNE and 4-ONE on HepG2 secreted and intracellular ApoB.
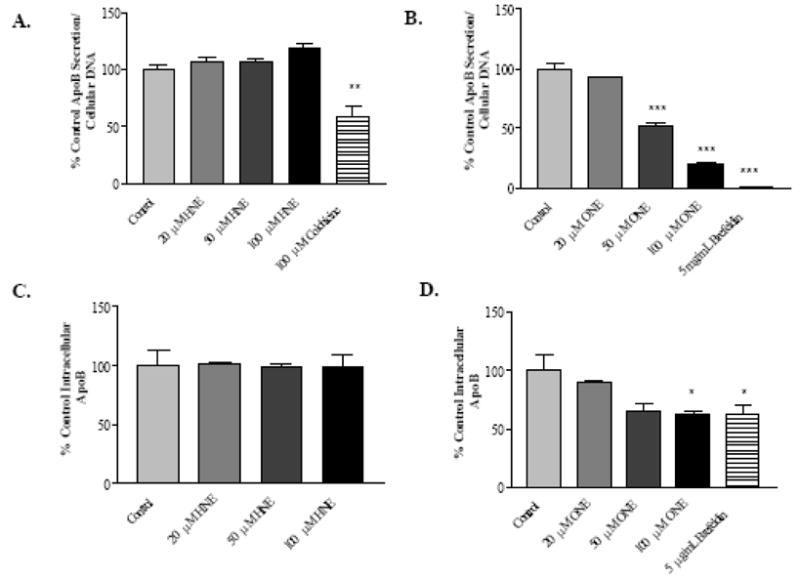
A, ApoB secretion from HepG2 cells following 1 hour 4-HNE or colchicine treatment and 24 hours recovery. B, ApoB secretion from HepG2 cells following 1 hour 4-ONE or brefeldin A treatment and 24 hours recovery. C, Intracellular levels of ApoB in HepG2 cells treated for 1 hour with 4-HNE and allowed to recover for 24 hours. D, Intracellular levels of ApoB in HepG2 cells treated for 1 hour with 4-ONE and allowed to recover for 24 hours. N = 3, * P < 0.05, ** P < 0.01, *** P < 0.001. Bars represent means +/− SEM.
In light of the observed differences in ApoB secretion mediated by 4-HNE and 4-ONE exposure, it was important to examine intracellular ApoB. As shown in Figure 4C, 1 hour 4-HNE treatment followed by a 24 hour recovery had no measurable effect on intracellular ApoB protein. However, brefeldin A caused a decrease in intracellular ApoB, and 4-ONE caused concentration-dependent loss of intracellular ApoB (Figure 4D).
Based on the differences in ApoB secretion and intracellular levels in cells treated with 4-HNE and cells treated with 4-ONE, and the possibility that ApoB adducts could alter secretion of VLDL, it was important to characterize ApoB adducts within HepG2 cells and using purified ApoB protein. Carbonyl measurement is a widely used and robust technique, robust technique for characterizing protein-aldehyde adducts [37]. Biotin hydrazide derivatization of lysates from HepG2 cells previously exposed to 4-HNE or 4-ONE was used to measure carbonylation of ApoB caused by aldehyde adduction. These experiments showed a significant increase in carbonylation of ApoB in 4-HNE treated cells, but no significant increase in ApoB modification in 4-ONE treated cells (Figure 5A).
Figure 5. Characterization of ApoB aldehyde adducts and HepG2 MTP activity.
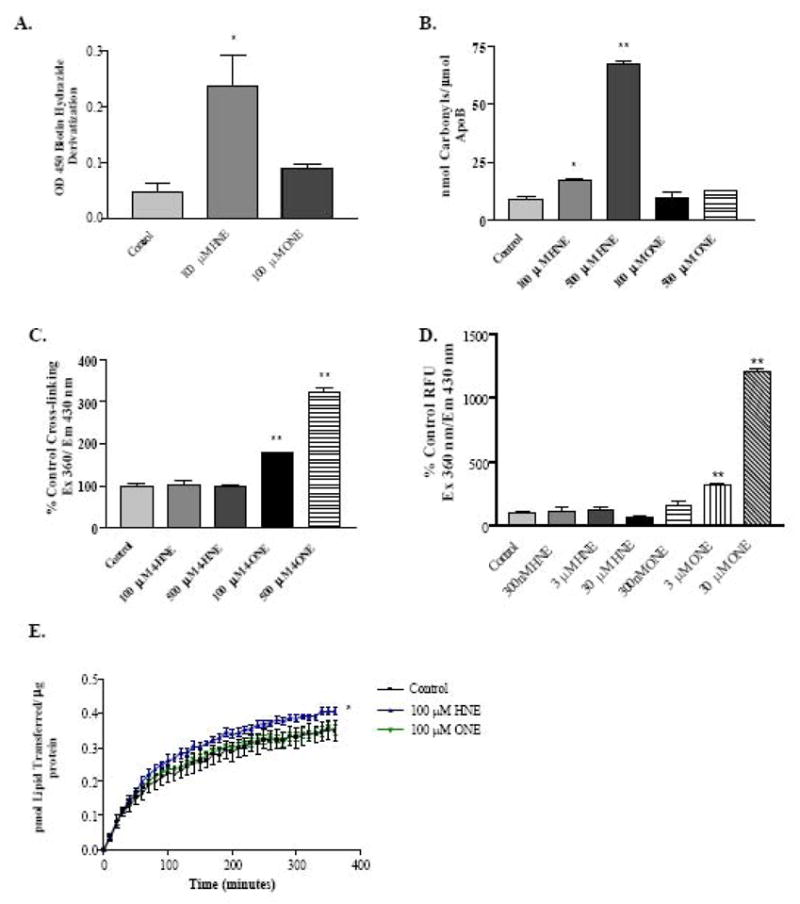
A, ApoB carbonylation in HepG2 cells treated for 30 minutes with 4-HNE or 4-ONE. B, Determination of simple adducts on purified human ApoB by measurement of ApoB carbonyl incorporation. C, Measurement of Lys-Lys ApoB adducts by fluorescent detection. D, Detection of ApoB Lys-Lys cross-links low aldehyde concentrations. E, MTP activity in HepG2 cells treated for 30 minutes with 4-HNE or 4-ONE. N = 3, * P < 0.05, **, P < 0.001. Bars represent means +/− SEM.
Because biotin hydrazide derivatization of ApoB for detection of aldehyde adducts measures only simple mono-adducts and will not detect cross-links, it was important to examine the relative importance of both simple adducts and cross-linking adducts on ApoB. The experiments in Figure 5B and 5C were performed using purified Human ApoB in a Tris-containing buffer. The primary amines present in Tris buffer can compete with the reaction of aldehydes with proteins and act as an aldehyde sink. These experiments were intended to demonstrate the ability of 4-HNE and 4-ONE to form ApoB adducts even in the presence of a competing target, as would occur in the cell. Figure 5B shows that 4-HNE significantly increases the detectable carbonyls in ApoB, while 4-ONE does not. However, an assay that specifically detects fluorescent aldehyde Lys-Lys cross-links [36, 46] shows that 4-ONE causes significant cross-linking of ApoB (Figure 5C). Figure 5D shows ApoB cross-linking by lower concentrations of 4-ONE in a sodium phosphate buffer that does not compete with protein adduct formation, indicating that cross-links occur at concentrations of 4-ONE as low as 3 μM. Thus, although ApoB is a target for adduction by 4-HNE and 4-ONE, the 4-HNE adducts are predominantly simple adducts, while the 4-ONE adducts predominantly form cross-links. This novel finding offers important mechanistic insight into the differing effects of 4-HNE and 4-ONE on HepG2 ApoB secretion.
The microsomal triglyceride transfer protein (MTP) is important in loading triglyceride onto ApoB to form VLDL particles for secretion by the hepatocyte. Impairment of MTP has been reported to cause intracellular lipid accumulation, and in this context it was important to investigate aldehyde effects on MTP. HepG2 cells treated for 30 minutes with 100 μM 4-HNE showed a modest, but statistically significant, increase in lipid transferred by MTP, while cells treated with 100 μM 4-ONE showed no change in MTP activity (Figure 5E).
4. Discussion
In view of the probable importance of lipid peroxidation products in the pathogenesis of ALD and NAFLD, it was important to investigate the effects of 4-HNE and 4-ONE on microtubule transport pathways and ApoB secretion in a model hepatic cell line. In this context, HepG2 cells are a well-characterized, widely used human hepatoma cell line. Although microtubules are dynamic cellular structures and can rapidly undergo changes in polymerization state, major changes in the overall microtubule polymerization state are abnormal except during cell division. In HepG2 cells, 4-HNE and 4-ONE were observed to rapidly and significantly disrupt cellular microtubules in a concentration-dependent manner (Figures 2 and 3).
Disruption of microtubule structures by 4-HNE and 4-ONE was observed at exposure times as early as 15 minutes, suggesting that the effect is likely due to a direct action of the aldehydes on either free dimer or on microtubules, or both. These results are in agreement with previously reported experiments using purified tubulin, showing impairment of polymerization by both 4-HNE and 4-ONE [30–32], as well as results reported for neuronal cells [28, 29]. Despite the changes in tubulin polymerization state, 4-HNE and 4-ONE do not measurably alter total levels of tubulin within HepG2 cells (Figure 3C). Tubulin cross-links were observed at an early time point (30 minutes) with 4-ONE treatment of HepG2 cells (Figure 3B). This observation agrees with our previous report of 4-HNE- and 4-ONE-mediated cross-linking of purified tubulin protein [32]. Covalent adducts of tubulin have previously been reported [28, 32], and the data presented here indicate that microtubule structures are among the protein targets of 4-HNE and 4-ONE within hepatocytes.
ApoB is the major protein component of VLDL, and was therefore used as an indicator of VLDL secretion. Despite causing similar levels of microtubule disruption, 4-HNE and 4-ONE had divergent effects on ApoB secretion. Impairment of ApoB secretion by 4-ONE is probably not a result of microtubule disruption, since colchicine causes a much more dramatic microtubule loss, but has a less marked effect on ApoB secretion, and cells recover more rapidly from 4-ONE induced microtubule disruption. This observation indicates that although microtubule structures play a role in ApoB secretion (Figure 4A), microtubule disruption alone cannot account for the 4-ONE-mediated impairment of ApoB secretion and suggests the involvement of additional pathways in mediating this effect. These results are consistent with previous reports indicating that although microtubules are normally used in many cellular transport and secretion pathways, disruption of microtubule structure does not necessarily result in impairment of secretion [47].
Based on its numerous solvent-accessible lysine residues, ApoB is a predictable target for adduction by reactive aldehydes, and can form Schiff base or Michael adducts, as well as intra- and intermolecular cross-links. Biotin hydrazide derivatization [48, 49] to detect aldehyde adducts of ApoB showed a significant increase in ApoB carbonylation in HepG2 cells treated with 4-HNE, but no statistically significant increase in cells treated with 4-ONE (Figure 5A). This likely reflects the difference in cross-linking capacity of 4-ONE compared to 4-HNE, since cross-links are not detected by this technique. Indeed, experiments using purified human ApoB demonstrated that 4-HNE forms simple adducts detectible by DNPH derivatization (Figure 5B), while 4-ONE adducts are present almost exclusively as cross-links (Figure 5C and 5D). This difference in the chemical nature of ApoB adducts formed by 4-HNE and 4-ONE provides a mechanistic basis for their different effects on ApoB secretion. 4-ONE caused a decrease in intracellular ApoB in a concentration-dependent manner (Figure 4C), from which it can be concluded that cross-linking of ApoB causes intracellular degradation, thereby inhibiting its secretion. 4-HNE did not affect intracellular ApoB levels, nor did it significantly affect ApoB secretion. Thus, the simple ApoB adduct that predominates with 4-HNE exposure does not significantly affect ApoB secretion, while the cross-linking adduct predominant with 4-ONE exposure significantly impairs secretion and accelerates degradation of ApoB. These results are consistent with previous reports implicating protein cross-links in the disruption of normal protein function [32, 50].
MTP is responsible for the co-translational lipidation of ApoB within the lumen of the endoplasmic reticulum, after which VLDL is transported to the Golgi where it undergoes additional lipidation prior to being secreted in a process that is at least partially microtubule-dependent [51, 52]. Failure of co-translational lipidation by MTP results in degradation of ApoB and impaired secretion of VLDL [45, 53, 54]. Thus, it was important to investigate the effects of 4-HNE and 4-ONE on MTP. Experiments in HepG2 cells treated with 4-ONE showed no significant change in lipid transfer by MTP, while 4-HNE treatment slightly increased lipid transfer (Figure 5E). Because ApoB is synthesized constitutively, the 4-ONE-mediated loss of secretion and decrease in intracellular ApoB is likely due primarily to accelerated degradation of ApoB at a later step in the VLDL maturation and secretion pathway, and not due to ApoB degradation caused by failure of cotranslational lipidation by MTP. Our findings are consistent with previous reports [23, 45].
In summary, important novel findings reported here include disruption of hepatic microtubules by 4-HNE and 4-ONE followed by rapid recovery of polymerization state, tubulin cross-linking by 4-ONE within a model hepatocyte cell line, 4-HNE adduction of ApoB, and a strong inhibition of ApoB secretion in hepatocytes exposed to 4-ONE. The overall differences between the cellular effects of 4-HNE and 4-ONE can be attributed to the fact that 4-ONE is a more potent protein cross-linking agent than 4-HNE [46]. These observations are significant, as they support the previously reported hypothesis that certain lipid peroxidation products generated in the liver may have cardiovascular-protective effects by inhibiting VLDL secretion and subsequent conversion to potentially atherogenic LDL [23]. In a broader sense, these results are significant because they indicate that chemically similar lipid aldehydes may in many cases have differing biological effects, and these effects can be mediated by subtle but important differences in the chemical nature of protein adducts that are formed.
Acknowledgments
Work supported by NIH 1 F31 AA015821-01 and NIH R37 AA 09300
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Peters TJ, Cairns SR. Alcohol. 1985;2:447–51. doi: 10.1016/0741-8329(85)90113-2. [DOI] [PubMed] [Google Scholar]
- 2.Paoletti R, Bolego C, Poli A, Cignarella A. Vasc Health Risk Manag. 2006;2:145–52. doi: 10.2147/vhrm.2006.2.2.145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Rector RS, Thyfault JP, Wei Y, Ibdah JA. World J Gastroenterol. 2008;14:185–92. doi: 10.3748/wjg.14.185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Browning JD, Szczepaniak LS, Dobbins R, Nuremberg P, Horton JD, Cohen JC, Grundy SM, Hobbs HH. Hepatology. 2004;40:1387–95. doi: 10.1002/hep.20466. [DOI] [PubMed] [Google Scholar]
- 5.El-Zayadi AR. World J Gastroenterol. 2008;14:4120–6. doi: 10.3748/wjg.14.4120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Esterbauer H, Schaur RJ, Zollner H. Free Radic Biol Med. 1991;11:81–128. doi: 10.1016/0891-5849(91)90192-6. [DOI] [PubMed] [Google Scholar]
- 7.Uchida K. Free Radic Res. 2000;33:731–7. doi: 10.1080/10715760000301251. [DOI] [PubMed] [Google Scholar]
- 8.Niemela O. Front Biosci. 1999;4:D506–13. doi: 10.2741/niemela. [DOI] [PubMed] [Google Scholar]
- 9.Sampey BP, Korourian S, Ronis MJ, Badger TM, Petersen DR. Alcohol Clin Exp Res. 2003;27:1015–22. doi: 10.1097/01.ALC.0000071928.16732.26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Girona J, Manzanares JM, Marimon F, Cabre A, Heras M, Guardiola M, Ribalta J, Masana L. Nutr Metab Cardiovasc Dis. 2008;18:380–7. doi: 10.1016/j.numecd.2007.04.002. [DOI] [PubMed] [Google Scholar]
- 11.Palmieri VO, Grattagliano I, Portincasa P, Palasciano G. J Nutr. 2006;136:3022–6. doi: 10.1093/jn/136.12.3022. [DOI] [PubMed] [Google Scholar]
- 12.Malhi H, Gores GJ. Semin Liver Dis. 2008;28:360–9. doi: 10.1055/s-0028-1091980. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Lee SH, Blair IA. Chem Res Toxicol. 2000;13:698–702. doi: 10.1021/tx000101a. [DOI] [PubMed] [Google Scholar]
- 14.Doorn JA, Petersen DR. Chem Biol Interact. 2003;143–144:93–100. doi: 10.1016/s0009-2797(02)00178-3. [DOI] [PubMed] [Google Scholar]
- 15.Lin D, Lee HG, Liu Q, Perry G, Smith MA, Sayre LM. Chem Res Toxicol. 2005;18:1219–31. doi: 10.1021/tx050080q. [DOI] [PubMed] [Google Scholar]
- 16.Parthasarathy S, Litvinov D, Selvarajan K, Garelnabi M. Biochim Biophys Acta. 2008;1781:221–31. doi: 10.1016/j.bbalip.2008.03.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Targher G, Arcaro G. Atherosclerosis. 2007;191:235–40. doi: 10.1016/j.atherosclerosis.2006.08.021. [DOI] [PubMed] [Google Scholar]
- 18.Jeppesen J, Hansen TW, Rasmussen S, Ibsen H, Torp-Pedersen C. Atherosclerosis. 2006;189:369–74. doi: 10.1016/j.atherosclerosis.2005.12.010. [DOI] [PubMed] [Google Scholar]
- 19.Holvoet P, Lee DH, Steffes M, Gross M, Jacobs DR., Jr Jama. 2008;299:2287–93. doi: 10.1001/jama.299.19.2287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Matsuura E, Hughes GR, Khamashta MA. Autoimmun Rev. 2008;7:558–66. doi: 10.1016/j.autrev.2008.04.018. [DOI] [PubMed] [Google Scholar]
- 21.Jurgens G, Lang J, Esterbauer H. Biochim Biophys Acta. 1986;875:103–14. doi: 10.1016/0005-2760(86)90016-0. [DOI] [PubMed] [Google Scholar]
- 22.Hoff HF, O’Neil J. J Lipid Res. 1993;34:1209–17. [PubMed] [Google Scholar]
- 23.Pan M, Cederbaum AI, Zhang YL, Ginsberg HN, Williams KJ, Fisher EA. J Clin Invest. 2004;113:1277–87. doi: 10.1172/JCI19197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Spiteller P, Kern W, Reiner J, Spiteller G. Biochim Biophys Acta. 2001;1531:188–208. doi: 10.1016/s1388-1981(01)00100-7. [DOI] [PubMed] [Google Scholar]
- 25.Zhu X, Sayre LM. Chem Res Toxicol. 2007;20:165–70. doi: 10.1021/tx600295j. [DOI] [PubMed] [Google Scholar]
- 26.Orci L, Le Marchand Y, Singh A, Assimacopoulos-Jeannet F, Rouiller C, Jeanrenaud B. Nature. 1973;244:30–2. doi: 10.1038/244030a0. [DOI] [PubMed] [Google Scholar]
- 27.Reaven EP, Reaven GM. J Cell Biol. 1980;84:28–39. doi: 10.1083/jcb.84.1.28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Neely MD, Boutte A, Milatovic D, Montine TJ. Brain Res. 2005;1037:90–8. doi: 10.1016/j.brainres.2004.12.027. [DOI] [PubMed] [Google Scholar]
- 29.Neely MD, Sidell KR, Graham DG, Montine TJ. J Neurochem. 1999;72:2323–33. doi: 10.1046/j.1471-4159.1999.0722323.x. [DOI] [PubMed] [Google Scholar]
- 30.Gabriel L, Miglietta A, Dianzani MU. Chem Biol Interact. 1985;56:201–12. doi: 10.1016/0009-2797(85)90006-7. [DOI] [PubMed] [Google Scholar]
- 31.Olivero A, Miglietta A, Gadoni E, Gabriel L. Cell Biochem Funct. 1990;8:99–105. doi: 10.1002/cbf.290080204. [DOI] [PubMed] [Google Scholar]
- 32.Stewart BJ, Doorn JA, Petersen DR. Chem Res Toxicol. 2007;20:1111–9. doi: 10.1021/tx700106v. [DOI] [PubMed] [Google Scholar]
- 33.Doorn JA, Petersen DR. Chem Res Toxicol. 2002;15:1445–50. doi: 10.1021/tx025590o. [DOI] [PubMed] [Google Scholar]
- 34.Labarca C, Paigen K. Anal Biochem. 1980;102:344–52. doi: 10.1016/0003-2697(80)90165-7. [DOI] [PubMed] [Google Scholar]
- 35.Kannarkat GT, Tuma DJ, Tuma PL. J Hepatol. 2006;44:963–70. doi: 10.1016/j.jhep.2005.07.007. [DOI] [PubMed] [Google Scholar]
- 36.Xu G, Sayre LM. Chem Res Toxicol. 1998;11:247–51. doi: 10.1021/tx980003d. [DOI] [PubMed] [Google Scholar]
- 37.Yuan Q, Zhu X, Sayre LM. Chem Res Toxicol. 2007;20:129–39. doi: 10.1021/tx600270f. [DOI] [PubMed] [Google Scholar]
- 38.Esterbauer H, Zollner H, Lang J. Biochem J. 1985;228:363–73. doi: 10.1042/bj2280363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Benedetti A, Comporti M, Esterbauer H. Biochim Biophys Acta. 1980;620:281–96. doi: 10.1016/0005-2760(80)90209-x. [DOI] [PubMed] [Google Scholar]
- 40.Cadenas E, Muller A, Brigelius R, Esterbauer H, Sies H. Biochem J. 1983;214:479–87. doi: 10.1042/bj2140479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Sampey BP, Carbone DL, Doorn JA, Drechsel DA, Petersen DR. Mol Pharmacol. 2007;71:871–83. doi: 10.1124/mol.106.029686. [DOI] [PubMed] [Google Scholar]
- 42.Baraona E, Leo MA, Borowsky SA, Lieber CS. J Clin Invest. 1977;60:546–54. doi: 10.1172/JCI108806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Adiels M, Olofsson SO, Taskinen MR, Boren J. Arterioscler Thromb Vasc Biol. 2008;28:1225–36. doi: 10.1161/ATVBAHA.107.160192. [DOI] [PubMed] [Google Scholar]
- 44.Venkatesan S, Ward RJ, Peters TJ. Biochim Biophys Acta. 1988;960:61–6. doi: 10.1016/0005-2760(88)90009-4. [DOI] [PubMed] [Google Scholar]
- 45.Cardozo C, Wu X, Pan M, Wang H, Fisher EA. Biochemistry. 2002;41:10105–14. doi: 10.1021/bi025749w. [DOI] [PubMed] [Google Scholar]
- 46.Sayre LM, Lin D, Yuan Q, Zhu X, Tang X. Drug Metab Rev. 2006;38:651–75. doi: 10.1080/03602530600959508. [DOI] [PubMed] [Google Scholar]
- 47.Bloom GS, Goldstein LS. J Cell Biol. 1998;140:1277–80. doi: 10.1083/jcb.140.6.1277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Mirzaei H, Regnier F. Anal Chem. 2005;77:2386–92. doi: 10.1021/ac0484373. [DOI] [PubMed] [Google Scholar]
- 49.Temple A, Yen TY, Gronert S. J Am Soc Mass Spectrom. 2006;17:1172–80. doi: 10.1016/j.jasms.2006.04.030. [DOI] [PubMed] [Google Scholar]
- 50.Roede JR, Carbone DL, Doorn JA, Kirichenko OV, Reigan P, Petersen DR. Chem Res Toxicol. 2008 doi: 10.1021/tx800244u. [DOI] [PubMed] [Google Scholar]
- 51.Davis RA. Biochim Biophys Acta. 1999;1440:1–31. doi: 10.1016/s1388-1981(99)00083-9. [DOI] [PubMed] [Google Scholar]
- 52.Le Marchand Y, Patzelt C, Assimacopoulos-Jeannet F, Loten EG, Jeanrenaud B. J Clin Invest. 1974;53:1512–7. doi: 10.1172/JCI107701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Benoist F, Grand-Perret T. J Biol Chem. 1997;272:20435–42. doi: 10.1074/jbc.272.33.20435. [DOI] [PubMed] [Google Scholar]
- 54.Hui TY, Olivier LM, Kang S, Davis RA. J Lipid Res. 2002;43:785–93. [PubMed] [Google Scholar]