

Abstract
To understand the mechanism behind aberrant Akt activation in T-ALL, PIK3CA, PTEN and SHIP1 expression and genotype were assessed. No cell lines or primary ALLs harbored PIK3CA mutations. PTEN was expressed in just one-third of the cell lines, but in two-thirds of the primary ALLs, though in the inactivated (phosphorylated) form. SHIP1 was undetectable in most primary ALL and in the T-ALL cell line Jurkat, which harbored a bi-allelic null mutation and a frame-shift deletion; primary ALL harbored the frame-shift as well as other translationally-inactivating deletions and insertions. The inactivation of SHIP1 could play a central role in the deregulation of Akt pathway and tumorigenesis, perhaps in conjunction with PTEN-inactivation.
Keywords: SHIP1, PTEN, PIK3CA, Acute Lymphoblastic Leukemia, Alternative Splicing
1. Introduction
Phosphatidylinositol-3,4,5-triphosphate (PI(3,4,5)P3), generated via the phosphorylation of PI(4,5)P2 by phosphatidylinositol-3-kinase (PIK3CA), is a critical component of the Akt signal transduction pathway. PI(3,4,5)P3) activation is balanced by phosphatases such as PTEN, a 3′-phosphatase that drives the hydrolysis of PI(3,4,5)P3 back to PI(4,5)P2, and SHIP1, a 5′ phosphatase that drives the hydrolysis of PI(3,4,5)P3 to PI(3,4)P2. By opposing the effects of PI3KCA activation, PTEN and SHIP1 function as crucial regulators of cell survival. Activating mutations in PIK3CA have been observed in many cancers and have been shown to exert oncogenic effects and promote tumorigenesis. Inactivation of PTEN is also common in many cancers and can also be oncogenic. Less studied is the role of SHIP1. While PIK3CA and PTEN are universally expressed, SHIP1 expression is restricted to cells of hematopoietic lineage, being highly expressed in CD4+ and CD8+ T-cells, and in both immature and mature T lymphocytes. SHIP1 can regulate cell growth, as supported by the finding that the expression of SHIP1 in SHIP1(-) cell lines induced growth suppression [1]. With recent reports of an inactivating SHIP1 mutation in AML [2], it appears SHIP1 may be a bona fide tumor suppressor gene and, coupled with its restricted expression in hematopoietic cells, merits further investigation as to its role in leukemogenesis.
Acute lymphoblastic leukemia (ALL) originates from normal lymphoid progenitor cells arrested at early stages of B- or T-lymphocyte ontogeny. Molecularly, p16/ARF inactivation, mutations of Notch1 and translocations involving the T-cell receptor are common in T-ALL. Akt has also been shown to be an essential component of maturation of thymocytes to T cells and in TCR signaling and activated Akt is common in ALL cell lines and primary tumor samples [3]. In T-ALL cell lines, the proliferative effects of IL-7 are dependent on PIK3CA, suggesting deregulation of this enzyme could be an oncogenic event in T-ALL. Furthermore, the ability of inhibitors of PIK3CA and inhibitors of proteins downstream of Akt to induce apoptosis in T-ALL cells further suggests a role for the Akt pathway in ALL. In this study, we considered the possibility of deregulation of the Akt pathway by assessing the genotype of PIK3CA and SHIP1, and the expression PTEN and SHIP1, in leukemia cell lines and primary T-ALL. We report near universal inactivation of SHIP1 and PTEN with sparing of PIK3CA in T-ALL and suggest deregulation of these phosphatases may be important in disease pathogenesis.
2. Materials and Methods
2.1 Sample Accrual and Preparation
Primary T-ALL samples were obtained from children treated on Children's Oncology Group (COG) ALL Biology protocols #8862, #9673, #9000 or #9400 with mononuclear cells (MNCs) isolated by Ficoll-Hypaque density gradient centrifugation. RNA was extracted using the Invitrogen Trizol® reagent, DNA using the Gentra DNA isolation kit, and protein as a cell lysate in RIPA buffer (50 mM Tris-HCl pH 7.5, 150 mM NaCl, 1 mM EDTA, 1% Triton X-100, 0.5% deoxycholate, 0.1% SDS). Samples are fully encoded to protect patient confidentiality and conform to HIPAA standards and are utilized under a UCSD-approved IRB protocol (#041429).
2.2 PCR amplification, sequencing and Western blot
For cDNA amplifications, 2μg RNA was reverse transcribed using Invitrogen Superscript III First Strand Synthesis System. SHIP1 amplification utilized primers SHIP1-5F (5′-AGGAAGTCAGTCAGTTAAGCTGGT-3′) or SHIP1-2409F (5′-CTCGAGCTGCTTGGAGAGTT-3′) and SHIP1-3863R (5′-CAGAAGCTAGGCCCTTTCCT-3′) in a 50-μl volume containing 1μl cDNA, 2 mM MgSO4, 140 μM dNTPs, 10 pmol each sense and antisense primer, and 3.75U Invitrogen Platinum Taq DNA Polymerase High Fidelity in 1X High Fidelity buffer. For sequence analysis, PCR products were purified on a Qiagen QiaQuick column and sequenced directly at the Moores-UCSD Cancer Center shared sequencing. Some PCR products were subcloned using the Invitrogen TA cloning kit and sequenced. PIK3CA amplification and resolution by dHPLC, SSCP and sequencing are presented as Supplemental Methods. SHIP1 codon numbering is based on variant 2 (GI:64085176). Antibodies used for Western blotting were as follows: PTEN, p-PTEN (Ser380/Thr382/383), Akt and p-Akt (Ser473) from Cell Signaling Technologies, SHIP1 N-terminal (#P1C1) from Santa Cruz Biotechnology, SHIP1 C-terminal (#SHIP-01) from Abcam and β-actin from Sigma-Aldrich.
3. Results
To assess the possibility of mutations to the PIK3CA gene in leukemia, we utilized a combination of SSCP, dHPLC and/or direct sequencing (See Supplemental Figure and Results). Of 81 primary T-ALL, one heterozygous conserved mutation and one heterozygous silent mutation were identified, suggesting PIK3CA mutation is not a player in T-ALL pathogenesis. Inactivation of PTEN and/or SHIP1 could have oncogenic effects analogous to PIK3CA activation. All three B-cell leukemia lines investigated in this study expressed PTEN protein, while only 2 of 6 T-ALL cell lines expressed the protein (Table 1, Fig 1B). SHIP1 transcript expression was detected in all leukemia cell lines examined, as well as in normal thymocytes and MNCs (Fig 1A). Using C- and N-terminal antibodies, SHIP1 was expressed in thymocytes, MNCs and in 5 of the 6 T-ALL cell lines. Despite expression of full-length transcript, SHIP1 was undetectable in the Jurkat T-ALL cell line (Table 1, Fig. 1B). In contrast, the three B-cell leukemia lines examined in this study all expressed high levels of SHIP1 protein, while normal thymocytes expressed an additional band detectable only with the N-terminal antibody (Fig. 1B). To address the possibility of functional mutations in SHIP1 expressing and null mutations in the SHIP1 non-expressing cell lines, we sequenced the entire coding region of the SHIP1 cDNA from all cell lines, identifying a number of known polymorphisms as well as unreported synonymous and non-synonymous alterations (Table 2). Three alterations unique to the Jurkat cell line were identified. At codon 345, a heterozygous alteration leading to a null mutation was detected; at codon 1133, a heterozygous CCG (Pro) → C(C/T)G (Pro/Leu) was present; and at the splice junction of exon 11/12 at codon 413, sequencing of PCR product revealed a double sequence that was determined by subcloning to be a deletion of the first 47 bps of exon 12 that results in a frameshift and premature termination of the allele wild-type at codon 345. Thus both alleles of SHIP1 in Jurkat have inactivating mutations. This finding is particularly notable as while it has been widely reported that the Jurkat cell line does not express SHIP1 protein, the mechanisms have not been previously known. No null mutations, deletions or other unique mutations were observed in SHIP1 cDNA from K562, which also do not express SHIP1 protein, demonstrating that alterative mechanisms of translational inactivation are also in play.
Table 1. Expression status of SHIP and PTEN and mutational status of SHIP1 and PIK3CA in cell lines and normal's.
| cell line | cell type | |----PIK3CA mutations----| | SHIP1 mRNA |
SHIP1 protein |
SHIP1 mutation (unique) |
PTEN protein |
p-PTEN protein |
p-Akt protein |
|
|---|---|---|---|---|---|---|---|---|---|
| PIK3CA exon 9 | PIK3CA exon 20 | ||||||||
| HSB | T-ALL | wt | wt | +ˆ | + | no | + | + | - |
| CEM | T-ALL | wt | wt | -/+ | -/+* | codon 278 | - | - | - |
| Molt4 | T-ALL | wt | wt | + | + | no | - | - | + |
| Molt16 | T-ALL | wt | wt | + | + | no | - | - | + |
| Jurkat | T-ALL | wt | wt | + | - | codon 345, 1133 & ×12 | - | - | + |
| Supt3 | T-ALL | nd | nd | + | + | no | + | + | - |
| SB | B-precursor ALL | wt | wt | + | + | no | + | + | + |
| WI-L2 | B-cell leukemia | nd | nd | + | + | no | + | + | - |
| RPMI 8866 | B-cell leukemia | nd | nd | + | + | no | + | + | - |
| K562 | CML | wt | wt | + | - | no | + | - | - |
| HL60 | AML | wt | wt | -/+ | +* | no | + | + | - |
| Ramos | B-cell leukemia | wt | wt | + | + | no | + | nd | nd |
| MNC | normal PBLs | wt | wt | + | + | no | + | nd | nd |
| Thy | normal thymocytes | wt | wt | + | +* | no | + | + | - |
a second band of lower molecular weight is detected in these samples;
-, -/+, +; undectable, weakly detectable and moderate to high gene or protein expression, respectively;
Fig 1. Expression of SHIP1 transcript and SHIP1 and PTEN protein in leukemia.
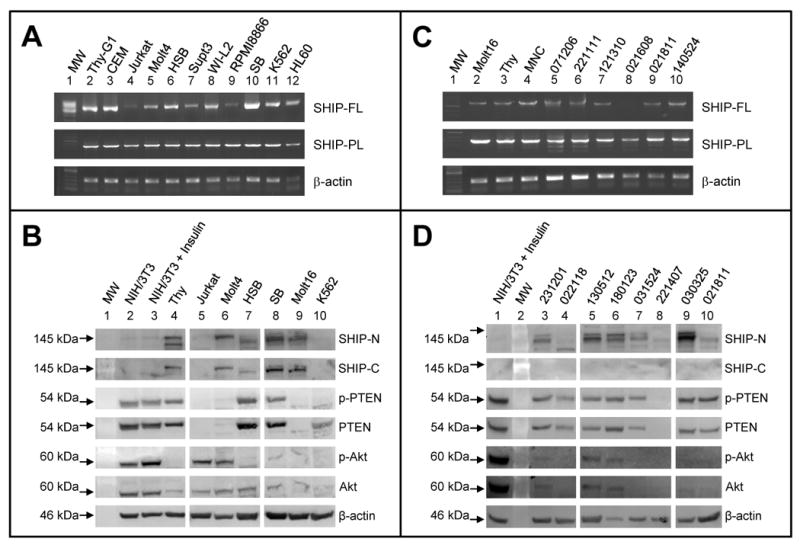
A. A single SHIP1 amplicon is observed from most leukemia cell lines and normal cells when amplifying with either a full-length (FL) or partial-length (PL) primer set.
B. SHIP1 protein was expressed at high levels in B-cell lines (SB, lane 8, Table 1), while T-cell lines have qualitatively lower levels of SHIP1 (lanes 5-7, 9), comparable to normal thymocytes (lane 4); no protein was detected in Jurkat or K562 cells. Also, consistent with its hematopoietic-specific expression profile, SHIP1 was not detected in NIH/3T3 cells. PTEN was phosphorylated in all cell lines where it was expressed except K562, but was undetectable in most T-cell lines. Akt at low levels was detectable in all cell lines, but activated in Jurkat, Molt4, SB, and Molt16. Insulin-treated and -untreated NIH/3T3 cells were used as controls for p-PTEN and p-Akt.
C. Full-length SHIP1 transcript was detectable in normal thymocytes and MNCs (lanes 3 and 4) and in many primary T-ALL (lanes 5-10), with shorter variants of weaker intensity also detectable in some samples (lanes 5 and 6). Some primary T-ALL that didn't amplify with the full-length primer set (i.e. lane 8) were amplifiable with a partial-length primer set, perhaps indicating usage of an alternative N-terminal.
D. SHIP1 protein was undetectable or only barely detectable in primary T-ALL when probed with a C-terminal antibody and undetectable or detected in truncated forms when probed with an N-terminal antibody (lanes 3-10), despite the presence of full-length transcript and its frequent expression in T-ALL cell lines. For comparison, SHIP1 expression in normal thymocytes is shown in Panel B (lane 4). PTEN protein was expressed in most primary T-ALL, unlike T-ALL cell lines, and present in the phosphorylated form. Akt was present in the activated phosphorylated form in ALL, in agreement with literature [3, 4]. MW, molecular weight marker.
Table 2. Mutational and Polymorphisms of SHIP1 in cell lines and normals.
The codon 345 null mutation found in Jurkat is found on the same allele as codon 1133 CCG (Pro) and exon 12 wild-type, while the codon 345 wild-type genotype cosegregated with codon 1133 CTG (Leu) and the exon 12 deletion.
| codon | change | conservative | reported? | cell line (genotype) |
|---|---|---|---|---|
| 162 | ACA (T)-->ACG (T) | yes | no | Molt4 (A/G) |
| 272 | TCA (S) --> TCG (S) | yes | no | SB (A/G), K562 (A), Ramos (G), Molt4 (A/G), Molt16 (G), CEM (A/G), Jurkat (G), Thy (G), MNC (A/G) |
| 278 | GTC (V) --> ATC (I) | yes | no | CEM (G/A) |
| 345 | CAT (Q) --> TAG (stop) | no | no | Jurkat (C/T) |
| 346 | CTC (L) -->CTG (L) | yes | yes | SB (C/G), K562 (C), Ramos (C), Molt4 (C/G), Molt16 (C/G), CEM (C), Jurkat (G), Thy (G), MNC (C/G) |
| 362 | ACA (T) --> ACG (T) | yes | no | SB (A/G), Jurkat (G), Thy (A/G) |
| 413 | 47bp deletion | no | no | Jurkat |
| 1036 | CAG (Q) --> AGG (R) | yes | no | MNC (A/G) |
| 1086 | ACG (T) --> ACA (T) | yes | yes | Ramos (G/A), Jurkat (G/A) |
| 1133 | CCG (P) --> CTG (L) | no | no | Jurkat (C/T) |
| 1168 | CAT (H) -->TAT (Y) | no | yes | Ramos (C/T), Jurkat (C/T) |
With the observation that some leukemia cell lines do not express SHIP1 protein and harbor inactivating as well as possible functional mutations, we sought to assess SHIP1 status in primary T-ALL (Fig 1C). Though not intended to be quantitative in nature, only 62% (38 of 61) of the tumor specimens expressed abundant levels of transcript; 11 samples (18%) expressed low levels of transcript while SHIP1 transcript was undetectable in 12 (20%). To address the possibility that usage of an alternative 5′ transcription site could result in the loss of detectable transcript expression, we also amplified SHIP1 using a sense primer located in the catalytic site. All full-length transcript expressing and many non-expressing T-ALL samples expressed this partial length SHIP1 amplicon, often in addition to amplicons of even smaller size (Fig 1C). To determine if levels of transcript corresponded with SHIP1 protein expression, we performed a Western blot analysis on select T-ALL samples with high, low and undetectable levels of SHIP1 transcript (Fig 1D, Table 1). To our surprise, when probed with a C-terminal antibody, SHIP1 protein was undetectable in 15 of 20 (75%) T-ALL samples analyzed and only barely detectable in the remaining 5 samples, far lower than that observed in normal thymocytes or most T-ALL cell lines and independent of the qualitatively observed levels of SHIP1 transcript. High, low and undetectable SHIP1-transcript expressing ALL samples were all among the samples negative for SHIP1 protein. When probed with an N-terminal antibody, no full-length SHIP1 was detected. Instead, lower molecular weight species of SHIP1 were detected in many ALL samples, though SHIP1 remained undetectable in 3 of 8 (∼38%) of the ALL (Fig 1D). To determine if ALL harbored inactivating sequence alterations of SHIP1, we sequenced, in its entirety, the transcript from 10 SHIP1 transcript-expressing T-ALL samples. There was no evidence of the codon 345 null or codon 1133 missense mutation. However, SHIP1 PCR products from primary T-ALL harbored regions of “dirty” sequence which upon subcloning were identified to include translationally inactivating alterations including the 47 bp deletion in exon 12, a 624 bp insertion of the entire intron 14 between exons 14 and 15, and a partial insertion of intron 10 between exons 10 and 11. Additionally, an in frame exon 8 deletion and clones lacking exon 25, exon 26, and exons 25 and 26, were identified. In a minority of clones full-length SHIP1 was detected (Fig 2).
Fig 2. Graphic representation of the SHIP1 splice alterations.

SHIP1 exons, introns and splice sites were identified by blasting the 5274bp SHIP1 cDNA (GI:64085176) against the chromosome 2 genomic sequence (GI:157724517), revealing that SHIP1 is a 145,886 base gene comprised of 27 exons and 1188 residues. Point mutations, splice deletions and insertions identified in T-ALL are indicated. T-ALL cells predominantly expressed translation-terminating alternatively spliced SHIP1 with wild-type SHIP1 being a minority species, and was reflected in the lack of protein expression. Normal cells predominantly expressed wild-type SHIP1, as reflected by the robust expression of SHIP1 protein, though alternative splicing at exon 25-26 and the exon 12 deletion/intron 14 insertion genotype were each found in one of 11 clones. (AA; Amino Acids)
Inactivation of PTEN and/or SHIP1 should result in Akt activation. In general, PTEN(-) cell lines expressed an activated (phosphorylated) Akt (Fig 1B, Table 1). Primary samples expressed Akt weakly, like normal thymocytes, but in the activated form (Fig 1D). However, many primary ALLs along with the SB cell line also expressed PTEN yet still harbored an activated Akt. Recently, phosphorylation of PTEN has been reported as an alternative mechanism of protein inactivation and Akt activation [3]. An examination of PTEN(+) cell lines and primary T-ALL revealed that the phosphorylated form was present in almost all PTEN-expressing samples. Thus all primary T-ALL harbored inactivating alterations of both PTEN and SHIP1, while leukemia cell lines primarily harbored only alterations of PTEN. We speculate that the presence of PTEN inactivation without Akt activation in normal thymocyte and some cell lines may be due to the presence of an intact SHIP1, which is not found in primary ALL.
4. Discussion
4.1 SHIP1 and PTEN, co-gatekeepers of the Akt signaling cascade in T-cells?
Deregulation of the Akt regulatory pathway is common in many cancers due to activating mutations of PIK3CA or inactivation of the phosphatase PTEN. In T-ALL, PIK3CA mutations were not found, and translational loss of PTEN, which was common in T-ALL cell lines, was infrequent though functional inactivation was common. The inactivation of SHIP1 could potentially function independently or in conjunction with PTEN inactivation in the deregulation of Akt. In general, T- and B- leukemic cell lines expressed full-length SHIP1 protein; the Jurkat T-ALL cell line lacked SHIP1 protein due to bi-allelic alterations. However, in contrast to leukemia cell lines and normal thymocytes, SHIP1 protein was low to undetectable in primary T-ALL when probed with a C-terminal antibody, although lower molecular weight species were frequently detected, but no FL SHIP1, when probed with an N-terminal antibody. Molecularly, primary T-ALL harbored frequent deletions, insertions and splicing variants that would lead to translational inactivation and are likely the source of these lower molecular weight proteins. It is noteworthy that most T-cell lines were predominantly translationally inactivated for PTEN while SHIP1 was intact. In contrast, primary T-ALL samples were generally functionally inactivated for PTEN but lacked full-length SHIP, a caution that ALL cell lines may not accurately mirror the roles PTEN and SHIP1 play in disease pathology.
The presence of SHIP1 protein species lacking a C-terminal have been speculated to be due to alternative splicing, partial deletion and/or protein truncation. Our detection of multiple SHIP1 transcripts from a single sample as well as alterations that include deletions and insertions occurring directly at splice sites indicate alternative splicing as the source of these species. It is notable that in both our sequencing of PCR product from primary T-ALL cells and subcloning of these PCR products, we found multiple sequence variants of SHIP1, which are stable as suggested by detection with an N-terminal antibody. Incredibly, subcloning suggests that full-length SHIP1 may be a minority species in primary T-ALL. The loss of SHIP1 expression has also been observed in both acute and chronic adult T cell leukemia (ATLL) [4]. These investigators further report that PTEN is generally lost in acute but retained in chronic ATLL, though the functional state of PTEN is unknown. Like our results, Akt was generally expressed only weakly, but in the activated state in both acute and chronic ATLL. The observation that SHIP1 knockout mice develop myeloproliferative disease and die early has helped lead to the hypothesis that SHIP1 may be a tumor suppressor gene. The discovery of a rare catalytically inactivating mutation of SHIP1 in a primary AML tumor sample further supports this hypothesis [2]. We hypothesize that inactivating splice alterations may also be frequent in AML, acting as a far greater mechanism of SHIP1 inactivation than point mutation. Taken together with our findings in childhood T-ALL, the data in ATLL and AML strongly support a role for SHIP1 in leukemia.
In addition to, or in lieu of, its role as a regulator of Akt activation, SHIP1 may play an important role in signal transduction. T cell receptor (TCR) activation by CD28 leads to phosphorylation of SHIP1, and SHIP1 transfection into the SHIP1-negative Jurkat cells slows proliferation and lengthens G1 transition [1]. However, SHIP1 knockout mice retain apparent normal thymic populations of CD4+ and CD8+ cells and continue to proliferate in response to TCR/CD28 stimulation. In mice where SHIP1 was specifically knocked out of the T cell population, T cell development, response to CD28 stimulation and Akt phosphorylation were all normal [5]. However, the T cells did show higher levels of cytotoxic activity. This suggests that inactivation of SHIP1 alone is not enough to result in neoplastic transformation and may require the co-inactivation of PTEN, either through translational inactivation or functional inactivation via phosphorylation, a scenario supported by our data in primary ALL versus cell lines, to tip the regulatory controls of a normal T cell into a leukemic one. In this regard, recent reports have suggested the role of PTEN as a “sentinel”, regulating basal/low receptor-stimulated levels of PI(3,4,5)P3, while SHIP1 acts as a “gatekeeper”, able to redirect signaling rather than stop it [6]. In T-ALL, the gatekeeper is missing, making the cells highly susceptible to neoplastic transformation after PTEN inactivation.
4.2 The significance of SHIP1 in the Notch1 and Ras signaling cascades
This is the first study to document that alternative splicing and translational inactivation of SHIP1 are widespread in T-ALL. We think it is unlikely to be inconsequential that such a high percentage of these cells harbor aberrant forms of the gene and lack detectable full-length protein expression. Given the recent emphasis on the PIK3CA/PTEN/Akt regulatory pathway in cancer, our data that SHIP1 is inactivated in primary T-ALL suggest it may be a largely overlooked player in disease pathobiology. Notch1 mutations are common and oncogenic in T-ALL and may contribute to more than half the cases of the disease. However, Notch1 inhibitors have shown only limited efficacy as single agents against cell lines harboring Notch1 mutations. Mutations and overexpression of Ras are common in many cancers, and in T-ALL we have shown that almost half harbor aberrantly activated Ras [7]. Oncogenic transformation by Ras and Notch1 appear to be cooperative. The tumors of Ras mutant-expressing mice frequently harbor Notch1 mutations [8], and transgenic mice with weakly oncogenic Notch1 mutations became strongly oncogenic when complemented with an activated Ras [9]. Notch1 also downregulates PTEN, with inhibition of Notch1 resulting in increased PTEN levels, an inhibition of Akt activation and cell cycle arrest, an ability lost in PTEN(-) tumor cells [10]. Ras can also downregulate PTEN [11], with activated Ras in T-ALL possibly acting against Notch1 inhibition in the suppression of T-ALL cell growth. In B-cells and possibly T-cells, SHIP1 is a known regulator of Ras. The lack of SHIP1 in T-ALL may thus result in increased availability of Ras and decreased levels of PTEN, thus exacerbating the influence of Notch1 mutations on T-ALL cell growth. The fact that SHIP1 can be connected to both the Notch and Ras pathways suggests a multi-targeted therapeutic approach directed at these two pathways may be warranted in T-ALL.
Supplementary Material
Acknowledgments
This work was supported in part by the San Diego Padres Cindy Matters Foundation (MBD), the Children's Oncology Group (ALY, MBD), the FDA (ALY) and the General Clinical Research Center program of the National Center for Research Resources, NIH. DNA sequencing was performed by the DNA Sequencing Shared Resource, UCSD Cancer Center, which is funded in part by a NCI Cancer Center Support Grant. YK was the recipient of an Undergraduate Scholastic Grant from the Associated Students of UCSD.
Footnotes
Conflict of Interest: The authors declare no conflict of interest.
Contributions: TCTL and LMB performed the SHIP1 cloning, sequencing and contributed to the manuscript writing; YK and EAN peformed the analyses on PIK3CA; ALY funded the research, contributed to project direction and manuscript writing, MBD funded the research, oversaw the entire project and wrote the manuscript.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Horn S, Endl E, Fehse B, Weck MM, Mayr GW, Jücker M. Restoration of SHIP activity in a human leukemia cell line downregulates constitutively activated phosphatidylinositol 3-kinase/Akt/GSK-3beta signaling and leads to an increased transit time through the G1 phase of the cell cycle. Leukemia. 2004;18:1839–1849. doi: 10.1038/sj.leu.2403529. [DOI] [PubMed] [Google Scholar]
- 2.Luo JM, Yoshida H, Komura S, Ohishi N, Pan L, Shigeno K, Hanamura I, Miura K, Iida S, Ueda R, Naoe T, Akao Y, Ohno R, Ohnishi K. Possible dominant-negative mutation of the SHIP gene in acute myeloid leukemia. Leukemia. 2003;17:1–8. doi: 10.1038/sj.leu.2402725. [DOI] [PubMed] [Google Scholar]
- 3.Silva A, Yunes JA, Cardoso BA, Martins LR, Jotta PY, Abecasis M, Nowill AE, Leslie NR, Cardoso AA, Barata JT. PTEN posttranslational inactivation and hyperactivation of the PI3K/Akt pathway sustain primary T cell leukemia viability. J Clin Invest. 2008 Nov 3;118(11):3762–3774. doi: 10.1172/JCI34616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Fukuda R, Hayashi A, Utsunomiya A, Nukada Y, Fukui R, Itoh K, Tezuka K, Ohashi K, Mizuno K, Sakamoto M, Hamanoue M, Tsuji T. Alteration of phosphatidylinositol 3-kinase cascade in the multilobulated nuclear formation of adult T cell leukemia/lymphoma (ATLL) Proc Natl Acad Sci USA. 2005;102:15213–152184. doi: 10.1073/pnas.0507184102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Tarasenko T, Kole HK, Chi AW, Mentink-Kane MM, Wynn TA, Bolland S. T cell-specific deletion of the inositol phosphatase SHIP reveals its role in regulating Th1/Th2 and cytotoxic responses. Proc Natl Acad Sci USA. 2007;104:11382–11387. doi: 10.1073/pnas.0704853104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Harris SJ, Parry RV, Westwick J, Ward SG. Phosphoinositide lipid phosphatases: natural regulators of phosphoinositide 3-kinase signaling in T lymphocytes. J Biol Chem. 2008;283:2465–2469. doi: 10.1074/jbc.R700044200. [DOI] [PubMed] [Google Scholar]
- 7.von Lintig FC, Huvar I, Law P, Diccianni MB, Yu AL, Boss GR. Ras activation in normal white blood cells and childhood acute lymphoblastic leukemia. Clin Cancer Res. 2000;6:1804–1810. [PubMed] [Google Scholar]
- 8.Kindler T, Cornejo MG, Scholl C, Liu J, Leeman DS, Haydu JE, Fröhling S, Lee BH, Gilliland DG. K-RasG12D-induced T-cell lymphoblastic lymphoma/leukemias harbor Notch1 mutations and are sensitive to gamma-secretase inhibitors. Blood. 2008;112:3373–3382. doi: 10.1182/blood-2008-03-147587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Chiang MY, Xu L, Shestova O, Histen G, L'heureux S, Romany C, Childs ME, Gimotty PA, Aster JC, Pear WS. Leukemia-associated NOTCH1 alleles are weak tumor initiators but accelerate K-ras-initiated leukemia. Clin Invest. 2008;118:3181–3194. doi: 10.1172/JCI35090. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Palomero T, Sulis ML, Cortina M, Real PJ, Barnes K, Ciofani M, Caparros E, Buteau J, Brown K, Perkins SL, Bhagat G, Agarwal AM, Basso G, Castillo M, Nagase S, Cordon-Cardo C, Parsons R, Zúñiga-Pflücker JC, Dominguez M, Ferrando AA. Mutational loss of PTEN induces resistance to NOTCH1 inhibition in T-cell leukemia. Nat Med. 2007;13:1203–1210. doi: 10.1038/nm1636. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Bahk YY, Cho IH, Kim TS. A Cross-talk between oncogenic Ras and tumor suppressor PTEN through FAK Tyr(861) phosphorylation in NIH/3T3 mouse embryonic fibroblasts. Biochem Biophys Res Commun. 2008;377:1199–1204. doi: 10.1016/j.bbrc.2008.10.157. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.