

Abstract
Reprogramming somatic cells to induced pluripotent stem (iPS) cells has been accomplished by expressing pluripotency factors and oncogenes1–8, but the low frequency and tendency to induce malignant transformation9 compromise the clinical utility of this powerful approach. We address both issues by investigating the mechanisms limiting reprogramming efficiency in somatic cells. We show that reprogramming factors can activate the p53 pathway. Reducing signaling to p53 by expressing a mutated version of one of its negative regulators, by deleting or silencing p53 or its target gene, p21, or by antagonizing apoptosis enhanced three factor (Oct4/Sox2/Klf4)-mediated reprogramming of mouse fibroblasts. Notably, decreasing p53 protein levels enabled fibroblasts to give rise to iPS cells capable of generating germline transmitting chimeric mice using only Oct4 and Sox2. Furthermore, silencing of p53 significantly increased the reprogramming efficiency of human somatic cells. These results provide insights into reprogramming mechanisms and suggest new routes to more efficient reprogramming while minimizing the use of oncogenes.
The p53 pathway reduces cancer initiation by inducing apoptosis or cell cycle arrest in response to a variety of stress signals, including over-expressed oncogenes such as c-Myc. Klf4 can either activate or antagonize p53, depending on the cell type used and expression level10. Consequently, reprogramming efficiency is likely reduced through oncogene-mediated activation of the p53 pathway. This is consistent with prior results showing that germ cells can be spontaneously reprogrammed in the absence of p5311 and a combination of p53 siRNA and UTF1 expression increased iPS cell formation.12
We first determined whether the reprogramming factors, individually or in combination, activate the p53 pathway in mouse embryo fibroblasts (MEFs). Relative to the GFP-retroviral control, c-Myc significantly increased p53 abundance and activity, manifested by increased expression of the cyclin-dependent kinase inhibitor p21 (Fig. 1a). This was achieved by induction of Arf, an antagonist of Mdm2, the E3-ubiquitin ligase responsible for p53 degradation13. Elevated p21 level was also observed in MEFs infected with Klf4, Oct4/Sox2 (2F) or Oct4/Sox2/Klf4 (3F) (Fig. 1a). Since introducing reprogramming factors increased γ-H2AX foci (Supplementary Fig. 1), we infer that the expression of reprogramming factors may induce p53 via DNA damage. We also compared p53 and p21 expression in a variety of mouse and human cell lines previously utilized for iPS cell production (Supplementary Fig. 2). Interestingly, keratinocytes, which have higher reprogramming efficiency, display lower p53 and p21 protein levels than other cell types. Moreover, p21 induction in keratinocytes is lower than in fibroblasts following infection with 3F (Supplementary Fig. 3). Together, these data imply that the p53 pathway is one determinant of reprogramming efficiency.
Fig. 1. Increased generation of iPS cells by blocking p53 and p21.
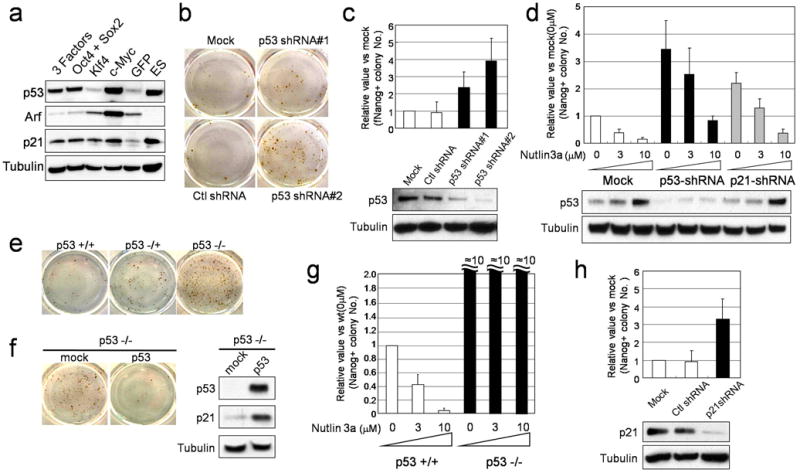
(a) MEFs were infected by retroviruses encoding 3 factors (Oct4/Sox2/Klf4), 2 factors (Oct4/Sox2), Klf4, c-Myc or GFP. Four days after infection, the protein levels of p53, Arf, and p21 were analyzed by Western Blotting. α-Tubulin was used as a loading control. (b) MEFs were infected by 3F (Oct4/Sox2/Klf4) in combination with mock, control shRNA (GFP) and p53 shRNA (#1 and #2). Emerging colonies of iPS cells were visualized by immunostaining with anti-Nanog antibody using an Avidin Biotin Complex (ABC) method. (c) The fold change in the number of Nanog-positive colonies compared to mock (n=4). For all figures in this study, error bars indicate s.d. p53 knockdown efficiency was examined by western blot. (d) MEFs were infected by 3F in combination with mock, p53 shRNA and p21 shRNA. Four days later, half of cells were treated with Nutlin 3a (0, 3, 10μM) and analyzed for p53 level. The remainder were stained for Nanog-positive colonies. (e) Immunostaining of Nanog positive colonies generated from p53(+/+), p53(+/−) and p53(−/−) MEFs by 3F showed p53 dose-dependent decrease of colony number. (f) Retroviral infection of p53 into p53(−/−) MEF decreased the number of Nanog-positive colonies induced by 3F. p53 and p21 levels on day 3 after infection were analyzed. (g) Nutlin 3a dramatically reduced reprogramming of p53(+/+) MEFs, but not on p53(−/−) MEF. (h) Fold change in the number of Nanog-positive colonies by p21 shRNA (n=4). p21 knockdown efficiency was examined by western blot.
We, therefore, tested the effects of reducing p53 signaling by determining reprogramming efficiencies in cells in which p53 function was reduced by shRNA or ablated by homologous recombination. Most cells were infected with shRNA (Supplementary Fig. 4), p53 mRNA and protein levels were reduced by 60–80% (Fig. 1c and Supplementary Fig. 5), iPS colony formation was increased by 2–4-fold using two different shRNAs (Fig. 1b, c). This likely under-estimates p53 suppressive capacity, since functional p53 protein clearly remained present, as indicated by the ability of the p53 activating agent Nutlin 3a14 to dose-dependently reduce iPS formation in MEFs treated with the most effective p53 shRNA (Fig. 1d). By contrast, reprogramming effiency was increased by at least 10-fold in p53-null MEFs, and this was not reduced by Nutlin 3a (Supplementary Table 1; Fig. 1e, g). p53+/− heterozygous MEFs also exhibited higher 3F reprogramming efficiency than wild-type MEFs. While culture stress can induce cellular senescence and activate p53, which would reduce reprogramming, fewer than 1% of the cells of all p53 genotypes stained with the senescence marker β-galactosidase (Supplementary Fig. 6). Since we did not detect loss of heterozygosity of the p53 gene in iPS colonies derived from p53+/− MEFs (Supplementary Fig. 7), the data suggest a p53 dosage-sensitivity to reprogramming (Fig. 1e). We were concerned that since p53 null MEFs are genetically unstable, increased reprogramming efficiency might result from expression of the 3F in variant cells. However, we found that re-expressing p53 in the p53-null MEFs dramatically reduced reprogramming efficiency (Fig. 1f).
Reducing factors downstream of p53 also increased reprogramming efficiency. For example, p21 shRNA increased reprogramming by approximately 3-fold (Fig. 1h). This likely underestimates the magnitude to which p21 induction suppresses reprogramming as p21 shRNA expressing cells still responded to Nutlin 3a treatment as discussed above. We also noted a modest induction of the pro-apoptotic factor BAX, another p53-inducible gene15 in 3F experiments (data not shown). Consistent with a limiting role of the p53 induced apoptotic response during reprogramming, overexpression of the BAX antagonist Bcl-2 suppressed apoptosis in 2F, 3F and 4F experiments and increased the frequency of colonies expressing the pluripotency factor Nanog by 4-fold (Supplementary Fig. 8). The 3F transduced colonies exhibit the characteristics of iPS cells, with mES cell morphology, high levels of alkaline phosphatase activity and expression of pluripotency-associated transcription factors and surface markers, and ability to differentiate into derivatives of all three embryonic germ layers in vitro (Supplementary Fig. 9–11). Taken together with the genetic and shRNA studies described above, these data show that complete loss of p53 function dramatically increases reprogramming efficiency, and that even low levels of p53 activity are all that is needed to compromise somatic cell reprogramming.
The ability of the 3F to increase p53 abundance suggests that controlling its stability might be crucial for p53-mediated reprogramming suppression. Thus we determined whether reducing Arf levels using Arf shRNA increases reprogramming efficiency, as lower Arf levels should decrease p53 stability16–18. Reducing Arf levels by 2–4 fold (Fig. 2a) engendered an approximately 2-fold increase in 3F reprogramming (Fig. 2a). Reducing Arf and p16Ink4a together increased iPS cell formation even more than Arf alone (4–5 fold, Fig. 2b), indicating that compromising retinoblastoma (Rb) tumor suppressor19 function by antagonizing p16Ink4a can collaborate with diminished p53 activity to improve reprogramming efficiency.
Fig. 2. Modulation of p53 activity alters reprogramming efficiency.
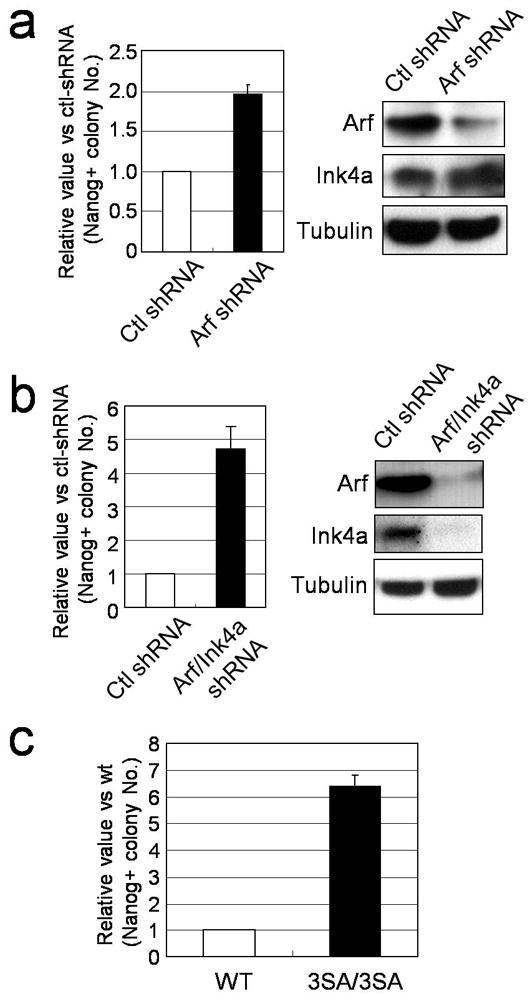
(a) and (b) Fold change in the number of 3F induced Nanog-positive colonies by Arf shRNA or by Arf/Ink4a shRNA compared to control shRNA (n=3). Protein knockdown efficiency was examined by western blot. (c) 3F induced Nanog-positive colonies from wild type (+/+) and homozygous (3SA/3SA) MEFs (n=3).
We next genetically modulated the activity of the E3 ligase that regulates p53 stability by generating a mouse encoding a mutant version of Mdmx, a critical negative regulator of p53. We substituted three serines with alanines (Mdmx S341, S367, S402, hereafter called Mdmx3SA) to prevent its phosphorylation and degradation in response to DNA damage or activated oncogenes20, 21. Importantly, MEFs or thymocytes derived from homozygous Mdmx3SA mice exhibited lower basal expression of p21, lower DNA damage induced p21 levels (Supplementary Fig. 12, and ref 21) and an impaired ability of c-Myc to activate p53 in vivo21. 3F reprogramming increased ~7 fold in Mdmx3SA MEFs, in agreement with their reduced sensitivity to activated oncogenes and DNA damage signaling (Fig. 2c).
Since Klf4 has been reported to have oncogenic properties when overexpressed22, and we showed that it alone can activate p53, we investigated whether cells with reduced p53 expression could be converted into iPS cells using only two factors, Oct4, and Sox2. We tested this hypothesis by transducing MEFs with a lentivirus expressing p53 shRNA plus retroviruses encoding Oct4 and Sox2 (hereafter designated as 2F-p53KD-iPS cells; Supplementary Fig. 13). Cells that developed into colonies exhibiting ES cell-like morphology were obtained by week four post-infection. Of six colonies selected for analysis, four grew using standard mouse ES cell culturing methods (Fig. 3a), and all of them were alkaline phosphatase positive (Fig. 3b) and expressed genes and cell surface markers characteristic of mouse ES cells including the pluripotency marker Nanog (Fig. 3c; Supplementary Fig. 14). 2F-p53KD-iPS cells and mouse ES cell lines exhibited indistinguishable gene expression patterns when maintained under similar conditions. Bisulfite sequencing of the Oct4 and Nanog promoters revealed nearly complete demethylation in 2F-p53KD-iPS cells when compared to MEFs (Supplementary Fig. 14). Consistent with this, we observed expression of the pluripotency-associated transcription factors Oct4 and Sox2 from the endogenous loci in 2F-p53KD-iPS cells, at levels that were comparable to those of ES cells (Supplementary Fig. 15). Also, like ES cells, the majority (70–80%) of cells were in S-phase (Supplementary Fig. 16). We tested the pluripotency of three 2F-p53KD-iPS clones in assays of embryoid body formation in vitro and/or teratoma induction in vivo.. The tested cell lines differentiated into the three germ layer derivatives, as shown by immunostaining and mRNA expression in vitro (Fig. 3d; Supplementary Fig. 17). Furthermore, these cells differentiated with high efficiency into beating cardiomyocytes (Supplementary Fig. 18 and movies 1–3). Upon injection into immunocompromised mice, two independent 2F-p53KD-iPS lines generated complex intratesticular and subcutaneous teratomas containing structures and tissues representative of the three embryonic germ layers (Fig. 3e). Microarray analyses demonstrate that gene expression patterns of these clones are similar to mouse ES cells (Supplementary Fig. 19). We also tested whether 2F-p53KD-iPS cells contribute to the formation of chimeric mice when injected into mouse blastocysts. One line (clone#6) contributed almost 100% to chimera formation, and the other line (clone#1) contributed 30–50%, as judged by coat color (Fig. 3f; Supplementary Fig. 20). We finally used the highest contribution chimera to test for germline competence by crossing it with wild-type C57BL6 mice. Importantly, the offspring of such crosses included agouti pups (Fig. 3g), indicating germline transmission of the 2F-iPS genome. Taken together, these results demonstrate that MEFs can be reprogrammed to pluripotency by the forced expression of only two factors, Oct4 and Sox2, when p53 levels are reduced.
Fig. 3. Generation and characterization of 2F-p53KD-iPS cells by p53 downregulation.
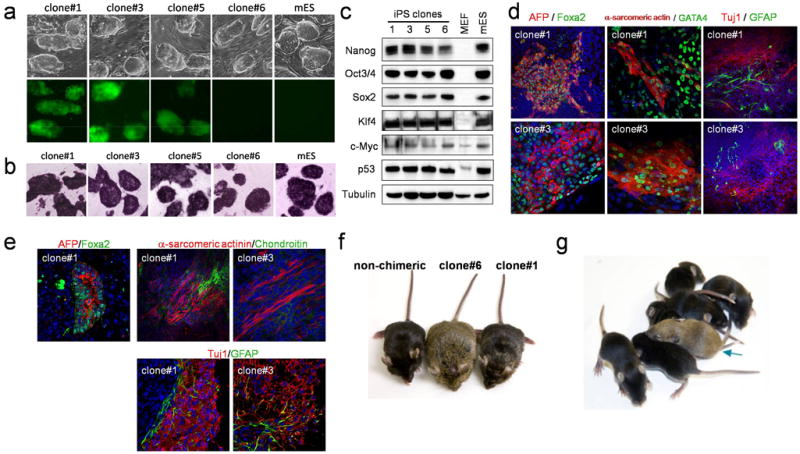
(a) Morphology and GFP fluorescence of 2F-p53KD-iPS cell lines. GFP expression is silenced in clone #6. (b) Alkaline phosphatase staining of 2F-p53KD-iPS cell lines. DAPI was used to visualize cell nuclei. (c) Protein levels of Nanog, Oct4, Sox2, Klf4, c-Myc, p53 in 2F-p53KD-iPS cell lines are shown. α-Tubulin was used as loading control. (d) Embryoid bodies (EBs) of 2F-p53KD-iPS cell clones on day 6 of differentiation. EBs were transferred to gelatinized dishes on day 3 to 5 for further differentiation. On day 14, EBs were subjected to immunofluorescence for α-fetoprotein (AFP)/Foxa2 (endoderm), α-sarcomeric actin/GATA4 (mesoderm) and Tuj1/GFAP (ectoderm). (e) Immunofluorescence of teratoma from 2F-p53KD-iPS cells by antibodies against AFP/Foxa2 (endoderm), α-sarcomeric actinin/Chondroitin (mesoderm), Tuj1/GFAP (ectoderm) showed spontaneous differentiation into all three germ layers. (f) Adult chimeric mice obtained from 2F-p53KD iPS lines (#1 and #6) and non-chimeric mouse in C57BL/6J host blastocysts. (g) As of the date of submission, the mating of offspring from clone #6 chimera to a C57BL/6J female generated 1 agouti pup (blue arrow), that together with PCR analysis (not shown) indicate germ line transmission of the 2F-iPS genome.
We next tested whether downregulating p53 activity had any effect on the reprogramming of human embryonic fibroblasts (HEFs) and juvenile epidermal keratinocytes. We could not obtain Nanog-positive colonies from HEFs with either 3F or 4F combined with control shRNA under our reprogramming conditions after up to 4 weeks. In contrast, cell reprogramming occurred rapidly (after 2 weeks) with high efficiency when p53 expression was reduced by shRNA in the system (Figure 4a and Supplementary Fig. 22, 23 and Table 2). p53 shRNA-induced ES-like colonies expressed human ES marker genes, could be successfully cloned and expanded, exhibited ES-like morphology, and differentiated in vitro in embryoid body (EB) formation assays (Supplementary Fig. 24, 25). We also obtained 2F induced iPS colonies from HEFs infected with p53 shRNA, although with lower efficiency (2 iPS colonies resulted from 6 independent attempts, Supplementary Fig. 26). Consistently, reprogramming efficiency was increased using either 3F or 4F in human primary keratinocytes when p53 activity was downregulated using a dominant negative mutant of p53 (p53-DD)23 (Fig. 4b, c) that inhibited p53 activity more effectively than p53 shRNA as Nutlin 3a did not reduce reprogramming of 3F or 4F p53-DD expressing iPS cells (Fig. 4c). 3F-p53DD-iPS grew robustly and strongly expressed pluripotency-associated transcription factors and surface markers (Fig. 4d and data not shown). Furthermore, 3F-p53DD-iPS cells readily differentiated in vitro into derivatives of the 3 embryonic germ layers as judged by cell morphology and specific immunostaining (Fig. 4e). These results show that p53 activity limits reprogramming of both mouse and human cells.
Fig. 4. Downregulation of p53 activity increases reprogramming efficiency of human somatic cells.
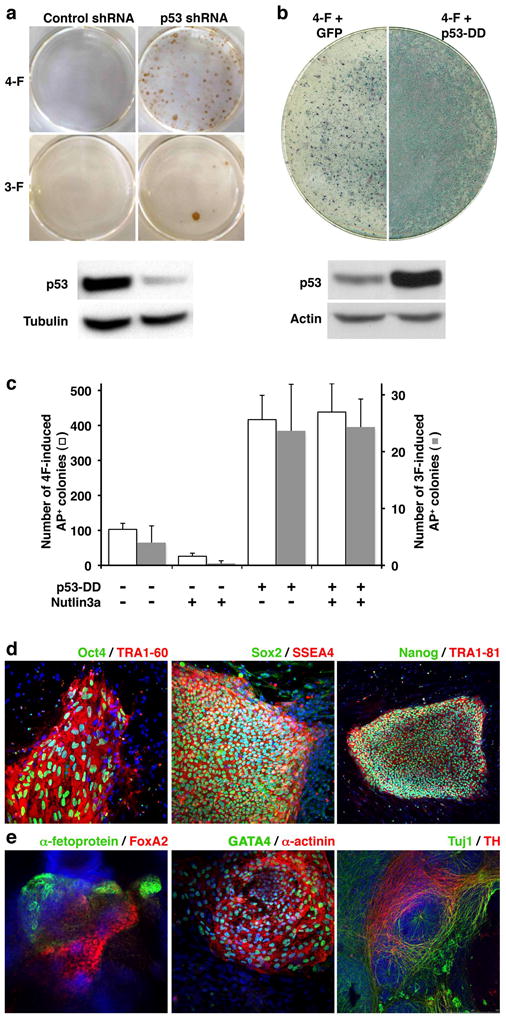
(a) Human embryonic fibroblasts were infected with retroviruses encoding Oct4/Sox2/Klf4 (3-F) or Oct4/Sox2/Klf4/c-Myc (4-F) factors in combination with lentiviruses expressing control-or p53-shRNA. Emerging colonies of iPS cells were immunostained with anti-Nanog antibody. p53 knockdown efficiency was examined by western blot. (b) Human primary keratinocytes were co-infected with 4-F and retroviruses expressing GFP or p53-DD. Two weeks later, cells were stained for AP activity. Expression of p53-DD resulted in stabilization of wild-type p53 (lower panels). Actin was used as a loading control. (c) The bars represent the average number of iPS-like colonies obtained from 104 keratinocytes reprogrammed with 3F or 4F and retroviruses encoding GFP or p53-DD, in the absence or presence of Nutlin3a (n=3). iPS-like colonies were scored as having hES-like morphology and positive AP staining. Due to the numerous colonies generated in 4F p53-DD keratinocytes, quantification was done using 104 cells. (d, e) Colonies of human keratinocyte-derived iPS cells generated by 3F and p53-DD display strong immunoreactivity for pluripotency-associated transcription factors and surface markers (d) and differentiate in vitro into cell types that express markers of endoderm (α-fetoprotein, FoxA2), mesoderm (GATA4, sarcomeric α-actinin), and ectoderm (Tuj1, TH) (e).
Oct4, Sox2 and Nanog interact with each other to enable the genome-wide chromatin remodeling required for induction of pluripotency. None of these factors are expressed at detectable levels in somatic cells. Previous work showed that p53 represses Nanog in response to DNA damage in ES cells24, raising the possibility that p53 might prevent Nanog expression in MEFs. However, we observed that Nanog mRNA was not expressed at detectable levels in either p53 wild type or p53-null MEFs (Supplementary Fig. 11a and data not shown). On the other hand, the oncogene Klf4 has been reported to induce Nanog25. It is possible that in the absence of Klf4 in 2F iPS, p53 elimination allowed Oct4 and Sox2 to remodel the chromatin to a threshold required for expression of sufficient Nanog to drive the subsequent events involved in iPS cell generation.
Our data show that reprogramming somatic cells to iPS cells is associated with activation of the p53 pathway, which serves as a barrier to reprogramming. The mechanisms by which p53 antagonizes reprogramming appear to involve both its ability to limit cell cycling through induction of the cyclin dependent kinase inhibitor p21 and its ability to induce apoptosis. This suggests that direct chemical inhibition of the apoptotic cascade may provide a useful tool for enhancing reprogramming efficiency without direct genetic manipulation of tumor suppressors. We also show that reprogramming in the absence of oncogenes such as c-Myc and Klf4 will require inactivating the p53 and Rb tumor suppressors. While p53 pathway inactivation will be key, this cannot be done on a permanent basis as this would increase the probability of malignant transformation and the generation of unstable genomes that would mitigate their use for understanding many diseases. Rather, transient inhibition using chemical antagonistsor reversible approaches that avoid genetic disruption will be required12,26. As Oct4 and Sox2 are oncogenic when overexpressed, using small molecules or proteins to transiently mimic their reprogramming functions27–29 may enable oncogene-free iPS cells to be obtained at acceptable frequencies. The mechanistic insights we provide (Supplementary Fig. 27) should shorten the road to developing clinically useful iPS cells.
Methods Summary
MEFs were isolated from ED13.5 embryos obtained from wild-type, p53 deficient, or Mdmx mutant mice. Retroviral and lentiviral viruses were produced in HEK293 cells, and 12 to 14 days after infection, MEFs were fixed for immunofluorescence. Reprogramming of human embryonic fibroblasts (IMR90) and keratinocytes was done as previously described2,3,6. Around 2 to 3 weeks after infection, cells were fixed for immunofluorescence studies. As for 2-F iPS cell formation, after a minimum of 4 weeks, colonies derived from infected MEFs or IMR90 cells were fixed for immunostaining or picked up for clonal expansion. Protein, mRNA, promoter methylation, microarray and immunofluorescence analysis were performed by standard methods30. In vitro differentiation of mouse iPS cells was established by the hanging drop method, while human iPS cells were differentiated by suspension culture. Teratomas were induced by injecting iPS cells subcutaneously and into the testicles, and analyzed three to four weeks post-injection chimeric mice, Chimeric mice were obtained by injecting iPS cells into C57BL/6J hosts. High-contribution chimeras were mated to C57BL/6J mice to test for germline transmission.
Methods
Reagents
Reagents were obtained from the following sources: Nutlin3a (Cayman Chemical); anti-Oct-3/4 (sc-5279), anti-GKLF (sc-20691), anti-p53 (sc-6243), anti-p21 (sc-53870), anti-p16Ink4a (sc-1207), anti-c-Myc (sc-764) and anti GATA4 (sc-9053) (Santa Cruz Biotechnology); anti-Sox2 (AB5603) (CHEMICON); anti-p53 antibody (1C12), anti-phospho-Histone H2A.X (Ser139) antibody (20E3) (Cell Signaling); anti-Arf (ab80) and anti-Nanog (ab21603) (Abcam); anti-Nanog (SC1000) and anti-p53 (DO-1) (Calbiochem); anti-Tuj1 antibody (MMS-435P-0) (Covance); anti-α–Tubulin (T5168), anti-α-actinin sarcomeric (A7811), anti-α-actin sarcomeric (A2172), anti-Actin (A2066) and anti-chondroitin (C8035) (SIGMA); anti-Foxa2 antibody (AF2400) (R&D systems); anti-alpha-1-fetoprotein (A008) and anti-GFAP (Z0334) (Dako); Anti-TRA-1-81 antibody (Millipore).
Mice
Wild-type MEFs used for iPS cell production were derived from embryos obtained by mating BDF1/ICR and ICR strains. p53 KO mice were purchased from Taconic Farms, Inc. p53−/−MEFs were obtained by heterozygous vs heterozygous mating. For genotyping, PCR primers are available on the company website. Mdmx mutant mice were generated from ES cells of 129Sv origin by homologous recombination21.
Plasmids
Mouse p53 and GFP cDNAs were cloned into pMXs retroviral vectors30. The cDNA of mouse Bcl-2 was cloned into HIV pBOBI lentiviral vector31. Human p53-DD (a kind gift from Oren, M.) is in pLXSN (Clonetech). The cDNAs of mouse p53 and p21, pMXs-Oct4, -Sox2, -Klf4 and -c-Myc were purchased from Addgene1,32,33. Human pMSCV-Oct4, -Sox2, -Klf4 and -c-Myc were constructed as previously described6. The short-hairpin RNA (shRNA) sequences against p53, p21, Arf and Ink4a were inserted into pLVTHM lentiviral vectors36. Sequences for shRNA are shown in Supplementary Table 3.
Production of retroviruses and lentiviruses and iPS cell formation
VSV-G viruses were produced in HEK293T cells. For pMX-based and pMSCV-based retroviruses, vectors were transfected using CaPO4 or lipofectamin, following the manufacturers’ directions. One day after transfection, culture medium was changed to new medium. For lentivirus, pBOBI-based32 or pLVTHM-based34 vectors were transfected by Lipofectamine 2000 (Invitrogen) according to the manufacturer’s protocol. Six hours after transfection, the DNA-lipofectamine-complex was removed and the medium was replaced the next day. Two days after transfection, the supernatant containing viruses was collected and filtered through a 0.45μm filter. Mouse iPS cells were induced as previously described35,36. Briefly, mouse embryonic fibroblasts (MEF, passage 3 to 5) were infected (day 0) with pMX-based retroviruses together with pLVTHM-based lentivirus for shRNAs or pBOBI-based lentivirus for Bcl-2. On day 2, cells were passed onto new gelatin-coated plates. Medium was changed every 2 days. On day 12 to 14, cells were fixed for immunofluorescence study. For the Nutlin3a experiments, cells were treated starting from day 4. Reprogramming of human embryonic fibroblasts (IMR90) was done as previously described2,3. Briefly, IMR90 fibroblasts (passage 7 to 9) were infected (day 0) with pMSCV-based retroviruses + pLVTHM-based lentiviruses for p53 shRNA. On day 4 or 5, cells were passed onto feeder MEFs. Medium was changed every other day. Around 3 weeks after infection, cells were fixed for immunofluorescence studies. Reprogramming of human primary keratinocytes was carried out essentially as described6. Cells were co-infected with retroviral supernatants containing 3 or 4 reprogramming factors and p53-DD or GFP at a 1:2 ratio. To assess the reprogramming efficiency, cells were trypsinized 3 days after retroviral infection and 104 cells were plated onto 6-cm tissue culture dishes on top of irradiated human foreskin fibroblasts with hES cell medium. After 2 weeks, the dishes were stained for alkaline phosphatase activity and colonies that displayed strong staining and showed hES-like morphology were scored positive. As for 2-F iPS cell formation, MEFs or IMR90 cells were co-infected with retroviral supernatants containing two factors (Oct4 and Sox2) and lentivirus supernatants (p53sh RNA) at a 1:1:3 ratio. Infected cells were passed onto new gelatin-coated plates at day2 (MEFs) or onto feeder MEFs at day 5 (IMR90). Medium was changed every other day, and after a minimum of 4 weeks, colonies were fixed for immunostaining or picked up for clonal expansion.
Protein and mRNA analysis
Cells were washed once in PBS and lysed in 2xSDS-PAGE sample buffer without 2-mercaptoethanol and glycerol. Lysates were briefly sonicated and cleared by centrifugation. The protein concentration was determined by a BCA Protein Assay Kit (Thermo Scientific). Lysates were then mixed with 2-mercaptoethanol, BPB and glycerol, and boiled. Equal amounts of proteins were subjected to SDS-PAGE. Total RNA was isolated using Trizol (Invitrogen) followed by cDNA synthesis using Superscript II Reverse Transcriptase (Invitrogen). Quantitative PCR was performed using SYBR GREEN PCR Master Mix (Applied Biosystems). Primer sequences are available upon request.
Promoter methylation analysis
Genomic DNA was isolated and bisulfite modification performed using the EZ DNA Methylation-Direct Kit (ZYMO RESEARCH). The promoter regions of Nanog and Oct4 were amplified by nested PCR using primer sets previously described37. The amplified PCR products were ligated into pCRII-TOPO (Invitrogen) and sequenced. Data was analyzed using Lasergene (DNASTAR).
In vitro and in vivo differentiation
For in vitro differentiation of mouse iPS cells, after dissociation with trypsin/EDTA, cells were cultured in suspension by the hanging drop method. For in vivo differentiation, cells were trypsinized, and injected subcutaneously into SCID mice. After 3 weeks, teratomas were dissected, fixed, and analyzed. Detailed methods for in vitro differentiation, teratoma formation and immunostaining are described in Supplementary Materials and Methods. In vitro differentiation of HEF-derived human iPS cells was induced by culturing cells in suspension and then transferring onto gelatine-coated dish. In vitro differentiation of keratinocytes-derived human iPS cells was carried out as previously described6.
Blastocyst injections for chimeric mice
iPS cells were injected into C57BL/6J hosts blastocysts and transferred into 2.5 dpc ICR pseudopregnant recipient females. Chimerism was ascertained after birth by the appearance of agouti coat color (from iPS cell) in black host pups. High-contribution chimeras were mated to C57BL/6J mice to test for germline transmission.
Supplementary Material
Acknowledgments
We are grateful to the CMRB Histology & Bioimaging and Cell culture Platforms for expert assistance, Stephanie Boue for microarray analysis, Sungtae Kim for help with maintenance of mouse colonies, Yvonne Richaud for excellent technical assistance, Motoshi Nagao for preparation of mouse neural stem cells, Kristen Brennand and Fred Gage for preparation of human neural stem cells, Inder Verma and Antonella Consiglio for advice and help with lentiviral transduction, Yelena Dayn in Salk Transgenic Core Facility for chimeric mouse production, and all members of the Gene Expression Laboratory and CMRB for helpful discussions and Manuel Serrano for sharing unpublished results. J.S. was partially supported by Astellas Pharma Inc. T.K. was partially supported by Japan Society for the Promotion of Science. Work in the laboratory of G.W. was supported by NIH grants (5 R01 CA061449 and CA100845). Work in the laboratory of J.C.I.B. was supported by grants from the NIH, Tercel, Marato, G. Harold and Leila Y. Mathers Charitable Foundation and Fundacion Cellex.
Footnotes
The authors declare that they have no competing financial interests.
References
- 1.Takahashi K, Yamanaka S. Induction of pluripotent stem cells from mouse embryonic and adult fibroblast cultures by defined factors. Cell. 2006;126:663–76. doi: 10.1016/j.cell.2006.07.024. [DOI] [PubMed] [Google Scholar]
- 2.Takahashi K, et al. Induction of pluripotent stem cells from adult human fibroblasts by defined factors. Cell. 2007;131:861–72. doi: 10.1016/j.cell.2007.11.019. [DOI] [PubMed] [Google Scholar]
- 3.Yu J, et al. Induced pluripotent stem cell lines derived from human somatic cells. Science. 2007;318:1917–20. doi: 10.1126/science.1151526. [DOI] [PubMed] [Google Scholar]
- 4.Park IH, et al. Reprogramming of human somatic cells to pluripotency with defined factors. Nature. 2008;451:141–6. doi: 10.1038/nature06534. [DOI] [PubMed] [Google Scholar]
- 5.Lowry WE, et al. Generation of human induced pluripotent stem cells from dermal fibroblasts. Proc Natl Acad Sci U S A. 2008;105:2883–8. doi: 10.1073/pnas.0711983105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Aasen T, et al. Efficient and rapid generation of induced pluripotent stem cells from human keratinocytes. Nat Biotechnol. 2008;26:1276–84. doi: 10.1038/nbt.1503. [DOI] [PubMed] [Google Scholar]
- 7.Nakagawa M, et al. Generation of induced pluripotent stem cells without Myc from mouse and human fibroblasts. Nat Biotechnol. 2008;26:101–6. doi: 10.1038/nbt1374. [DOI] [PubMed] [Google Scholar]
- 8.Wernig M, Meissner A, Cassady JP, Jaenisch R. c-Myc is dispensable for direct reprogramming of mouse fibroblasts. Cell Stem Cell. 2008;2:10–2. doi: 10.1016/j.stem.2007.12.001. [DOI] [PubMed] [Google Scholar]
- 9.Okita K, Ichisaka T, Yamanaka S. Generation of germline-competent induced pluripotent stem cells. Nature. 2007;448:313–7. doi: 10.1038/nature05934. [DOI] [PubMed] [Google Scholar]
- 10.Rowland BD, Bernards R, Peeper DS. The KLF4 tumour suppressor is a transcriptional repressor of p53 that acts as a context-dependent oncogene. Nat Cell Biol. 2005;7:1074–82. doi: 10.1038/ncb1314. [DOI] [PubMed] [Google Scholar]
- 11.Kanatsu-Shinohara M, et al. Generation of pluripotent stem cells from neonatal mouse testis. Cell. 2004;119:1001–12. doi: 10.1016/j.cell.2004.11.011. [DOI] [PubMed] [Google Scholar]
- 12.Zhao Y, et al. Two supporting factors greatly improve the efficiency of human iPSC generation. Cell Stem Cell. 2008;3:475–9. doi: 10.1016/j.stem.2008.10.002. [DOI] [PubMed] [Google Scholar]
- 13.Cleveland JL, Sherr CJ. Antagonism of Myc functions by Arf. Cancer Cell. 2004;6:309–311. doi: 10.1016/j.ccr.2004.09.020. [DOI] [PubMed] [Google Scholar]
- 14.Vassilev LT, et al. In vivo activation of the p53 pathway by small-molecule antagonists of MDM2. Science. 2004;303:844–8. doi: 10.1126/science.1092472. [DOI] [PubMed] [Google Scholar]
- 15.Miyashita T, Reed JC. Tumor suppressor p53 is a direct transcriptional activator of the human bax gene. Cell. 1995;80:293–9. doi: 10.1016/0092-8674(95)90412-3. [DOI] [PubMed] [Google Scholar]
- 16.Kamijo T, et al. Functional and physical interactions of the ARF tumor suppressor with p53 and Mdm2. Proc Natl Acad Sci USA. 1998;95:8292–8297. doi: 10.1073/pnas.95.14.8292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Pomerantz J, et al. The Ink4a tumor suppressor gene product, p19Arf, interacts with MDM2 and neutralizes MDM2’s inhibition of p53. Cell. 1998;92:713–723. doi: 10.1016/s0092-8674(00)81400-2. [DOI] [PubMed] [Google Scholar]
- 18.Zhang Y, Xiong Y, Yarbrough WG. ARF promotes MDM2 degradation and stabilizes p53: ARF-INK4a locus deletion impairs both the Rb and p53 tumor suppression pathways. Cell. 1998;92:725–734. doi: 10.1016/s0092-8674(00)81401-4. [DOI] [PubMed] [Google Scholar]
- 19.Knudsen ES, Knudsen KE. Tailoring to RB: tumour suppressor status and therapeutic response. Nature Rev Cancer. 2008;8:714–24. doi: 10.1038/nrc2401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Marine JC, Dyer MA, Jochemsen AG. MDMX: from bench to bedside. J Cell Sci. 2007;120:371–8. doi: 10.1242/jcs.03362. [DOI] [PubMed] [Google Scholar]
- 21.Wang YV, et al. Increased radioresistance and accelerated B cell lymphomas in mice with Mdmx mutations that prevent modifications by DNA-damage-activated kinases. Cancer Cell. 2009;16:33–43. doi: 10.1016/j.ccr.2009.05.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Foster KW, et al. Oncogene expression cloning by retroviral transduction of adenovirus E1A-immortalized rat kidney RK3E cells: transformation of a host with epithelial features by c-MYC and the zinc finger protein GKLF. Cell Growth Differ. 1999;10:423–34. [PubMed] [Google Scholar]
- 23.Shaulian E, Zauberman A, Ginsberg D, Oren M. Identification of a minimal transforming domain of p53: negative dominance through abrogation of sequence-specific DNA binding. Mol Cell Biol. 1992;12:5581–92. doi: 10.1128/mcb.12.12.5581. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Lin T, et al. p53 induces differentiation of mouse embryonic stem cells by suppressing Nanog expression. Nat Cell Biol. 2005;2:165–71. doi: 10.1038/ncb1211. [DOI] [PubMed] [Google Scholar]
- 25.Jiang J, et al. A core Klf circuitry regulates self-renewal of embryonic stem cells. Nat Cell Biol. 2008;10:353–60. doi: 10.1038/ncb1698. [DOI] [PubMed] [Google Scholar]
- 26.Komarov PG, et al. A chemical inhibitor of p53 that protects mice from the side effects of cancer therapy. Science. 1999;285:1651–53. doi: 10.1126/science.285.5434.1733. [DOI] [PubMed] [Google Scholar]
- 27.Shi Y, et al. Induction of pluripotent stem cells from mouse embryonic fibroblasts by Oct4 and Klf4 with small-molecule compounds. Cell Stem Cell. 2008;6:568–74. doi: 10.1016/j.stem.2008.10.004. [DOI] [PubMed] [Google Scholar]
- 28.Huangfu D, et al. Induction of pluripotent stem cells from primary human fibroblasts with only Oct4 and Sox2. Nat Biotechnol. 2008;26:1269–75. doi: 10.1038/nbt.1502. [DOI] [PubMed] [Google Scholar]
- 29.Kim D, et al. Generation of human induced pluripotent stem cells by direct delivery of reprogramming proteins. Cell Stem Cell. 2009;4:472–6. doi: 10.1016/j.stem.2009.05.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Gonzalez F, et al. Generation of mouse-induced pluripotent stem cells by transient expression of a single nonviral polycistronic vector. Proc Natl Acad Sci U S A. 2009;106:8918–8922. doi: 10.1073/pnas.0901471106. [DOI] [PMC free article] [PubMed] [Google Scholar]
Additional References
- 30.Kitamura T, et al. Retrovirus-mediated gene transfer and expression cloning: powerful tools in functional genomics. Exp Hematol. 2003;31:1007–14. [PubMed] [Google Scholar]
- 31.Miyoshi H, Blömer U, Takahashi M, Gage FH, Verma IM. Development of a self-inactivating lentivirus vector. J Virol. 1998;72:8150–7. doi: 10.1128/jvi.72.10.8150-8157.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Sherley JL. Guanine nucleotide biosynthesis is regulated by the cellular p53 concentration. J Biol Chem. 1991;266:24815–28. [PubMed] [Google Scholar]
- 33.Huppi K, et al. Molecular cloning, sequencing, chromosomal localization and expression of mouse p21 (Waf1) Oncogene. 1994;9:3017–20. [PubMed] [Google Scholar]
- 34.Wiznerowicz M, Trono D. Conditional suppression of cellular genes: lentivirus vector-mediated drug-inducible RNA interference. J Virol. 2003;77:8957–61. doi: 10.1128/JVI.77.16.8957-8961.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Blelloch R, Venere M, Yen J, Ramalho-Santos M. Generation of induced pluripotent stem cells in the absence of drug selection. Cell Stem Cell. 2007;1:245–247. doi: 10.1016/j.stem.2007.08.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Takahashi K, Okita K, Nakagawa M, Yamanaka S. Induction of pluripotent stem cells from fibroblast cultures. Nat Protoc. 2008;2:3081–9. doi: 10.1038/nprot.2007.418. [DOI] [PubMed] [Google Scholar]
- 37.Blelloch R, et al. Reprogramming efficiency following somatic cell nuclear transfer is influenced by the differentiation and methylation state of the donor nucleus. Stem Cells. 2006;24:2007–13. doi: 10.1634/stemcells.2006-0050. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.