

Abstract
Microarray data can be used to derive understanding of the relationships between the genes involved in various biological systems of an organism, given the availability of databases of gene expression measurements from the complete spectrum of experimental conditions and materials. However, there have been no reports, to date, of such a database being constructed for rice (Oryza sativa). Here, we describe the construction of such a database, called RiceArrayNet (RAN; http://www.ggbio.com/arraynet/), which provides information on coexpression between genes in terms of correlation coefficients (r values). The average number of coexpressed genes is 214, with sd of 440 at r ≥ 0.5. Given the correlation between genes in a gene pair, the degrees of closeness between genes can be visualized in a relational tree and a relational network. The distribution of correlated genes according to degree of stringency shows how each gene is related to other genes. As an application of RAN, the 16-member L7Ae ribosomal protein family was explored for coexpressed genes and gene expression values within and between rice and Arabidopsis (Arabidopsis thaliana), and common and unique features in coexpression partners and expression patterns were observed for these family members. We observed a correlation pattern between Os01g0968800, a drought-responsive element-binding transcription factor, Os02g0790500, a trehalose-6-phosphate synthase, and Os06g0219500, a small heat shock factor, reflecting the fact that genes responding to the same biological stresses are regulated together. The RAN database can be used as a tool to gain insight into a particular gene by examining its coexpression partners.
Microarray technology provides high-throughput genome-wide measurements of gene transcription levels and promises to yield insights into the biological processes involved in gene regulation. This technology has revolutionized biological research by providing opportunities for researchers to inspect gene expression across the entire genome of the organism of interest (Schena et al., 1995; Boldrick et al., 2002; Clark et al., 2002). The information obtained from this technology has been employed in a wide variety of specific applications such as genetic dissection, drug discovery, and disease diagnostics. In the area of plant biology, genome-wide analyses with microarrays have been used to dissect gene expression during organogenesis, in response to stress, and in interactions with microorganisms, among other things (Jiao et al., 2005; Li et al., 2006; Mangelsen et al., 2008; Qin et al., 2008).
A number of analytical tools have been developed to extract gene relationships and functions from microarray data. Most of these tools provide clustering methods to group genes that show similar expression patterns under well-designed experimental conditions. The methods are based on algorithms to calculate pairwise relations and similarity measures, assuming that those clustered genes are related, and they greatly simplify the analysis of huge microarray data sets (Eisen et al., 1998). Algorithms such as hierarchical clustering, K-mean clustering, and self-organizing maps partition genes into mutually exclusive clusters based on pairwise measures. Other algorithms, such as information-based clustering, model-based approaches, and projection methods, have been proposed based on collective notions of similarity of gene expression (Bussemaker et al., 2001; Lee and Batzoglou, 2003; Slonim et al., 2005).
Centralized data storage systems for genome-wide expression profiling have been constructed both for individual organisms and for multiple species. As an example of the former, AtGenExpress contains more than 500 data sets from experiments examining Arabidopsis (Arabidopsis thaliana) development and responses to stress, light, pathogens, and hormone responses, based on the Affymetrix ATH1 GeneChip (Schmid et al., 2005; Kilian et al., 2007; Goda et al., 2008). As an example of the latter, the Gene Expression Omnibus represents the largest public repository of high-throughput gene expression data (Barrett et al., 2009; http://www.ncbi.nlm.nih.gov/geo/). The database contains genome-wide data generated by the research community based on microarrays and, increasingly, on next-generation sequencing. Users can explore, analyze, and download expression data according to their interests in genes or specific biological themes, such as development and abiotic stresses. There are several databases to support efficient access and data mining of collections of microarray data for Arabidopsis, For example, the comprehensive systems biology database CSB.DB (Steinhauser et al., 2004), Botany Array Resource (Toufighi et al., 2005), Arabidopsis Co-expression Tool (Manfield et al., 2006), GeneCat (Mutwil et al., 2008), Plant Gene Expression Database (Horan et al., 2008), and the Arabidopsis trans-factor and cis-element prediction database ATTED-II (Obayashi et al., 2009) provide various tools for comparative gene analysis such as cis-element prediction and coexpression analysis. Genevestigator (http://www.genevestigator.ethz.ch) provides an analysis toolbox for databases of several model organisms, but it is not completely publicly available (Zimmermann et al., 2004). Other methods have been suggested to identify coexpressed genes from microarray data (Rawat et al., 2008). These sources serve not only as the main references for the transcriptome but also as principal information resources for data mining. For example, the tolerance responses of plants to many abiotic stresses, such as heat, cold, drought, salt, high osmolarity, UV-B light, and wounding, were explored using the AtGenExpress database (Kilian et al., 2007).
In reality, a gene may be part of several biological processes, and its expression is subject to controls for maximum efficiency. For example, cells have evolved efficient gene expression mechanisms in response to external signals in order to adapt to changing environments. In these concerted processes, many genes are likely to be subject to coregulation: they may be induced or repressed together or inversely. The accumulation of microarray data has provided good opportunities to correlate and understand patterns of gene expression simultaneously, both individually and in relation to other genes. Efforts to evaluate gene expression in the biological context of an organism, for the complete spectrum of experimental conditions and materials reported in the database, have been promising. In the case of Arabidopsis, a model plant for dicotyledons, strong evidence suggests that related genes, such as those involved in cell wall synthesis, are coregulated (Manfield et al., 2006). In addition, coexpression analysis has been used as a “primary screen” to identify novel genes of OPCL1 named OPC-8:0 CoA Ligase1, Myb transcription factors as regulators of aliphatic glucosinolate biosynthesis, AtPS1 (for Arabidopsis parallel spindle 1) involved in meiosis, or subunits of NAD(P)H dehydrogenase in biosynthetic processes (Koo et al., 2006; Hirai et al., 2007; d'Erfurth et al., 2008; Takabayashi et al. 2009).
Rice (Oryza sativa) is a major, and financially important, crop worldwide and has been used as a model plant for monocotyledons because of the availability of its complete genomic sequence and full-length cDNA libraries. A map-based, finished-quality sequence that covers 95% of the 389-Mb genome, including virtually all of the euchromatin and two complete centromeres, is an invaluable source for research (International Rice Genome Sequencing Project, 2005). The recent release of RAP2 (for the Rice Annotation Project version 2) contains 31,439 expressed and 22,022 ab initio predicted loci (http://rapdb.dna.affrc.go.jp/). A significant proportion of the genes appear in clustered gene families. The comparison of transposable elements found in the rice genome with those of the maize (Zea mays) and sorghum (Sorghum bicolor) genomes allows us to find evidence for the hypothesis that the syntenic regions are expanding in these grasses.
We analyzed transcriptome profiles using the Rice 60k Microarray (Jung et al., 2005), which contains 60,727 70-mer oligonucleotides representing 58,417 genes, including predicted genes. As of April 2008, expression data from 183 microarrays for 20 organs and 50 different treatments have been gathered. In support of this, we built a database, RiceArrayNet (RAN; http://www.ggbio.com/arraynet/), which provides information on coexpression between genes in terms of correlation coefficients (r values) using the accumulated data. A statistical analysis of correlation is applied both to the gene(s) of interest and the coregulated genes, and their correlations are visualized in a relational tree and a relational network. Furthermore, additional information, which suggests likely biochemical pathways and cis-regulatory elements of clustered genes, is provided through links to a pathway map in the KEGG database and the PLACE database, respectively.
We employed the RAN database to study coexpression patterns in rice. The distribution of the correlation coefficients according to the degree of stringency shows how closely a given gene is coexpressed with other genes in the genome. Across the entire genome, the average number of coexpressed genes is 214, with sd of 440 at r ≥ 0.5. For the 16-member L7Ae ribosomal protein family, between 20 and 590 genes are coexpressed, with a broad range of variation across the subgroups under the criterion of r ≥ 0.5. A member of the subgroup, Os10g0124000, has 314 coexpressed genes, many of which are ribosomal proteins, so they may be expressed in stoichiometric ratios for efficient translation, as suggested in Arabidopsis (Jen et al., 2006). Interestingly, a comparison by selecting the top-ranked 5% of coexpressed genes of the family from the RAN and Arabidopsis coexpression databases (ACT; http://www.arabidopsis.leeds.ac.uk/act/) identifies 360 to 460 coexpressed genes, respectively, a narrower range of numbers among phylogenetically conserved members. Regardless of the subgroup, the gene expression values and their ratios of the whole rice L7Ae family are comparable to those obtained from the Arabidopsis microarray collections, such as AFGN (http://www.uni-tuebingen.de) and Genevestigator. These data show that the gene family is undergoing its own evolutionary paths related to developmental stages or in response to abiotic stresses along with each gene's unique biological functions. In RAN, we also observe correlation patterns in stress-related genes such as Os01g0968800, drought-responsive element-binding transcription factors (DREB), with Os02g0790500 and Os06g0219500 coding for a trehalose-6-phosphate synthase (T6pS) and a small heat shock protein (SHSP), respectively. The method could thus be used to identify the functional equivalents of a given set of genes in model organisms, and this information could be applied to identify the gene functions in other organisms.
RESULTS
Database Content
Expression data from 183 microarrays were collected from the samples of either wild-type or mutant rice organs, such as leaf, root, flower, and callus, at various developmental stages (Supplemental Table S1). The experiments were performed to test how gene expression is modulated and reprogrammed in response to various biotic and abiotic stresses and hormone treatments. RAN was designed to be flexible in terms of choosing query genes and finding coexpressed genes. Users can directly input the oligomer identifiers (IDs) or spot numbers used to design the Rice 60k Microarray, or their gene IDs as annotated by TIGR or RAP, in order to search for coexpressed genes. Additionally, because RAN stores oligomer matches against various sequence databases, such as GenBank NR, Swiss-Prot, and the National Center for Biotechnology Information Conserved Domain Database (Marchler-Bauer et al., 2005), users can identify genes by keywords. In the case of a hypothetical gene, users can search for an oligomer of interest in the microarray sequences using BLAST with a known sequence. Users can also choose the cutoff value for the correlation coefficient (r) and the number of genes to be shown.
The correlation information between genes is presented in three different ways. First, the gene coexpression relationships can be visualized in a cluster diagram or network, where genes that have close expression relationships form a cluster (and close network), so researchers can easily detect groups of coexpressed genes. For example, given the oligomer ID Os056379_01, representing the ribosomal protein L7Ae, Os10g0124000, a relational tree of gene expression (Fig. 1A) represents the global degrees of correlation between genes, while a network (Fig. 1B) shows how the 36 genes are correlated with each other. In the network, genes are denoted as filled circles and located such that their proximity represents the closeness of their relationships, with colored edges showing the sign of the r values. A red edge denotes a gene pair with a positive r value, while green denotes a negative value. In addition, the color contrast and line thickness of an edge are deeper and thicker, respectively, as the absolute values of the r values increase. If a gene in the network has more correlations, then it has more edges resulting in subnetworks. Thus, the researcher can observe the gene relationships in perspective with the graphs. Below the tree view (or network view), the coexpressed genes are listed (Supplemental Table S2). Each row in the list contains the spot number, RAP2 ID, TIGR ID, the most similar Arabidopsis gene, and a cluster or group number to which the oligomer belongs. The spot numbers of genes used as seeds (input for a retrieval) in the previous section are marked with asterisks. Statistical information for each gene in the tree (or network) and the other genes is provided on separate pages as a list, with particular information for each correlation between genes in a gene pair (Table I). Statistics on the correlation coefficients between genes are given in descending order of r values, including the significance level or P value, calculated based on a t distribution (Manfield et al., 2006), and the standard score or Z score, calculated based on a distribution made from the r values of all pairs of a query gene and any other gene (a total of 58,416, including predicted genes). The statistical information list also contains KEGG (Kanehisa and Goto, 2000) pathways for which at least two genes in the list are involved in the pathways. Additionally, a list of frequent cis-elements of the genes in the statistical information list is provided on the page. Detailed information is provided in separate information pages for each correlation between genes in a gene pair (Fig. 2). The page contains a scatterplot of log ratios representing expression levels as well as supplementary data, such as common cis-elements between the promoters of the gene pair. A distribution of log ratios to calculate r values is presented based on all the log2 ratios in the data, displayed as a scatterplot. Using the scatterplot (Fig. 2A), a researcher can compare correlation coefficients between gene pairs and/or can select the source of a particular microarray result, represented as outlined in the plot. The log2 ratio in the individual microarray experiment is represented by a dot. The r value shows positive correlation. The coinduced and corepressed log2 ratios are located in the first and third quadrants, respectively, of the coordinate plane. This is typical of positive correlation coefficients. Some information on representative microarray batches and their values is listed on the page, and a full list is available as a file. Additionally, the page provides data, such as the histogram of r values of the gene, along with statistical analysis by Z score (Fig. 2B). The biological context of coexpressed genes can be related to cis-acting regulatory elements, so the page provides cis-elements that are common between the promoters of the gene pair (Supplemental Table S3). Thus, a researcher can use these data to select biologically significant cis-elements versus all previously published ones.
Figure 1.

Graphical presentation of coexpressed genes. A, Relational tree of gene expression with a ribosomal protein, B11032220 (oligomer ID Os056379_01; RAP2 ID Os10g0124000; marked with asterisks). The 36 genes are retrieved under the parameters of r ≥ 0.6 and depth = 1. B, The network is drawn with the same parameters, using B11032220 (asterisk). The network shows how the 36 genes are correlated with each other. A microarray spot representing a gene is denoted as a filled circle, and their proximity represents the closeness of their relationship in the network, with colored edges showing the sign of the r values. A red edge denotes a positive r value of the gene pair, while green denotes a negative value. In addition, the color contrast and line thickness of an edge are deeper and thicker, respectively, as the absolute value of the r value increases. [See online article for color version of this figure.]
Table I.
Partial list of coexpressed genes of a ribosomal protein L7Ae, Os10g0124000
This list includes the significance level, calculated by t test, for the correlation coefficient and the standard score, calculated based on a distribution made from the r values of all pairs of a query spot, B11032220 (oligomer ID Os056379_01; RAP2 ID Os10g0124000), to all other spots (genes; 58,416). The top-ranked 20 of the 36 genes are retrieved from RAN with r ≥ 0.6 and depth = 1. The page is displayed by clicking spot number B11032220 shown in Supplemental Table S2.
| No. | Target Spot | Oligomer ID | TAIR Locus | r | P | Z Score | Description by BLAST Analysis |
|---|---|---|---|---|---|---|---|
| 1 | A03012214 | Os025798_01 | 0.698 | 3.22e-28 | 4.03 | Unknown | |
| 2 | A01010825 | Os009333_01 | 0.694 | 9.56e-28 | 4.00 | 60S ribosomal protein L2, putative, expressed | |
| 3 | B07032307 | Os057000_01 | 0.693 | 1.17e-27 | 4.00 | Similar to 40S ribosomal protein S3a (CYC07 protein) | |
| 4 | A10020810 | Os008770_01 | AT1G22780 | 0.669 | 3.02e-25 | 3.86 | 40S ribosomal protein S18, putative, expressed |
| 5 | A08022109 | Os024792_01 | 0.668 | 3.20e-25 | 3.86 | Unknown | |
| 6 | A05041513 | Os017613_01 | AT4G36195 | 0.666 | 5.13e-25 | 3.85 | Prolyl carboxypeptidase-like protein, putative, expressed |
| 7 | A04031020 | Os011464_01 | 0.660 | 1.82e-24 | 3.82 | Unknown | |
| 8 | A02031001 | Os010699_01 | AT2G39700 | 0.659 | 2.59e-24 | 3.81 | α-Expansin 6 precursor, putative, expressed |
| 9 | A09021815 | Os021369_01 | 0.651 | 1.28e-23 | 3.76 | Peptidyl-prolyl cis-trans trans-isomerase, FKBP-type family protein, expressed | |
| 10 | A10020824 | Os009498_01 | AT3G09630 | 0.650 | 1.66e-23 | 3.76 | 60S ribosomal protein L4, putative, expressed |
| 11 | A08040105 | Os000342_01 | AT1G73230 | 0.641 | 1.03e-22 | 3.71 | Transcription factor BTF3, putative, expressed |
| 12 | A03040817 | Os009061_01 | AT4G34670 | 0.639 | 1.38e-22 | 3.70 | 40S ribosomal protein S3a, putative, expressed |
| 13 | B10022015 | Os053964_01 | 0.636 | 2.44e-22 | 3.68 | Unknown | |
| 14 | A07021717 | Os020233_01 | 0.634 | 3.51e-22 | 3.67 | Conserved hypothetical protein | |
| 15 | A09021718 | Os020238_01 | AT1G33120 | 0.630 | 8.40e-22 | 3.64 | 60S ribosomal protein L9, putative, expressed |
Figure 2.

Distribution of log ratios to calculate r values and statistical analysis of the r value. A, At the top, all the log2 ratios in the data are drawn in a scatterplot. The log2 ratio in each individual microarray experiment is represented by a dot. The r value shows a positive correlation between two genes represented by spot numbers. The coinduced and corepressed log2 ratios are positioned in the first and third quadrants, respectively, of the xy coordinate plane. This is a typical positive correlation coefficient. The page is displayed by clicking the r value, 0.694, between B11032220 (oligomer ID Os056379_01; RAP2 ID Os10g0124000) and A01010825 (oligomer ID Os009333_01; RAP2 ID AK060350; 60S ribosomal protein L2) shown in Table I. A table of brief experimental descriptions of microarrays is given below the log ratio scatterplot. B, At the top, the distribution of r values is shown between B11032220 and other spots (genes). At the bottom, the r values are shown in the ACT-type presentation of a scatterplot. At the bottom right is the distribution of r values between A01010825 and other spots (genes). [See online article for color version of this figure.]
The Distribution of Correlated Genes According to Degree of Stringency
To examine the distribution of r values, we calculated the average (μ) and sd (σ) of about 1.9 × 109 r values, obtained from combinations of all 60,727 oligomers in the microarray. We tested and confirmed that the distribution of the r values was approximately normal, with an average of 0.003 and sd of 0.159. Consequently, 95.0% and 99.4% of the r values are within the ranges 0.003 ± 0.318 (μ ± 2σ) and 0.003 ± 0.477 (μ ± 3σ), respectively. We also projected the data in RAN according to the r values (Supplemental Table S4). In the tallied r values of the first row in the table, the closer the values are to 1, the more likely it is that the two genes are coinduced, while the closer the values are to −1, the more likely it is that the two genes are regulated inversely. The column in Supplemental Table S4 was divided into eight r value bins and displayed in a histogram, depending on the number of genes in r value bins (Fig. 3A). As the r value bins decrease in the coinduced region (r = 0.3–0.7 in Fig. 3A, right half of the graph) to 0.2 from 0.7, the skew in the number of eight r value bins shifts from right to left; the number of genes in the (0, 100) region increases from 13,643 at r ≥ 0.7, reaches a maximum of 33,214 at r ≥ 0.5, and decreases to 1,145 at r ≥ 0.3, while the number of genes in the (101, 200) region reaches a maximum of 8,613 at r ≥ 0.4 and drastically decreases to 1,920 at r ≥ 0.4 in the same region. This is a general phenomenon where the number of correlated genes decreases with higher stringency or correlation coefficients. A similar distribution was observed with the inversely regulated region (r values of approximately −0.6 to approximately −0.2 in Fig. 3A, left half of the graph). With absolute value r ≥ 0.5, 53% of the genes have fewer than 100 correlations, and the average number of correlated genes is 214 with sd of 440. Almost 70% of the genes have fewer than 300 correlated genes, and 6% (3,702) of the genes have more than 1,000 coexpressed genes in this condition. Out of the 3,702, genes, 2,630 of them could be assigned to the RAP2 database. For example, Os09g0491740, a permease, Os02g0536300, an F-box domain protein, and Os05g0178300, the FAD-dependent oxidoreductase family of proteins, are among those with around 3,200 correlated genes. Interestingly, the histogram is roughly symmetric between the positively and inversely coexpressed regions, suggesting that the numbers of genes in the eight r value bins are very similar. To see how many genes are coexpressed for the individual gene levels, the numbers of genes in columns ∼0.5 and ∼−0.5 in Supplemental Table S4 were plotted in xy Cartesian coordinates (Fig. 3B). The slope is 0.7 and its associated P value is <2e−16, confirming the symmetric appearance of the graphical distribution. These data suggest that if a certain number of genes are coinduced with a specific gene, an almost equivalent number of genes are inversely expressed. This tendency is even stronger for genes that have more coexpressed genes than for those that have fewer.
Figure 3.

Distribution of r values in RAN. A, Distribution of number of genes according to the eight population regions in the r value bins. The x axis represents the r value bins and the eight r value bins. The y axis denotes the number of genes. The r value bins closer to 1 suggest that the two genes are more strongly coinduced, while coefficients closer to −1 suggest that the two genes are more strongly regulated inversely. The histogram is roughly symmetric. The average number of coexpressed genes is 214, with sd of 440 and r ≥ 0.5. B, The number of genes in columns ∼0.5 and ∼−0.5 in Supplemental Table S4 are drawn in xy Cartesian coordinates. The slope is 0.7, and its associated value is less than 2e-16, suggesting that a gene has regulation mechanisms in which almost equivalent coinduction and inverse expression are exerted on a gene.
The Rice Ribosomal Protein L7Ae Shows Both Similar and Unique Patterns of Coexpression in a Member-to-Member Comparison with the Arabidopsis Family
If RAN accurately reflects the coexpression of genes, then ribosomal proteins might be good test candidates, as these proteins should be under a coordinated mechanism of regulation to maintain stoichiometric ratios for efficient gene expression in Arabidopsis (Barakat et al., 2001; Jen et al., 2006). Using the keyword L7Ae, 12 and six genes from the ribosomal protein L7Ae family were retrieved from RAP2 and TAIR8 (Rhee et al., 2003), respectively. The long domains of the 95-amino acid domains were compared with each other by BLASTp. The BLASTp results ranged from 1e−39 to 1e−75. The OrthoMCL (Li et al., 2003; http://www.orthomcl.org) test showed that these proteins are divided into four groups. At this step, AT4G01790.1 and AT5G20160.1 were excluded by the program (Supplemental Table S5). In the four groups (MCL0, -1, -2, and -3) grouped by the OrthoMCL test, MCL0 has eight genes, with four each from rice and Arabidopsis. The groups MCL1, MCL2, and MCL3 have three, three, and two members, respectively, and ClustalW analysis showed that these proteins were well aligned (Supplemental Fig. S1). Phylogenetic analysis was also performed as described in “Materials and Methods,” and the analysis generally confirmed the OrthoMCL results, except with two clusters for MCL0 (Supplemental Fig. S2). Os10g0124000 showed high percentage identity, ranging from a minimum of 90.2% for Os03g0241200 to a minimum of 40.8% for Os02g0728600, as expected from their relative branch lengths. Os10g0124000 has similar values of 89.1% and 93.7% with AT4G12600 and AT4G22380 in Arabidopsis, respectively. It has the lowest similarity, 40.8%, with AT5G08180 and Os02g0728600 in the same MCL0 group. As grouped in MCL3, Os07g0150200 showed percentage identity of 100% with Os07g0229900 in the same MCL3 subgroup, while it showed a low value of 34.8% with Os02g0728600 in the MCL0 subgroup. Each member has differing numbers of coexpressed genes at a cutoff of r ≥ 0.5 (Supplemental Table S6). For example, Os03g0241200 and Os10g0124000, in the MCL0 group, have 345 and 315 coexpressed genes, respectively, at r ≥ 0.5. In contrast, Os07g0150200, in the MCL3 group, has 19 coexpressed genes for r ≥ 0.5.
In the 315 genes retrieved with Os10g0124000 under the parameters of r ≥ 0.5 and depth = 1 from RAN, the 20 top-ranked genes show r values from 0.62 (P value of 4.8e−22) to 0.7 (3.2e−28), and seven of these genes are ribosome components (Supplemental Table S2). A total of 242 out of the 315 genes have RAP2 loci (Supplemental Table S7), and enriched Gene Ontology (GO) terms were tested using GoMiner (http://discover.nci.nih.gov/gominer/). The false discovery rate (fdr) values were calculated by 100 simulations. In molecular function, 45 of these genes are in the GO category GO:0003735 (structural constituent of ribosome; fdr = 0). Thirteen and four genes have been given the categories GO:0003723 (RNA binding; fdr = 0.0025) and GO:0003899 (DNA-directed RNA polymerase activity; fdr = 0.0358), respectively. In biological process, 49 genes are classified as GO:0006412 (translation; fdr = 0). Four and three genes are GO:0006364 (rRNA processing; fdr = 0.0078) and GO:0006270 (DNA replication initiation; fdr = 0.0365), respectively. In cellular component, 11 and four genes are given to GO:0022627 (cytosolic small ribosomal subunit; fdr = 0.0005) and GO:0022625 (small nucleolar ribonucleoprotein complex; fdr = 0.0023), respectively. Most of the GO terms are predominantly involved in protein synthesis, as protein binding, ribosomal constituents, and ribosomal RNA synthesis are included in these categories. The analysis is also consistent with the coexpressed genes in Arabidopsis (Jen et al., 2006). In Arabidopsis, the top 15 genes that were most highly correlated with the L7Ae protein AT4G12600 were mixtures of 60S and 40S subunits. Coexpressed genes were retrieved from ACT with the initial condition r ≥ 0.5. AT4G22380, AT5G20160, AT4G12600, and AT5G08180 have 1,218, 1,072, 934, and 1,503 coexpressed genes, respectively.
Group-to-Group Comparison between Ribosomal Protein L7Ae Families in Rice and Arabidopsis
The correlation coefficient distribution for genes varies depending on the gene, and performing coexpression analysis on different data sets could yield spurious coexpression results. It is likely that different suites of genes get turned on in response to stress or at different developmental stages. To avoid a biased analysis when performed under strict conditions for two different data sets, we selected the top-ranked 5% of the coexpressed genes for the members of the family in each database, around 1,700 out of the 33,689 genes identifiable from RAN and 1,150 out of the 22,765 genes from ACT, as described in “Materials and Methods.” In the subsequent analysis, all of the members of the MCL0 group and one of the other subgroups were used. A DREB transcription factor, Os01g0968800 (DREB1F), was chosen as an external control. When rice coexpressed genes are directly compared, members in MCL0, MCL1, and MCL2 showed values of 354 to 869, and the most commonly expressed genes were from Os05g0490100 in MCL1 and Os09g0507800 in MCL2 (Table II). In contrast, the members showed relatively low numbers, 155 to 323, with MCL3. This may reflect the divergence of members of MCL3 from the others (Supplemental Fig. S2). Compared with Os01g0968800 (DREB1F), an external control that is likely to be expressed in response to drought, the coexpressed genes produced even lower numbers, 75 to 105. The commonly coexpressed genes of MCL0, -1, and -2 groups (Os10g0124000, Os03g0241200, Os02g0728600, Os05g0490100, and Os09g0507800) included 197 genes. This number drops to 31 when the MCL3 member Os07g0150200 is considered. A similar analysis was performed for Arabidopsis. Interestingly, the members of this family from Arabidopsis belong to MCL0 and show 712 to 908 coexpressed genes among them (Table II). This average is higher than that of rice members. There are 629 commonly coexpressed genes of Arabidopsis among all the MCL0 members: AT4G12600, AT4G22380, AT5G20160, and AT5G08180. Next, we asked how many coexpressed genes of rice have homologs in the Arabidopsis counterparts. As it is difficult to define the exact counterpart in the comparison between species, we first did BLASTp analysis and considered the genes with scores of 100 or higher to be the tentative counterparts (Supplemental Table S4; Supplemental Methods). Like other members in rice, Arabidopsis members (MCL0) showed more coexpressed genes, 372 to 452, with MCL0, MCL1, and MCL2 and lower numbers, 169 to 174, with the MCL3 member Os07g0150200. Like other rice members, they also showed lower numbers, 131 to 156, with Os01g0968800 (DREB1F). Lastly, the number of commonly coexpressed genes by both rice MCL0, -1, and -2 and Arabidopsis MCL0 is 118 (Supplemental Table S7). This table shows that almost 60% of the coexpressed genes of the rice MCL0, -1, and -2 groups have counterparts in Arabidopsis. A simulation test under the same conditions was performed 100 times, and the average is around 50 and the sd is 16.6. Significance analysis showed a P value of 0, suggesting that the value 118 is very significant. Enriched GO terms were tested for the 118 genes. In molecular function, 39 genes have been given the identifier GO:0003735 (structural constituent of ribosome; fdr = 0). Fifteen and five genes have been given the codes GO:0003723 (RNA binding; fdr = 0) and GO:0051082 (unfolded protein binding; fdr = 0.0111), respectively. In the cellular component, 12, three, and three genes have been given GO:0022627 (cytosolic small ribosomal subunit; fdr = 0), GO:0005732 (small nucleolar ribonucleoprotein complex; fdr = 0.0183), and GO:0022625 (cytosolic large ribosomal subunit; fdr = 0.0110), respectively. In biological processes, 43, seven, and four genes have been given the IDs GO:0006412 (translation; fdr = 0), GO:0006457 (protein folding; fdr = 0.0081), and GO:0006364 (rRNA processing; fdr = 0.0042), respectively. As is shown with an individual member, Os10g0124000, under the condition of r ≥ 0.5, most of these GO terms are predominantly involved in protein synthesis, as protein binding, ribosomal constituents, and ribosomal RNA synthesis are included in these categories. The comparison, either member-to-member using a gene with a medium range of coexpressed genes, or group-to-group for the L7Ae family, shows that similar GO terms are enriched.
Table II.
In- and between-species comparisons of coexpressed genes of a ribosomal protein L7Ae in rice and Arabidopsis
The top-ranked 5% of genes in each database (1,684/33,689 for rice in RAN and 1,138/22,765 for Arabidopsis in ACT) are compared. Os01g0968800 (DREB), known to be expressed in response to drought, is used as an external control. In the between-species comparison, the coexpressed genes are shown in bold. BLASTp analysis is performed for the two species, and the genes with scores of 100 or higher were considered to be the tentative counterparts (Supplemental Table S4). As in the case of other members of the rice family, Arabidopsis members show more coexpressed genes (372–452) with MCL0, MCL1, and MCL2 of rice and lower numbers (169–174) with MCL3. They also show lower numbers (131–156) with Os01g0968800, as did other rice members.
| Os03g0241200 (MCL0) | Os02g0728600 (MCL0) | Os05g0490100 (MCL1) | Os09g0507800 (MCL2) | Os07g0150200 (MCL3) | Os01g0968800 (DREB) | AT4G12600 (MCL0) | AT4G22380 (MCL0) | AT5G20160 (MCL0) | AT5G08180 (MCL0) | |
|---|---|---|---|---|---|---|---|---|---|---|
| Os10g0124000 (MCL0) | 544 | 688 | 765 | 802 | 323 | 75 | 444 | 445 | 445 | 463 |
| Os03g0241200 (MCL0) | 354 | 548 | 808 | 155 | 105 | 392 | 373 | 397 | 397 | |
| Os02g0728600 (MCL0) | 504 | 489 | 190 | 78 | 360 | 372 | 375 | 383 | ||
| Os05g0490100 (MCL1) | 869 | 259 | 86 | 446 | 417 | 429 | 451 | |||
| Os09g050780 (MCL2) | 190 | 109 | 452 | 418 | 437 | 456 | ||||
| Os07g0150200 (MCL3) | 93 | 169 | 174 | 172 | 172 | |||||
| Os01g0968800 (DREB) | 131 | 156 | 156 | 146 | ||||||
| AT4G12600 (MCL0) | 726 | 712 | 877 | |||||||
| AT4G22380 (MCL0) | 908 | 948 | ||||||||
| AT5G20160 (MCL0) | 904 |
Comparison of Gene Expression of Ribosomal Protein L7Ae in Rice and Arabidopsis at Developmental Stages and in Response to Abiotic Stresses
Although it is difficult to compare gene expression directly between rice, a monocot, and Arabidopsis, a dicot, we assumed that the expression pattern(s) of a gene(s) in a gene family in rice would be comparable to that (those) of Arabidopsis. A distribution of log ratios to calculate r values as shown in Figure 2A using Os10g0124000 suggested that the genes in the ribosomal protein L7Ae consistently decreased in specific organs, compared with the callus or anther (Supplemental Table S8). These values were also compared with those of Arabidopsis, obtained from microarray collections in AFGN and Genevestigator. In the comparison, microarray sets from samples as described above in the rice database were compared with those performed with samples of similar tissues at similar developmental stages of Arabidopsis, as described in Supplemental Tables S9 and S10. The log ratios of the rice genes Os10g0124000, Os03g0241200, and Os02g0728600 in MCL0, Os05g0490100 in MCL1, Os09g0507800 in MCL2, and Os07g0150200 in MCL3 were compared with those of AT4G12600, AT4G22380, AT5G20160, and AT5G08180 in MCL0 (Fig. 4). These genes were strongly decreased in the leaf, root, and flower, compared with the callus and in response to abscisic acid (ABA) treatment, and were relatively weakly decreased in seeds compared with the callus and in response to abiotic stresses such as drought and cold. The comparisons between rice members show positive Pearson correlations, even though they are less significant (Supplemental Table S11). Interestingly, Os07g0150200 in MCL3, which showed lower numbers of coexpressed genes with other members of the L7Ae family, shows significant correlation with Os10g0124000 in MCL0 and Os09g0507800 in MCL2. Many Arabidopsis members show positive Pearson correlations not only with other Arabidopsis members but also with rice members. In contrast, DREB1F (Os01g0968800) was compared as a control, and the gene is induced by a plant hormone, ABA, and abiotic stresses such as drought and cold, unlike the ribosomal protein L7Ae family. Os01g0968800 shows even negative correlation with most ribosomal protein L7Ae members. These data show that gene expression of ribosomal protein L7Ae in rice and Arabidopsis could be under similar control mechanisms at various developmental stages and in response to abiotic stresses.
Figure 4.

Log2-based ratios of a ribosomal protein L7Ae obtained from microarray sets of rice and Arabidopsis. The samples for comparison are indicated above the histogram. Microarray sets from samples in RAN are compared with those performed with samples of similar tissues or developmental stages from the Arabidopsis databases AFGN and Genevestigator, as described in “Materials and Methods.” The expression of these genes is strongly decreased in the leaf, root, and flower compared with the callus and in treatment with ABA, while it relatively weakly decreases in seeds compared with the callus and in response to abiotic stresses such as drought and cold. In treatments with ABA, drought, and cold, the gene expression values at the initial condition of each experiment were used as controls. The log ratios of rice genes Os10g0124000, Os03g0241200, and Os02g0728600 in MCL0, Os05g0490100 in MCL1, Os09g0507800 in MCL2, and Os07g0150200 in MCL3 are compared with those of AT4G12600, AT4G22380, AT5G20160, and AT5G08180 in MCL0. Os01g0968800 (DREB) is also compared as a control, and gene expression is found to increase in organs such as leaf and root. As expected, it also increases in response to drought and ABA.
Drought-Related Genes Might Be Coregulated
We further applied the RAN database to dissect coexpression patterns in stress-related genes, such as DREB, T6pS, and SHSP genes (Jang et al., 2003; Kotak et al., 2007; Wang et al., 2008). The 16 DREB genes were retrieved from RAP2. The long AP2 domains of 58 amino acids were extracted from these genes, and OrthoMCL analysis was performed. The analysis suggested that all of these DREB transcription factors belong to the same group, except Os08g0521600. AP2 domains within the group showed percentage identity from 44.8 to 100. Among the rice members, Os01g0968800 (DREB1F) showed the highest identity (72.4%) with Os04g0572400 and the lowest identity (50.9%) with Os06g0165600 (data not shown). Although the top 5% ranked genes among the coexpressed genes show functions identical to those of the ribosomal protein L7Ae family, the number of coexpressed genes at a certain correlation coefficient might represent the unique biological functions for individual genes. The numbers of coexpressed genes for DREB at r ≥ 0.4 are listed in Supplemental Table S12. Among these members, Os02g0752800 had the fewest correlated genes, with only three, and Os01g0968800 had the most, with 849 correlated genes. Genes with a GO term among the 849 correlated genes of Os01g0968800 include the following biological processes (Supplemental Table S13): 11 GO:0009408 (response to heat; fdr = 0), 12 GO:0006979 (response to oxidative stress; fdr = 0.0367), six GO:0009644 (response to high light intensity; fdr = 0), and six GO:0042542 (response to hydrogen peroxide; fdr = 0). Many GO terms suggest that these coexpressed genes are involved in drought stress. These genes include a chaperonin clpA/B family protein, a heat shock protein Hsp70 family protein, an Armadillo-like helical domain-containing protein, a cytokine-induced apoptosis inhibitor 1, a zinc finger transcription factor, a NAC transcription factor, a DUF563 family protein, a heat shock 22-kD protein, a CCR4-associated factor 1, and a Y19 protein, implying a drought stress response.
The 11 T6pS genes were extracted from rice RAP2. OrthoMCL analysis suggests that these genes belong to a group (Supplemental Table S14; nine members shown on the chip). Os01g0730300 showed percentage identities ranging from 59.2% to 70.9% with Os05g0517200, Os09g0397300, and Os03g0224300. It was also highly identical with genes from Arabidopsis, such as AT1G23870, AT1G68020, AT1G70290, AT2G18700, and AT4G17770, with percentage identities ranging from 27.8% to 66.6% (data not shown). Os01g0730300, Os02g0790500, and Os09g0397300 have 3,789, 2,679, and 2,498 identical genes, respectively. A total of 2,622 out of the 3,789 genes of Os01g0730300 have GO terms (Supplemental Table S15). Notable GO terms include 214 GO:0050896 (response to stimulus; fdr = 0.0367) and 10 GO:0009832 (plant-type cell wall biogenesis; fdr = 0.10) as biological processes. Fifty-seven genes are given GO:0044428 (nuclear part; fdr = 0) as a cellular component. Thus, these coexpressed genes are involved in more varied biological functions than DREB. The individual genes include a CBS domain-containing protein, a PGPD14 protein, a β-galactosidase precursor, an extensin, and a protein prenyltransferase domain-containing protein, among others.
The DREBs are important transcription factors that induce a set of abiotic stress-related genes and impart stress endurance to plants. From the preceding analysis, we observed that drought-related genes might be coexpressed, with one of the highest numbers of coexpressed genes for DREB, Os01g0968800 (data not shown). We observed a similar partitioning of members of a gene family according to coexpression patterns in stress-related genes, even including SHSP genes (data not shown). Since RAN can make a relational tree from multiple seeds, we used three keywords: Os01g0968800, a DREB1; Os02g0790500, a T6pS; and Os06g0219500, a SHSP. RAN produced a dense relational tree with these three keywords (Fig. 5A). The tree shows not only the close relationship between these three genes with short edges but also the genes correlated with each. The relational tree shows that T6pS and SHSP have a close expressional correlation with DREB1 but that their expressional correlation with each other is relatively distant. In fact, the r values between T6pS and DREB1 and between SHSP and DREB1 are 0.529 and 0.614, respectively, while the r value between T6pS and SHSP is 0.394. For Os10g0124000, a ribosomal protein, L7Ae also has a relatively lower r value, 0.348, with Os01g0968800. Fewer coexpressed genes at r ≥ 0.5 are found for Os01g0968800 and Os06g0219500, (849 and 354, respectively), while many more coexpressed genes at r ≥ 0.5 are found for Os02g0790500 (a total of 2,679). There are 459 genes coexpressed with Os01g0968800 and Os02g0790500, while 192 genes are coexpressed with Os01g0968800 and Os06g0219500 (Supplemental Table S16). These relationships are even clearer when the relational network is constructed with these three genes as seeds (Fig. 5B). These results suggest that genes involved in the same biological pressures (e.g. drought response) are closely regulated. Additionally, these data reveal that multiple seeds are as well handled in RAN as in single-seed analysis.
Figure 5.

Multiple seeded oligomers are used in RAN. A, B11031413 (oligomer ID Os046292_01; RAP2 ID Os01g0968800; DREB) is coexpressed with A03031616 (oligomer ID Os018703_01; RAP2 ID Os02g0790500; Tre6ps) and B07011319 (oligomer ID Os045474_01; RAP2 ID Os06g0219500; SHSF) at r ≥ 0.8 and depth = 1. B, Relational network of three coexpressed genes. DREB is coexpressed with Tre6ps and SHSF at r ≥ 0.7 and depth = 1. The pattern of edges of these genes shows their corelational subnetworks. The genes are denoted as filled circles, and their proximity represents the closeness of their relationship in the network, while the color of the edge shows the sign of the r value. A red edge denotes a positive r value of the gene pair, while green denotes a negative one. In addition, the color contrast and line thickness of an edge are deeper and thicker, respectively, as the absolute value of the r value is increased. [See online article for color version of this figure.]
DISCUSSION
Following the introduction of microarray technology in biology, much interest and effort have been devoted to describing the gene expression patterns of individual genes or groups of genes across genomic scales (Boldrick et al., 2002; Li et al., 2006). Furthermore, employing the complete spectrum of experimental conditions and materials in microarray databases and associated data, it has been possible to explore the relationships between genes involved in various biological systems of an organism, beyond the confines of the primary design of the individual experiments (Jen et al., 2006). We built the RAN database, which provides coexpression information between genes in terms of correlation coefficients for rice. Given the correlation between a pair of oligomers, the degrees of closeness between genes are visualized in a relational tree and a relational network. In addition, RAN supports scatterplots of log ratios between genes and links them to pathway maps, while providing a common cis-element list of promoter regions that are involved. We applied the RAN database to study the coexpression patterns in rice. For example, for Os10g0124000 and Os03g0241200, two of the 16-member L7Ae ribosomal protein family, 314 and 344 genes, respectively, were identified as being coexpressed, with correlation coefficients of r ≥ 0.5; many of these coexpressed genes encode ribosomal proteins, suggesting that these proteins are expressed in stoichiometric ratios for efficient translation, as seen in Arabidopsis. In contrast, other members of this gene family show lower numbers of coexpressed genes than seen for Os10g0124000 and Os03g0241200, indicating that coexpression can be partitioned across a gene family. To avoid bias in the analysis when performed under strict conditions for two different data sets, we selected the top-ranked 5% of the correlated genes of a “seed gene” in each database and compared the coexpressed genes within and between species. Major rice groups (MCL0–MCL2) have around 350 to 870 (1%−2%) genes within the species, while Arabidopsis has 710 to 910 (3%−4%) within the species. Rice has 12 members in the family and likely evolved more divergently between members. It is notable that an MCL3 member, Os07g0150200, which is further diverged in the phylogenetic tree, shares the lowest number of coexpressed genes not only with rice members but also with Arabidopsis members. Indeed, in comparison with Os01g0968800 (DREB1F), an external control that is likely to be expressed in response to drought, the coexpressed genes showed even lower numbers, while there are 118 commonly coexpressed genes by both rice major MCL groups and Arabidopsis MCL0. Analyses of the coexpressed genes by GO enriched terms, either by an individual gene (e.g. Os10g0124000) or by groups of genes, show that most of these GO terms are predominantly involved in protein synthesis, as protein binding, ribosomal constituents, and ribosomal RNA synthesis are included in these categories.
To evaluate the significance of the r value, a P value was generally used, taking a statistical perspective (Manfield et al., 2006), since it would have been difficult or impossible to analyze all of the actual data. However, the P value is determined from the r value and the data size alone, so the P value cannot represent the actual significance of the r value. Thus, we added the standard Z score, which reflects the actual distributional character of the r values of a gene. Since calculating Z scores takes a long processing time due to the huge number of calculations, we developed a calculation module in the C programming language. In an analysis of randomly selected sample data, we found that many data pairs of P values and Z scores show variations, although the two data sets have similar tendencies in general (Supplemental Fig. S3). For example, the r value between conserved hypothetical proteins (Os01g0642200 and Os021588_01) and metallothionein-like proteins (Os01g0974200 and Os000443_01) is 0.905, so its P value is highly significant, at 7.17e-69, while the Z scores of the r value are 3.32 and 3.14 for Os021588_01 and Os000443_01, respectively. This result shows that the P value does not reflect the significance of the r value as well as the Z score. Thus, if the long calculating time for Z scores can be overcome, it is more reasonable to use Z scores to evaluate the significance of the r value.
It is a challenge to determine how well the r value reflects the real correlation. We first adopted an r value from the distribution of the numbers of genes among eight r value bins. As the r value bins decrease from 0.7 to 0.2 in the coinduced regions (r values of 0.3–0.7 in Fig. 4), the skew in the number of genes in each of the eight r value bins shifts from right to left. In particular, the number of genes with coexpressed genes in the region (0, 100) is maximized at r ≥ 0.5, and the number of genes with coexpressed genes in the region (1,000, 4,000) begins to increase exponentially. We cautiously propose that the simple number of coexpressed genes at r ≥ 0.5 could be used as an indicator of the degree to which the gene is coexpressed with other genes within the genome. Interestingly, the histogram is roughly symmetric between the positive and inversely coexpressed regions, implying that the numbers of genes in the eight r value bins are very similar. This was manifested by comparing the number of coexpressed genes at r ≥ 0.5 and r ≤ −0.5 for the individual genes (Fig. 3B). Statistical inference strongly suggests that these numbers are correlated. This implies that, in general, if a gene is associated with a certain number of coinduced genes, then it is also associated with an almost equivalent number of genes that are inversely expressed. This may reflect the fact that many cellular signaling and expression machinery elements are known to have both positive and negative regulators (Li et al., 1994; Levitzki and Gazit, 1995; Torchia et al., 1997).
At least 18 genes in the family of rice and Arabidopsis were retrieved and first grouped with OrthoMCL. The program identifies identical proteins based on sequence similarities and distinguishes orthologs from paralog relationships without intensive computational phylogenetic analysis. The result suggests that the 16 members consist of four groups. For each individual gene, the coexpressed genes were compared with the Arabidopsis genes. While the numbers of correlated genes of rice in MCL0 range from 55 to 345 at r ≥ 0.5, those of the Arabidopsis genes range from 934 to 1,503. As a direct comparison is not easy, we first considered the number of coexpressed rice genes of Os10g0124000 at r ≥ 0.5. Most of these GO terms for the 315 coexpressed genes are predominantly involved in protein synthesis, and this is also consistent with the coexpressed genes in Arabidopsis (Jen et al., 2006). Because of differences between organisms, numbers of microarrays, methods of data presentation, and methods of hybridization and statistical handling, it is not easy to compare RAN for rice and ACT for Arabidopsis directly. Still, comparisons by different correlation thresholds and by considering the top-ranked 5% correlated genes in the two data sets reveal the similarity of coexpressed genes, which can probably be attributed to their similar biological functions. This similarity may imply that the functions overlap with each other in the identical groups. It is notable that the proportion of commonly expressed genes is at most 50%, even for the most commonly expressed genes (869), between Os05g0490100 in MCL1 and Os09g0507800 in MCL2. From analyses of coexpressed genes using RAN for rice and ACT for Arabidopsis, it is clear that the number of coexpressed genes varies from one gene to another, even if they belong to the same gene family or to identical groups within a species. The differences seen for the remainder of the genes (around 50%) may reflect the unique biological functions of each gene.
Beyond revealing the coexpressed gene characteristics of gene families, RAN can also be used for comparisons of gene expression patterns between rice and Arabidopsis. The gene expression values, as exemplified in the case of Os10g0124000 (Fig. 2), suggest the gene expression of the family is comparable with those values of genes from the Arabidopsis microarray collections such as AFGN and Genevestigator. The results show that, for both species, transcripts from the gene encoding a ribosomal protein L7Ae consistently decreased in specific organs compared with callus or anther tissues and decreased only slightly in seeds compared with callus and in response to abiotic stresses such as drought and cold (Fig. 4). These data show that RAN not only reveals the coexpressed gene characteristics of the gene family but can also be used in the comparison of gene expression patterns between rice and Arabidopsis.
As a model dicot plant, the Arabidopsis genome was sequenced (Arabidopsis Genome Initiative, 2000). Phylogenetic analysis by comparison of whole genome sequences showed that flowering plants, such as Arabidopsis and rice, followed their own evolutionary paths when monocot-dicot divergence occurred from a common ancestral angiosperm, roughly approximately 170 to 235 million years ago (Bowers et al., 2003; Freeling and Thomas, 2006). During the evolutionary path, the descendants of each angiosperm experienced different levels of chromosomal duplication and subsequent gene loss. Although Arabidopsis and rice have chromosomal synteny and there are many identical genes between these species, evolutionary events on the chromosomes have obscured the ortholog and paralog relations among genes. Orthologs supposedly had the same function or similar functions after the speciation event, and paralogs arose subsequently by duplications of orthologs, resulting in the production of gene families. Paralogous genes may or may not have the same functions, depending on selective pressure or their positioning on the genome. Without selective pressure, paralogous genes might evolve quickly and be endowed with the capacity to participate in and regulate a multitude of transcriptional programs. These sometimes make it complicated to determine which identical genes are orthologs or paralogs within or between organism(s). Information on orthologs and paralogs is a key point in the taxonomic classification of organisms. The comparison of coexpressed genes between rice and Arabidopsis in this analysis may reveal orthologs and homologs. The comparative analysis of coexpressed genes using L7Ae in RAN and ACT revealed that many of the coexpressed genes in rice are also ribosomal constituents or are involved in protein translation. Still, many of the correlated genes are unique to each species.
We also analyzed genes involved in the drought stress response in rice. As stress responses are complicated processes involving transcription factors, enzymes, and effectors (Agarwal et al., 2006; Kilian et al., 2007), we selected DREB, T6pS, and small heat shock factors (SHSF) as representative genes. The DREB, T6pS, and SHSF were retrieved from RAP2. The 143 members of the 20- to 26-kD SHSF family were also retrieved (data not shown) and grouped into 10 groups. Group 1 has 29 members. The number of coexpressed genes of DREB transcription factors ranged from 3 to 849, that of T6pS from 1 to 3,789, and that of SHSF from 1 to 1,032 at r ≥ 0.5. The coexpressed genes of Os01g0968800 (DREB1F) include Os02g0790500, a T6pS, and Os06g0219500, a SHSF. Interestingly, these genes are the representatives, within each group, that have the largest number of coexpressed genes, suggesting that genes with a high r value are under common coregulation mechanisms. The GO analysis of these genes is even more suggestive, as many of the terms are related to water homeostasis, protein folding, and response to heat.
The DREBs are important transcription factors that induce a set of abiotic stress-related genes and impart stress endurance to plants. The two DREB transcription factors, DREB1 and DREB2, are involved in two separate signal transduction pathways that respond under conditions of low temperature and dehydration, respectively (Agarwal et al., 2006). Thus, Os01g0968800, a DREB1F (belonging to a subfamily of DREB1), was thought to be involved in cold stress. In this study, it appears that this gene might also be involved in drought response. Recently, OsDREB1F was isolated and proven to be involved in both dehydration (drought, salt) and cold stresses (Wang et al., 2008), supporting our analysis. Overexpressed OsDREB1F in rice resulted in increased tolerance for drought, salt, and low temperatures, without any abnormality in the phenotype under nonstressed conditions (Wang et al., 2008). Besides, from the analysis of the DREB family in rice and Arabidopsis, we observed that coexpressed genes of OsDREB1F had the highest identity levels with the coexpressed genes of AtDREB2A (T.T.M. Pham and Y.-K. Kim, unpublished data). In previous studies, AtDREB2A was linked to functions in drought- and salinity-responsive gene expression but not cold stress. The overexpression of constitutively active DREB2A resulted in significant drought stress tolerance but only slight freezing tolerance in transgenic Arabidopsis plants (Sakuma et al., 2006). OsDREB1F and AtDREB2A are identical and function similarly. Thus, we hypothesized that OsDREB1F and AtDREB2A might be orthologs.
Several Web-based microarray analysis tools are currently available (Owen et al., 2003; Zimmermann et al., 2004; Srinivasasainagendra et al., 2008). Each Web site employs distinct algorithms to provide distinct functionalities. RAN might be a helpful data resource on coexpressed genes in correlated gene groups in rice, which are easily depicted in a relational tree and a network.
Future Developments
Phylogenetic analysis by comparison of whole genome sequences suggests that flowering plants such as Arabidopsis and rice followed their own evolutionary paths after the monocot-dicot divergence from a common ancestral angiosperm (Bowers et al., 2003; Freeling and Thomas, 2006). Although Arabidopsis and rice have chromosomal synteny in gene orders, and there are many identical genes between these species, evolutionary events in the chromosomes have confounded the ortholog and paralog relationships among the genes. If genes are involved in similar functions, the lists of genes that are coexpressed might be expected to overlap. Therefore, the RAN database could be used to evaluate coexpression patterns and provide valuable information regarding the orthologs and paralogs of given genes. In this way, the functional equivalents in model organisms could be identified, and the resulting information could be applied to other organisms to identify gene functions. To help the analysis, we are developing the function to quantify overlap in the lists of genes. As both rice and Arabidopsis are model organisms for monocotyledons and dicotyledons, respectively, coexpression information in RAN can be compared with the coexpression information from Arabidopsis. Future research will focus on which genes have more comparable coexpression patterns with which others.
CONCLUSION
RAN is a data resource that provides information on coexpressed genes in rice, based on two-dye microarray data. The closeness of coexpression between two genes is represented by the correlation coefficient and the statistical significance of the r value. The correlated gene groups are conveniently depicted in a relational tree and a relational network. RAN not only reveals the coexpressed gene characteristics of the gene family but can also be used in the comparison of gene expression between rice and Arabidopsis. Coexpression patterns in stress-related genes responding to the same biological pressures are shown to be regulated together. These results show that data obtained from a given experimental design could be cross-checked in RAN, and a new experiment could then easily be designed. Moreover, RAN is designed to be extended to other related plants, and the same database structure can be applied to construct a comprehensive resource on expression correlation between genes in any organism.
MATERIALS AND METHODS
Microarray Data
As of April 2008, expression data from 183 microarrays were collected using either wild-type or mutant rice (Oryza sativa) organs, such as the leaf, root, flower, and callus, at various developmental stages. Various treatments were applied to the plant under research conditions. These include biotic and abiotic stresses and hormones (Supplemental Table S1). The microarray data are processed as described previously (Jung et al., 2005). Briefly, the expression profiling is conducted with the 60k Rice Whole Genome Microarray. The 60k microarray is designed to represent all of the genes in rice. In total, 60,727 oligomers are designed from gene-specific regions of both japonica and indica subspecies. These include 58,417 from known and predicted genes and 66 randomized DNA oligomers. Out of these genes, 2,310 are also designed as antisense oligomers. The oligomer sequences are extracted by Qiagen-Operon based on rice genome information from the Beijing Genomics Institute. The oligomers are synthesized and purified by Qiagen-Operon and spotted on SuperAmine slides using the facilities of David Galbraith at the University of Arizona (http://ag.arizona.edu/microarray/deconvolution.html). A set of two slides of the 60k microarray has 64,896 spot addresses. Each slide is formatted with 48 12 × 4 blocks composed of spots (4,099), which are also included for easy scanning alignment. An oligomer 70 nucleotides long with an average melting temperature of 78°C is printed in each spot address, with a diameter of 100 μm.
Noncorrelation of the signal and background intensities is confirmed by plotting base 2 log background intensity on the x axis and base 2 log intensity subtracted from background intensity on the y axis. Before normalization, the normal distribution and linear relations of the Cy3 and Cy5 intensities are tested by qqplot and a linear regression model, respectively, in the R statistical language (http://cran.r-project.org). The spatial effects on the chip during the hybridization process are checked with spatial.func in the SMA package. The variance differences between the Cy3 and Cy5 intensities within the microarray are tested with the t test under the assumptions of both uniform and nonuniform variances. One- and two-way ANOVAs of the signal intensity differences between microarrays were performed. The median pixel intensities are transformed as log ratios with base 2 and then adjusted by block-by-block Lowess normalization for each slide (Yang et al., 2002). To improve the specificity of our statistical hypothesis in low-intensity regions, we adopt the following empirical criteria: a spot is selected if it is not flagged for its morphology, its diameter is larger than 51 pixels, and the intensities of both signals are higher than 500. Multivariate statistical tests, such as clustering, principal component analysis, and multidimensional scaling, are performed with Acuity 3.1 (Axon Instruments).
RAN Implementation
Microarray data are collected and processed as described previously (Jung et al., 2005). In order to evaluate the expression relationships between genes of interest and their targets, RAN calculates the Pearson correlation coefficients (denoted as r) of the log ratios between a pair of oligomers representing genes. When all the oligomers are matched to the recently annotated RAP2 database, the 33,689 oligomers unanimously matched to genes in the database. However, as the 60k microarray contains oligomers that are highly correlated with other oligomers but do not match to the currently annotated genes, we calculated the r value based on this assumption. It is known that the two microarray data sets in a gene pair are independent samples from a bivariate normal distribution in which the variables of two data sets have zero covariance (Jen et al., 2006). Thus, the P value of a correlation coefficient is calculated based on the Student's t distribution, with the numerical formula:
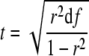 |
An additional standard score, the Z score, of the r value of a gene pair (designated “query pair”) is calculated from the r values of all gene pairs including a given gene to determine the Z score in the query pair and the rest of the 58,416 genes. According to the central limit theorem, the distribution of all possible r values of a gene should be a normal distribution because the sample size is large enough, and we confirmed that most of the distributions are normal (Supplemental Table S4). Thus, the Z score is calculated with the mean (μ) and sd (σ) of the distribution, with the numerical formula:
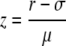 |
To draw a relational tree, RAN first searches relational genes with a minimum absolute r value and a depth set by a researcher from each gene (denoted as the seed) among the query genes selected by the researcher and makes a list from all the genes together. An option for “depth” is provided that determines the genes directly coexpressed: primary with the input gene and secondary with the genes coexpressed with the primary gene. Subsequently, a distance table, based on the r values between all the listed genes, is created. The distance (designated as d) between any two genes is determined by the formula d = 1 − | r | (D'haeseleer, 2005). Next, RAN transforms the distance table into a tree data file containing branch length values representing relative degrees of relationship between genes, using the “neighbor” program in the Phylip package (version 3.67; Felsenstein, 1989), with the option of a neighbor-joining method (Saitou and Nei, 1987). Based on the tree file, a tree is constructed by the A Tree Viewer (ATV) java applet program (version 4.1.04; Zmasek and Eddy, 2001) at the RAN Web site. The first two processes in drawing the relational network, namely listing the relational genes and calculating the r values between all the listed genes, are the same in the process of drawing the relational tree. However, the network is drawn with the r values between genes in a gene pair by Networkx (http://networkx.lanl.gov/) and Matplotlib (http://matplotlib.sourceforge.net), without calculating distances with the r values. Correlated genes are denoted by filled circles in a network graph and are closely located in the graph with a colored edge that represents the sign and degree of the r value, by a force-directed algorithm (spring algorithm). The color of the edge between genes is red when the r value between the genes is positive and green when the r value is negative. In addition, the color and thickness of an edge are deeper and thicker, respectively, as the absolute value of the r value is increased. To link the retrieved genes to a biological pathway, BLASTp is performed against the Swiss Protein database; each gene was mapped to its EC number and linked to a pathway map in the KEGG database (Kanehisa and Goto, 2000). To compare the cis-elements between correlated genes, we first mapped all oligomers to genes in Pseudomolecule 5 (TIRG release), extracted the 1-kb upstream region from the first codon of each mapped gene, and stored the data in the database. Next, RAN predicted the cis-elements using the Signal Scan program against the PLACE database (Higo et al., 1999) and listed the frequent cis-elements with their ranks, whose frequency among the promoters of coexpressed genes is greater than half the number of promoters.
Analysis of Ortholog Groups
Genes in a family were retrieved from RAP2 rice genome annotation (http://rapdb.dna.affrc.go.jp/) or TAIR Arabidopsis (Arabidopsis thaliana) genome annotation version 8 (http://www.arabidopsis.org/) using keywords. To draw the phylogenic tree, a two-step analysis is applied. First, ortholog groups are tested with OrthoMCL (http://www.orthomcl.org/). This analysis compartmentalizes the family into two groups. Second, the amino acid sequences of each group are aligned with ClustalW, and then a distance matrix of the alignment is calculated using the ‘protdist’ program in the Phylib package. The matrix is transformed into a tree by the neighbor program. The tree is tested by bootstrap 1,000 by the ‘seqboot’ program. The bootstrapping values are reported in place of the branch lengths.
Comparison of Coexpressed Genes of Ribosomal Protein L7Ae
To test coexpressed genes with a ribosomal protein L7Ae, Os10g0124000 (oligomer ID Os056379_01; spot no. B11032220), lists of clustered genes were retrieved using Os10g0124000 (oligomer ID Os056379_01; spot no. B11032220) as the input word or “seed,” as shown in Figure 1.
To test coexpressed genes in the ribosomal protein L7Ae family within and between rice and Arabidopsis, the top-ranked 5% of coexpressed genes within each family were retrieved from RAN and ACT. To reduce the variation caused by oligomers from predicted genes in the Rice 60k Microarray, only the 33,689 oligomers that unanimously matched the recently annotated RAP2 are used. In the Arabidopsis design, ATH1 contains 22,765 genes that are positively identified by the chip design. The numbers of the top-ranked 5% of genes are 1,684 and 1,138 for rice and Arabidopsis, respectively. These numbers of genes are used to compare coexpressed genes with the ribosomal protein L7Ae family within and between rice and Arabidopsis. Os01g0968800 (DREB1F), known to be expressed in response to drought, is used as an external control. In the comparison of the coexpressed genes between species, BLASTp analysis is performed for the two species, and genes with scores of 100 or higher are considered to be the tentative counterparts (Supplemental Table S4). GO analysis was performed with GoMiner (Ashburner et al., 2000; Zeeberg et al., 2003). The fdr values were obtained from 100 randomizations.
Comparison of Gene Expression of Ribosomal Protein L7Ae in Rice and Arabidopsis
Rice microarray log ratios were retrieved for those used in the calculation of r values as shown in Figure 2A. Initial analysis using Os10g0124000 suggests that the gene for the ribosomal protein L7Ae consistently decreased in specific organs, compared with the callus or anther. We also searched the expression values of the gene under conditions of plant hormone, ABA, and abiotic stresses such as drought and cold to test the ribosomal protein expression. A drought-responsive transcription factor, Os01g0968800, was also searched. The microarray sets ranged from 3 to 12. The distribution of the correlation coefficients for these genes varies in response to stress or at different developmental stages. To avoid bias in the analysis, microarray collections in AFGN and Genevestigator were searched for those sets performed with similar organs at similar developmental stages and experimental conditions (Supplemental Table S9). For example, organs and tissues are directly retrieved from the average values from the Web site. The lemma and palea of rice are sepal equivalents in Arabidopsis that nourish and protect florets and developing kernels. As experiments for drought stress, RNA samples are prepared from rice leaves after stress treatment for 2 to 6 h, and the values for drought for Arabidopsis are retrieved from the values marked as “Stress: drought_green_early,” in which leaves are harvested at 0.5, 1, and 3 h after the onset of treatment. As microarray data in RAN are prepared by the two-dye method depicted in Figure 4, the microarray values of Arabidopsis are compared with the control rice data set and transformed by log2 transformation.
Supplemental Data
The following materials are available in the online version of this article.
Supplemental Figure S1. ClustalW results of 16 ortholog genes of ribosomal protein L7Ae.
Supplemental Figure S2. Phylogenetic tree of 16 ortholog genes of ribosomal protein L7Ae.
Supplemental Figure S3. Scatterplot between P value and Z score.
Supplemental Table S1. Microarray collection in the RiceArrayNet.
Supplemental Table S2. List of genes correlated with Os10g0124000 in Figure 1.
Supplemental Table S3. Partial list of common cis-elements between DREB1 and T6pS.
Supplemental Table S4. The number of genes, depending on tallied r values, for every oligomer on the microarray (Partial List).
Supplemental Table S5. Groups of a ribosomal protein L7Ae in rice and Arabidopsis.
Supplemental Table S6. Number of genes coexpressed with a ribosomal protein in rice and Arabidopsis.
Supplemental Table S7. List of the genes retrieved with a ribosomal protein L7Ae.
Supplemental Table S8. Log2-based ratios of a ribosomal protein L7Ae in RAN.
Supplemental Table S9. Comparison of tissues or developmental stages between rice and Arabidopsis.
Supplemental Table S10. The expression values of the Arabidopsis ribosomal proteins L7Ae.
Supplemental Table S11. Comparison of log2-based ratios of a ribosomal protein L7Ae obtained from microarray sets of rice and Arabidopsis.
Supplemental Table S12. List of genes r ≥ 0.5 among DREB genes.
Supplemental Table S13. List of 559 genes identified in RAP2, among 849 correlated genes of Os01g0968800 (a DREB).
Supplemental Table S14. The 11 trehalose-6-phosphate synthase genes extracted from rice RAP2.
Supplemental Table S15. List of 3,789 coexpressed genes of a trehalose-6-phosphate synthase.
Supplemental Table S16. List of coexpressed genes by both a DREB1 and a T6pS.
Supplementary Material
Acknowledgments
We thank Drs. I.W. Manfield and J.R. Bradford of ACT at the University of Leeds for their helpful advice.
This work was supported by the Crop Functional Genomics Center of the Frontier Research Program, funded by the Ministry of Science and Technology (grant no. CG1210 to M.K. and grant no. CG1122 to B.H.N.), by the BioGreen21 Program (grant no. 20070401034008 to Y.-K.K. and grant no. 20090101060028 to B.H.N.), by the Rural Development Administration of the Republic of Korea, and by the Brain Korea 21 Project (grants to T.-H.L. and B.H.N.).
The author responsible for distribution of materials integral to the findings presented in this article in accordance with the policy described in the Instructions for Authors (www.plantphysiol.org) is: Baek Hie Nahm (bhnahm@mju.ac.kr).
Some figures in this article are displayed in color online but in black and white in the print edition.
The online version of this article contains Web-only data.
Open Access articles can be viewed online without a subscription.
References
- Agarwal PK, Agarwal P, Reddy MK, Sopory SK (2006) Role of DREB transcription factors in abiotic and biotic stress tolerance in plants. Plant Cell Rep 25 1263–1274 [DOI] [PubMed] [Google Scholar]
- Arabidopsis Genome Initiative (2000) Analysis of the genome sequence of the flowering plant Arabidopsis thaliana. Nature 408 796–815 [DOI] [PubMed] [Google Scholar]
- Ashburner M, Ball CA, Blake JA, Botstein D, Butler H, Cherry JM, Davis AP, Dolinski K, Dwight SS, Eppig JT, et al (2000) Gene Ontology: tool for the unification of biology. The Gene Ontology Consortium. Nat Genet 25 25–29 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barakat A, Szick-Miranda K, Chang IF, Guyot R, Blanc G, Cooke R, Delseny M, Bailey-Serres J (2001) The organization of cytoplasmic ribosomal protein genes in the Arabidopsis genome. Plant Physiol 127 398–415 [PMC free article] [PubMed] [Google Scholar]
- Barrett T, Troup DB, Wilhite SE, Ledoux P, Rudnev D, Evangelista C, Kim IF, Soboleva A, Tomashevsky M, Marshall KA, et al (2009) NCBI GEO: archive for high-throughput functional genomic data. Nucleic Acids Res 37 D885–D890 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boldrick JC, Alizadeh AA, Diehn M, Dudoit S, Liu CL, Belcher CE, Botstein D, Staudt LM, Brown PO, Relman DA (2002) Stereotyped and specific gene expression programs in human innate immune responses to bacteria. Proc Natl Acad Sci USA 99 972–977 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bowers JE, Chapman BA, Rong J, Paterson AH (2003) Unravelling angiosperm genome evolution by phylogenetic analysis of chromosomal duplication events. Nature 422 433–438 [DOI] [PubMed] [Google Scholar]
- Bussemaker HJ, Li H, Siggia ED (2001) Regulatory element detection using correlation with expression. Nat Genet 27 167–171 [DOI] [PubMed] [Google Scholar]
- Clark TA, Sugnet CW, Ares M Jr (2002) Genomewide analysis of mRNA processing in yeast using splicing-specific microarrays. Science 296 907–910 [DOI] [PubMed] [Google Scholar]
- d'Erfurth I, Jolivet S, Froger N, Catrice O, Novatchkova M, Simon M, Jenczewski E, Mercier R (2008) Mutations in AtPS1 (Arabidopsis thaliana parallel spindle 1) lead to the production of diploid pollen grains. PLoS Genet 4 e1000274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- D'haeseleer P (2005) How does gene expression clustering work? Nat Biotechnol 23 1499–1501 [DOI] [PubMed] [Google Scholar]
- Eisen MB, Spellman PT, Brown PO, Botstein D (1998) Cluster analysis and display of genome-wide expression patterns. Proc Natl Acad Sci USA 95 14863–14868 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Felsenstein J (1989) PHYLIP: Phylogeny Inference Package (version 3.2). Cladistics 5 164–166 [Google Scholar]
- Freeling M, Thomas BC (2006) Gene-balanced duplications, like tetraploidy, provide predictable drive to increase morphological complexity. Genome Res 16 805–814 [DOI] [PubMed] [Google Scholar]
- Goda H, Sasaki E, Akiyama K, Maruyama-Nakashita A, Nakabayashi K, Li W, Ogawa M, Yamauchi Y, Preston J, Aoki K, et al (2008) The AtGenExpress hormone and chemical treatment data set: experimental design, data evaluation, model data analysis and data access. Plant J 55 526–542 [DOI] [PubMed] [Google Scholar]
- Higo K, Ugawa Y, Iwamoto M, Korenaga T (1999) Plant cis-acting regulatory DNA elements (PLACE) database: 1999. Nucleic Acids Res 27 297–300 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hirai MY, Sugiyama K, Sawada Y, Tohge T, Obayashi T, Suzuki A, Araki R, Sakurai N, Suzuki H, Aoki K, et al (2007) Omics-based identification of Arabidopsis Myb transcription factors regulating aliphatic glucosinolate biosynthesis. Proc Natl Acad Sci USA 104 6478–6483 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Horan K, Jang C, Bailey-Serres J, Mittler R, Shelton C, Harper JF, Zhu JK, Cushman JC, Gollery M, Girke T (2008) Annotating genes of known and unknown function by large-scale coexpression analysis. Plant Physiol 147 41–57 [DOI] [PMC free article] [PubMed] [Google Scholar]
- International Rice Genome Sequencing Project (2005) The map-based sequence of the rice genome. Nature 436 793–800 [DOI] [PubMed] [Google Scholar]
- Jang IC, Oh SJ, Seo JS, Choi WB, Song SI, Kim CH, Kim YS, Seo HS, Choi YD, Nahm BH, et al (2003) Expression of a bifunctional fusion of the Escherichia coli genes for trehalose-6-phosphate synthase and trehalose-6-phosphate phosphatase in transgenic rice plants increases trehalose accumulation and abiotic stress tolerance without stunting growth. Plant Physiol 131 516–524 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jen CH, Manfield IW, Michalopoulos I, Pinney JW, Willats WG, Gilmartin PM, Westhead DR (2006) The Arabidopsis co-expression tool (ACT): a WWW-based tool and database for microarray-based gene expression analysis. Plant J 46 336–348 [DOI] [PubMed] [Google Scholar]
- Jiao Y, Ma L, Strickland E, Deng XW (2005) Conservation and divergence of light-regulated genome expression patterns during seedling development in rice and Arabidopsis. Plant Cell 17 3239–3256 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jung KH, Han MJ, Lee YS, Kim YW, Hwang I, Kim MJ, Kim YK, Nahm BH, An G (2005) Rice Undeveloped Tapetum1 is a major regulator of early tapetum development. Plant Cell 17 2705–2722 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kanehisa M, Goto S (2000) KEGG: Kyoto Encyclopedia of Genes and Genomes. Nucleic Acids Res 28 27–30 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kilian J, Whitehead D, Horak J, Wanke D, Weinl S, Batistic O, D'Angelo C, Bornberg-Bauer E, Kudla J, Harter K (2007) The AtGenExpress global stress expression data set: protocols, evaluation and model data analysis of UV-B light, drought and cold stress responses. Plant J 50 347–363 [DOI] [PubMed] [Google Scholar]
- Koo AJ, Chung HS, Kobayashi Y, Howe GA (2006) Identification of a peroxisomal acyl-activating enzyme involved in the biosynthesis of jasmonic acid in Arabidopsis. J Biol Chem 281 33511–33520 [DOI] [PubMed] [Google Scholar]
- Kotak S, Larkindale J, Lee U, von Koskull-Doring P, Vierling E, Scharf KD (2007) Complexity of the heat stress response in plants. Curr Opin Plant Biol 10 310–316 [DOI] [PubMed] [Google Scholar]
- Lee SI, Batzoglou S (2003) Application of independent component analysis to microarrays. Genome Biol 4 R76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Levitzki A, Gazit A (1995) Tyrosine kinase inhibition: an approach to drug development. Science 267 1782–1788 [DOI] [PubMed] [Google Scholar]
- Li L, Stoeckert CJ Jr, Roos DS (2003) OrthoMCL: identification of ortholog groups for eukaryotic genomes. Genome Res 13 2178–2189 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li M, Moyle H, Susskind MM (1994) Target of the transcriptional activation function of phage lambda cI protein. Science 263 75–77 [DOI] [PubMed] [Google Scholar]
- Li X, Duan X, Jiang H, Sun Y, Tang Y, Yuan Z, Guo J, Liang W, Chen L, Yin J, et al (2006) Genome-wide analysis of basic/helix-loop-helix transcription factor family in rice and Arabidopsis. Plant Physiol 141 1167–1184 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Manfield IW, Jen CH, Pinney JW, Michalopoulos I, Bradford JR, Gilmartin PM, Westhead DR (2006) Arabidopsis Co-expression Tool (ACT): Web server tools for microarray-based gene expression analysis. Nucleic Acids Res 34 W504–W509 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mangelsen E, Kilian J, Berendzen KW, Kolukisaoglu UH, Harter K, Jansson C, Wanke D (2008) Phylogenetic and comparative gene expression analysis of barley (Hordeum vulgare) WRKY transcription factor family reveals putatively retained functions between monocots and dicots. BMC Genomics 9 194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marchler-Bauer A, Anderson JB, Cherukuri PF, DeWeese-Scott C, Geer LY, Gwadz M, He S, Hurwitz DI, Jackson JD, Ke Z, et al (2005) CDD: a Conserved Domain Database for protein classification. Nucleic Acids Res 33 D192–D196 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mutwil M, Obro J, Willats WG, Persson S (2008) GeneCAT: novel webtools that combine BLAST and co-expression analyses. Nucleic Acids Res 36 W320–W326 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Obayashi T, Hayashi S, Saeki M, Ohta H, Kinoshita K (2009) ATTED-II provides coexpressed gene networks for Arabidopsis. Nucleic Acids Res 37 D987–D991 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Owen AB, Stuart J, Mach K, Villeneuve AM, Kim S (2003) A gene recommender algorithm to identify coexpressed genes in C. elegans. Genome Res 13 1828–1837 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qin F, Sakuma Y, Tran LS, Maruyama K, Kidokoro S, Fujita Y, Fujita M, Umezawa T, Sawano Y, Miyazono K, et al (2008) Arabidopsis DREB2A-interacting proteins function as RING E3 ligases and negatively regulate plant drought stress-responsive gene expression. Plant Cell 20 1693–1707 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rawat A, Seifert GJ, Deng Y (2008) Novel implementation of conditional co-regulation by graph theory to derive co-expressed genes from microarray data. BMC Bioinformatics (Suppl 9) 9 S7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rhee SY, Beavis W, Berardini TZ, Chen G, Dixon D, Doyle A, Garcia-Hernandez M, Huala E, Lander G, Montoya M, et al (2003) The Arabidopsis Information Resource (TAIR): a model organism database providing a centralized, curated gateway to Arabidopsis biology, research materials and community. Nucleic Acids Res 31 224–228 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saitou N, Nei M (1987) The neighbor-joining method: a new method for reconstructing phylogenetic trees. Mol Biol Evol 4 406–425 [DOI] [PubMed] [Google Scholar]
- Sakuma Y, Maruyama K, Osakabe Y, Qin F, Seki M, Shinozaki K, Yamaguchi-Shinozaki K (2006) Functional analysis of an Arabidopsis transcription factor, DREB2A, involved in drought-responsive gene expression. Plant Cell 18 1292–1309 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schena M, Shalon D, Davis RW, Brown PO (1995) Quantitative monitoring of gene expression patterns with a complementary DNA microarray. Science 270 467–470 [DOI] [PubMed] [Google Scholar]
- Schmid M, Davison TS, Henz SR, Pape UJ, Demar M, Vingron M, Schölkopf B, Weigel D, Lohmann JU (2005) A gene expression map of Arabidopsis thaliana development. Nat Genet 37 501–506 [DOI] [PubMed] [Google Scholar]
- Slonim N, Atwal GS, Tkacik G, Bialek W (2005) Information-based clustering. Proc Natl Acad Sci USA 102 18297–18302 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Srinivasasainagendra V, Page GP, Mehta T, Coulibaly I, Loraine AE (2008) CressExpress: a tool for large-scale mining of expression data from Arabidopsis. Plant Physiol 147 1004–1016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Steinhauser D, Usadel B, Luedemann A, Thimm O, Kopka J (2004) CSB.DB: a comprehensive systems-biology database. Bioinformatics 20 3647–3651 [DOI] [PubMed] [Google Scholar]
- Takabayashi A, Ishikawa N, Obayashi T, Ishida S, Obokata J, Endo T, Sato F (2009) Three novel subunits of Arabidopsis chloroplastic NAD(P)H dehydrogenase identified by bioinformatic and reverse genetic approaches. Plant J 57 207–219 [DOI] [PubMed] [Google Scholar]
- Torchia J, Rose DW, Inostroza J, Kamei Y, Westin S, Glass CK, Rosenfeld MG (1997) The transcriptional co-activator p/CIP binds CBP and mediates nuclear-receptor function. Nature 387 677–684 [DOI] [PubMed] [Google Scholar]
- Toufighi K, Brady SM, Austin R, Ly E, Provart NJ (2005) The Botany Array Resource: e-northerns, expression angling, and promoter analyses. Plant J 43 153–163 [DOI] [PubMed] [Google Scholar]
- Wang Q, Guan Y, Wu Y, Chen H, Chen F, Chu C (2008) Overexpression of a rice OsDREB1F gene increases salt, drought, and low temperature tolerance in both Arabidopsis and rice. Plant Mol Biol 67 589–602 [DOI] [PubMed] [Google Scholar]
- Yang YH, Dudoit S, Luu P, Lin DM, Peng V, Ngai J, Speed TP (2002) Normalization for cDNA microarray data: a robust composite method addressing single and multiple slide systematic variation. Nucleic Acids Res 30 e15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zeeberg BR, Feng W, Wang G, Wang MD, Fojo AT, Sunshine M, Narasimhan S, Kane DW, Reinhold WC, Lababidi S, et al (2003) GoMiner: a resource for biological interpretation of genomic and proteomic data. Genome Biol 4 R28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zimmermann P, Hirsch-Hoffmann M, Hennig L, Gruissem W (2004) Genevestigator: Arabidopsis microarray database and analysis toolbox. Plant Physiol 136 2621–2632 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zmasek CM, Eddy SR (2001) ATV: display and manipulation of annotated phylogenetic trees. Bioinformatics 17 383–384 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
{kind=link}
{kind=link}
{kind=link}