

Abstract
Functionalized, dendrimer-stabilized gold nanoparticles (Au DSNPs) are of scientific and technological interest for biological applications. In this work, we show that acetamide-functionalized Au DSNPs can be formed by acetylation of amine-terminated poly(amidoamine) (PAMAM) dendrimers of generation 5 (G5.NH2) complexed with Au(III) ions (AuCl4−). In addition, hydroxyl-functionalized Au DSNPs can be formed by simply mixing the glycidol hydroxyl-terminated G5 dendrimers (G5.NGlyOH) with HAuCl4. In both cases, no additional reducing agents were needed and the reactions were completed at room temperature. We also show that Alexa Fluor 594 dye-functionalized Au DSNPs can be formed by acetylation of Alexa Fluor 594-conjugated, amine-terminated G5 dendrimers complexed with HAuCl4. All of these functionalized Au DSNPs are water-soluble and stable. Fluorescence spectroscopy studies reveal that the Alexa Fluor 594-functionalized Au DSNPs retain similar fluorescence intensity to the Alexa Fluor 594-functionalized dendrimers that lack Au nanoparticles. These preparations of Au DSNPs provide a straightforward approach to synthesizing functionalized metal nanoparticles for biomedical applications.
Introduction
Gold (Au) nanoparticles (NPs) have recently received interest for applications in biology, catalysis, and nanotechnology because of their unique optical, electronic, and quantum size-related properties.1–9 The surface of Au NPs requires modification with different molecules to function in various applications. In most cases, this surface modification is often performed onto preformed Au NPs through thiol derivatization.10,11 However, the relatively poor long-term stability of Au–S bonds12 and the multiple synthetic steps required for the final functionalization of Au NPs limit their biomedical applications.
It is well-demonstrated that poly(amidoamine) (PAMAM) dendrimers can be used as either templates or stabilizers to prepare Au or other inorganic NPs.13–30 In general, the dendrimer-entrapped Au NPs (Au DENPs) involve a nanostructure where one dendrimer molecule entraps one or more Au NPs with the metal core size usually smaller than 5 nm.13,26–28,31 In contrast, the dendrimer-stabilized Au NPs (Au DSNPs) refer to a nanostructure where one Au NP is stabilized by multiple dendrimer molecules (the metal NP diameter is usually larger than 5 nm).15,19,22 Both Au DENPs and Au DSNPs are commonly prepared by using amine-terminated PAMAM dendrimers as templates or stabilizers. To overcome the toxicity and nonspecific cell membrane binding in biological systems, it is necessary to neutralize the amine surface of Au NPs. In a previous work, we have shown that Au DENPs (2.1 nm in diameter) can be functionalized with acetamide and hydroxyl groups by reacting them with acetic anhydride and glycidol, respectively, thereby significantly improving their biocompatibility.28 We have also shown that biofunctionalized Au DENPs with neutral surface charges (surface potential close to zero) can be used as a platform for cancer cell targeting and imaging.27 It is expected that functionalization of Au DSNPs with a neutral surface may also be of biological importance for cell labeling or tracking studies.
Au NPs with different surfaces can be prepared by using functionalized dendrimers as templates or stabilizers.32,33 However, in these cases, additional reducing agents or physical treatments are necessary to assist the formation of NPs. Recent reports suggest that certain polymers or copolymers can simultaneously act as both a reducing agent and a stabilizer for the synthesis of Au NPs.12,34–41 In some cases, Au NPs can be formed by simply mixing the aqueous solutions of copolymers and HAuCl4 at room temperature.12 This implies that by appropriately manipulating the dendrimer surface chemistry, it may be possible to synthesize Au NPs through essentially a single-step approach.
In this present study, we have developed an approach to the formation and functionalization of Au DSNPs that essentially occurs as a spontaneous process (Scheme 1). Au DSNPs with acetamide surfaces were formed by acetylation of amine-terminated PAMAM dendrimers of generation 5 (G5.NH2) complexed with AuCl4− ions, while hydroxyl-functionalized Au DSNPs were formed by simply mixing the glycidol hydroxyl-terminated G5 dendrimers (G5.NGlyOH) with AuCl4− ions. In both cases, no additional reducing agents or physical treatment were needed and the reactions were performed at room temperature. To further prove the concept of the spontaneous formation of functionalized Au DSNPs, fluorescent Alexa Fluor 594 (AF594)-functionalized Au DSNPs were formed by acetylation of AF594-conjugated, amine-terminated G5 dendrimers complexed with HAuCl4. The spontaneously formed functionalized Au DSNPs are stable and water-soluble. This simple approach may be used to directly synthesize and functionalize Au DSNPs for biomedical applications. In addition, recent advances show that dendrimers or dendrimer-protected Au NPs can be assembled to form complex layered structures.42–44 Our work with the spontaneous formation of Au DSNPs could also be useful for the assembly of Au NP-related complex architectures for various applications.
SCHEME 1. Schematic Representation of the Approaches to Synthesizing {(Au0)7-G5.NHAc} (a), {(Au0)7-G5.NGlyOH} (b), and {(Au0)10-G5.NHAc-AF594} DSNPs (c).

Experimental Section
Materials
Ethylenediamine core amine-terminated PAMAM dendrimers of generation 5 (G5.NH2) with a polydispersity index less than 1.08 were purchased from Dendritech (Midland, MI). Hydroxyl-terminated G5 dendrimers (G5.NGlyOH) and acetamide-terminated G5 dendrimers (G5.NHAc) were prepared by reacting G5.NH2 dendrimers with glycidol and acetic anhydride, respectively, and were characterized as previously reported.45,46 AF594 succinimidyl ester was purchased from Molecular Probes (Eugene, OR). All other chemicals were obtained from Aldrich and were used as received. Water used in all of the experiments was purified with use of a Milli-Q Plus 185 water purification system (Millipore, Bedford, MA) with resistivity higher than 18 MΩ·cm. Regenerated cellulose dialysis membranes (MWCO = 10000) were acquired from Fisher.
Synthesis of Au DSNPs
Synthesis of Acetamide-Functionalized Au DSNPs (Scheme 1a)
G5.NH2–Au(III) complexes were first prepared by mixing a methanol solution of HAuCl4 (3 mL, 1.7 mM) with a methanol solution of the G5.NH2 dendrimers (3 mL, 0.24 mM) at a 1:7 molar ratio of G5.NH2/Au atoms. The formed complex was identified as {(Au3+)7-G5.NH2}. The primary amine groups of the {(Au3+)7-G5.NH2} complexes were acetylated as previously reported.47 Briefly, 65 μL of triethylamine was added to the formed {(Au3+)7-G5.NH2} solution. A methanolic solution (0.5 mL) of acetic anhydride (48 mg, 500% molar excess of the total primary amines of {(Au3+)7-G5.NH2}) was added dropwise into the {(Au3+)7-G5.NH2}/triethylamine mixture while it was being stirred vigorously, and the mixture was allowed to react for 24 h. The mixture was spontaneously changed to pink after 6 h, indicating the formation of Au NPs. The methanol and the excess of reactants and byproduct were removed from the mixture by extensive dialysis against PBS buffer (3 times, 4 L) and water (3 times, 4 L) for 3 days, followed by lyophilization to obtain the {(Au0)7-G5.NHAc} DSNPs.
Synthesis of Hydroxyl-Functionalized Au DSNPs (Scheme 1b)
Hydroxyl-functionalized Au DSNPs were prepared by mixing a methanol solution of G5.NGlyOH dendrimers (3 mL, 0.24 mM) with a methanol solution of HAuCl4 (3 mL, 1.7 mM). The mixture was stirred vigorously for 24 h. After 12 h, the solution changed to a deep red color, suggesting the spontaneous formation of Au NPs. The mixture was then dried under a gentle N2 stream and subsequently redissolved into water, followed by lyophilization. The formed Au NPs were denoted as {(Au0)7-G5.NGlyOH}.
Synthesis of AF594-Functionalized Au DSNPs (Scheme 1c)
AF594-conjugated amine-terminated G5 dendrimers were synthesized according to previous reports with slight modification.48 Briefly, 3 molar equiv of AF594 succinimidyl ester (5 mg, 6.10 μmol) dissolved in methanol (4 mL) were added dropwise to a solution of G5.NH2 dendrimer (54.46 mg, 2.03 μmol) in methanol (4 mL) in a nitrogen atmosphere under vigorous magnetic stirring. After 24 h, the reaction mixture (8 mL) was divided into 2 aliquots of equal volume (4 mL). For aliquot 1, the AF594-labeled G5.NH2 dendrimers complexed with HAuCl4 were acetylated to convert the remaining amino groups of G5 dendrimer into acetamide groups, according to a procedure described in the literature.47 Briefly, aliquot 1 was added to a 4-mL methanol solution of HAuCl4 (4.02 mg, 10.2 μmol) then the solution was mixed well for 30 min. Next, triethylamine (66.0 mg, 0.65 mmol) was added to the mixture and the solution mixed well, followed by dropwise addition of an acetic anhydride solution (66.7 mg, 0.65 mmol, in 0.5 mL of methanol) under vigorous stirring. After 24 h, the reaction mixture was extensively dialyzed against PBS buffer (3 times, 4 L) and water (3 times, 4 L), using a regenerated cellulose dialysis membrane (MWCO = 10000), for 3 days to remove the excess of reactants and byproduct, followed by lyophilization to obtain a product of {(Au0)10-G5.NHAc-AF594} DSNPs. Aliquot 2 was processed by using the same procedure without adding HAuCl4 to obtain the control product of G5.NHAc-AF594 dendrimers.
Note that the number 7 or 10 in the nomenclature is the number of Au atoms per dendrimer molecule, according to the reaction stoichiometry. The denotation of {(Au0)7-G5.NHAc}, {(Au0)7-G5.NGlyOH}, and {(Au0)10-G5.NHAc-AF594} does not necessarily represent any single particle. Assuming that the reaction to convert Au(III) to Au(0) is complete, the ratio of 1:7 or 1:10 could be regarded as the ligand-to-metal stoichiometry for the final products.
Characterization Techniques
UV–Vis Spectrometry
UV–vis spectra were collected with a Perkin-Elmer Lambda 20 UV–vis spectrometer. All Au DSNP and dendrimer–Au (III) complex samples were dissolved in water at the concentration of 3 mg/mL.
NMR Spectroscopy
1H NMR spectra of Au DSNPs were recorded on a Bruker DRX 500 nuclear magnetic resonance spectrometer. Samples were dissolved in D2O before NMR measurements.
ζ Potential
ζ potential measurements were performed using a Malvern Zetasizer Nano ZS model ZEN3600 (Worcestershire, UK) equipped with a standard 633 nm laser.
TEM Measurements
A JEOL 2010F analytical electron microscope was performed at 200 kV with an energy dispersive spectroscopy (EDS) system attached. A 5-μL aqueous solution of Au DSNPs (3 mg/mL) was dropped onto a carbon-coated copper grid and air-dried before the measurements. The size distribution histograms of Au DSNPs were measured with use of ImageJ software (http://rsb.info.nih.gov/ij/download.html). For each sample, 200 NPs were randomly selected to analyze the size distribution.
Matrix-Assisted Laser Desorption/Ionization-Time-of-Flight (MALDI-TOF) Mass Spectrometry
The control G5.NHAc-AF594 dendrimers were characterized by MALDI-TOF mass spectrometry, using a Waters Tofspec-2E mass spectrometer (Beverly, MA) run in linear mode with the high mass PAD detector. Briefly, 1 mg of dendrimer samples was dissolved in 1 mL of methanol and then diluted with methanol to obtain the final concentration of 0.2 mg/mL. Equal volumes of the dendrimer solution (0.2 mg/mL) and the matrix trihydroxyacetophenone (THAP) solution (10 mg/mL, dissolved in 50/50 water/acetonitrile) were well mixed. Then, a 1-μL solution of this mixture was injected onto the target spots and evaporated to dryness. The instrument was calibrated with bovine serum albumin (BSA) in THAP. The data were acquired and processed with use of MassLynx 3.5 software.
Fluorescence Spectroscopy
Fluorescence spectroscopy of dye-conjugated dendrimers and Au DSNPs were performed with a Fluoromax-2 fluorimeter. Samples were dissolved in water at the concentration of 40 μM. The excitation wavelength was set at 594 nm. The fluorescence spectra were collected at the wavelength range of 615–900 nm.
Results and Discussion
As compared with metal DENPs, metal DSNPs are formed under mild reduction conditions, such as using mild reducing agents (e.g., hydrazine),22 UV- irradiation,17,30 or thermo-treatment.38,39 In this work, we found that Au NPs could be spontaneously formed and functionalized by acetylation of the {(Au3+)7-G5.NH2} complex or by mixing G5.NGlyOH dendrimers with HAuCl4. In either case, no additional reducing agents were added and the reactions were performed at room temperature.
Figure 1 shows the UV–vis spectra of the formed {(Au3+)7-G5.NH2} complexes (curve 1), {(Au0)7-G5.NHAc} DSNPs (curve 2), and {(Au0)7-G5.NGlyOH} DSNPs (curve 4). The broad shoulder of absorbance at 280 nm for {(Au3+)7-G5.NH2} complexes (curve 1, Figure 1) is indicative of an ion pair formation between AuCl4− and G5.NH2.16,18 After acetylation of the {(Au3+)7-G5.NH2} complexes, the formed {(Au0)7-G5.NHAc} DSNPs display a surface plasmon band at 540 nm (curve 2, Figure 1), which is attributed to collective oscillation of free electrons in Au NPs.49 The {(Au0)7-G5.NGlyOH} DSNPs formed by simply mixing preformed G5.NGlyOH dendrimers with HAuCl4 display a surface plasmon band at 525 nm (curve 4, Figure 1), implying that Au DSNPs with different sizes can be prepared by using the aforementioned two different approaches. The slightly red-shifted surface plasmon band of {(Au0)7-G5.NHAc} DSNPs when compared to that of {(Au0)7-G5.NGlyOH} DSNPs is ascribed to the larger size of the former particles, which were further confirmed by TEM imaging (see below).
Figure 1.
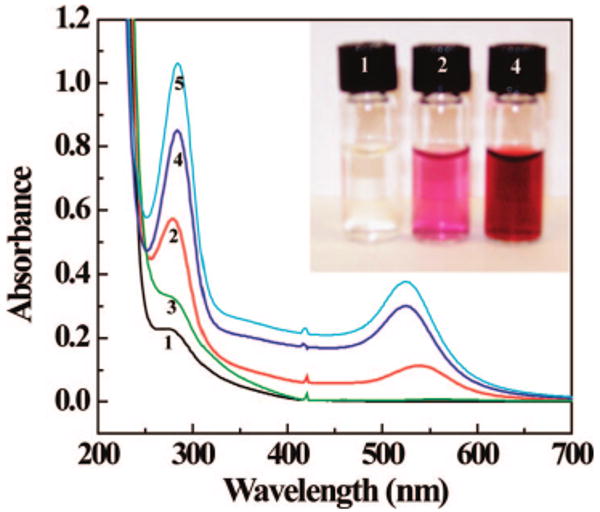
UV–vis spectra of the formed {(Au3+)7-G5.NH2} complexes (curve 1); {(Au0)7-G5.NHAc} DSNPs (curve 2); {(Au3+)7-G5.NH2} complexes after reaction with glycidol for 24 h (curve 3); {(Au0)7-G5.NGlyOH} DSNPs (curve 4); and {(Au0)7-G5.NGlyOH} DSNPs formed by addition of additional glycidol (curve 5). The inset shows the photographs of the corresponding solutions of {(Au3+)7-G5.NH2} complexes (1); {(Au0)7-G5.NHAc} DSNPs (2); and {(Au0)7-G5.NGlyOH} DSNPs (4).
Since acetylation of {(Au3+)7-G5.NH2} complexes spontaneously induces the formation of {(Au0)7-G5.NHAc} DSNPs, it is reasonable to consider the possibility of forming {(Au0)7-G5.NGlyOH} DSNPs by reaction of {(Au3+)7-G5.NH2} complexes with glycidol molecules. We found that this reaction did not induce the formation of the {(Au0)7-G5.NGlyOH} DSNPs, as no characteristic surface plasmon band appeared after the reaction (curve 3, Figure 1). Similar to {(Au3+)7-G5.NH2} complexes, only the broad shoulder of absorbance at 280 nm appears, which is ascribed to the ion pair formation between dendrimers and AuCl4− ions. This is potentially due to the natural differences between the acetylation and hydroxylation reactions. As compared with a hydroxylation reaction, acetylation is a faster reaction and this may generate high local energy levels (probably a heating effect) to assist the reduction of Au(III). We note that the dendrimer terminal acetyl groups do not take part in the redox reaction, as the chemical shift of the acetyl protons in the 1HNMR spectrum does not change after the formation of Au NPs when compared with G5.NHAc dendrimers (see below). Regarding the reaction mechanism to form Au NPs, it is not clear that this is an Au(I) disproportionation reaction assisted by the dendrimer as described by Ceroni and co-workers.50 It is believed that the tertiary amine groups of G5 dendrimers in this case play a major role in the reduction of Au(III) to Au(0), which is further confirmed by later control experiments (see below). The amine groups of G5.NH2 dendrimers do not readily reduce Au(III) ions to form Au NPs at room temperature, as a slight yellow solution remained for at least 3 months (inset, Figure 1). This is potentially due to the strong complexation between dendrimer terminal amines and AuCl4− ions, which inhibits the effective reduction of AuCl4− ions to form zerovalent Au. This was confirmed by NMR studies (see below). However, when the {(Au3+)7-G5.NH2} complexe was stored in 4 °C for over 8 months, we started to observe that the solution changed to a slightly red color, suggesting the partial reduction and formation of Au NPs. On the basis of literature reports and our findings in this work, we can briefly conclude that under certain conditions, such as UV irradiation,17 thermo-treatment,39 laser irradiation,21 or long-term aging process, the amine-terminated dendrimers are able to reduce and stabilize Au NPs. Our results imply that an acetylation reaction not only converts dendrimer terminal amines to acetamide groups (this was confirmed by NMR studies, see below), but also plays a major role to promote the formation of Au DSNPs. We note that in both cases of Au NP formation (acetylation of {(Au3+)7-G5.NH2} complexes and mixing of G5.NGlyOH dendrimers with HAuCl4), the reactions to reduce Au3+ are complete. The addition of NaBH4 (a 3 molar excess of the HAuCl4) into the respective Au NP solution did not induce any changes in the surface plasmon band of the Au NPs. It should be noted that the small peak at 280 nm of curves 2, 4, and 5 (information for 5 can be seen below) in Figure 1 is different than the broad shoulder absorbance at 280 nm for curves 1 and 3 (Figure 1), which is assigned to the ion pair formation between dendrimers and AuCl4− ions.16,18 The small peak at 280 nm for curves 2, 4, and 5 has a controversial assignment in literature regarding its origin.17,51 However, it is reproducibly observed during Au DENP52 and DSNP syntheses,22 using more molar excess of strong or mild reducing agents.
To further understand the formation mechanism of the synthesized {(Au0)7-G5.NHAc} DSNPs, we attempted to use preformed G5.NHAc dendrimers as stabilizers. Three control experiments were performed in parallel to synthesize acetamide-functionalized Au NPs under conditions similar to those used to synthesize {(Au0)7-G5.NHAc} DSNPs (e.g., the molar ratio of dendrimer/Au atom, the amount of triethylamine and/or acetic anhydride added, and the reaction solvent): (1) simply mixing G5.NHAc dendrimers with HAuCl4; (2) the mixture of G5.NHAc dendrimers and HAuCl4 was added with triethylamine; and (3) the mixture of G5.NHAc dendrimers, HAuCl4, and triethylamine was added with acetic anhydride. In all three cases, black precipitates appeared on the bottom of the vials (Figure S1, Supporting Information). This implies that preformed G5.NHAc dendrimers are not able to stabilize Au NPs, while in all cases the G5.NHAc dendrimers with or without triethylamine are able to reduce Au(III) to Au(0). This suggests that the dendrimer tertiary amines are able to reduce AuCl4− ions to form Au NPs. In sharp contrast, for amine-terminated G5 dendrimers a strong complexation between the terminal amine groups and the AuCl4− ions impedes the effective reduction of the AuCl4− ions by either the dendrimer tertiary amines or the dendrimer terminal amines.
The formation of {(Au0)7-G5.NGlyOH} DSNPs by simply mixing G5.NGlyOH dendrimers with Au(III) ions is quite surprising. We think that the tertiary or terminal secondary amine groups of G5.NGlyOH dendrimers are able to reduce Au(III), and the tertiary or terminal secondary amine and hydroxyl groups may simultaneously help stabilize the formed Au NPs. We also found that the addition of a 5-fold molar excess of glycidol into the G5.NGlyOH dendrimers before mixing with the Au(III) ions resulted in the formation of {(Au0)7-G5.NGlyOH} DSNPs (curve 5, Figure 1) with a size similar to that formed in the absence of free glycidol molecules (A TEM image of {(Au0)7-G5.NGlyOH} DSNPs prepared in the presence of free glycidol molecules is shown in the Supporting Information, Figure S2). However, simply mixing free glycidol molecules (in a molar equivalent similar to that of the terminal groups of G5.NGlyOH dendrimers) with Au(III) ions under similar conditions did not induce the formation of Au NPs. This implies that both glycidol terminal groups and dendrimer tertiary and secondary amine groups are essential for the formation of Au NPs. In this case, G5.NGlyOH dendrimers serve a dual role (both as reductants and as stabilizers) for the Au NP synthesis.
The formation of {(Au3+)7-G5.NH2} complexes and {(Au0)7-G5.NHAc} DSNPs with acetamide groups was further confirmed by 1H NMR measurements. Figure 2 shows the 1H NMR spectra of {(Au3+)7-G5.NH2} complexes and {(Au0)7-G5.NHAc} DSNPs. The chemical shift of protons related to the {(Au3+)7-G5.NH2} (Figure 2a) is quite different from those of the G5.NH2 dendrimers (Figure S3a, Supporting Information), presumably due to the strong complexation between dendrimer terminal amines and AuCl4− ions. It is clear that protons 5 and 6 in the G5.NH2 dendrimers display a downfield shift and merge with protons 3 and 1, respectively, after the formation of the {(Au3+)7-G5.NH2} complexes. After acetylation of the {(Au3+)7-G5.NH2} complexes, the formed {(Au0)7-G5.NHAc} DSNPs (Figure 2b) exhibit similar 1H NMR signals to the G5.NHAc dendrimers in the absence of Au NPs (Figure S3b, Supporting Information). This suggests the successful conversion of the amine groups of the {(Au3+)7-G5.NH2} complexes to acetamide groups. The formation of Au DSNPs and the conversion of the dendrimer terminal amines to acetamide groups diminish the strong electrostatic interaction between the dendrimer terminal amines and the AuCl4− ions in the original complexes, leading to similar 1H NMR signals for both the G5.NHAc dendrimers and the {(Au0)7-G5.NHAc} DSNPs.
Figure 2.
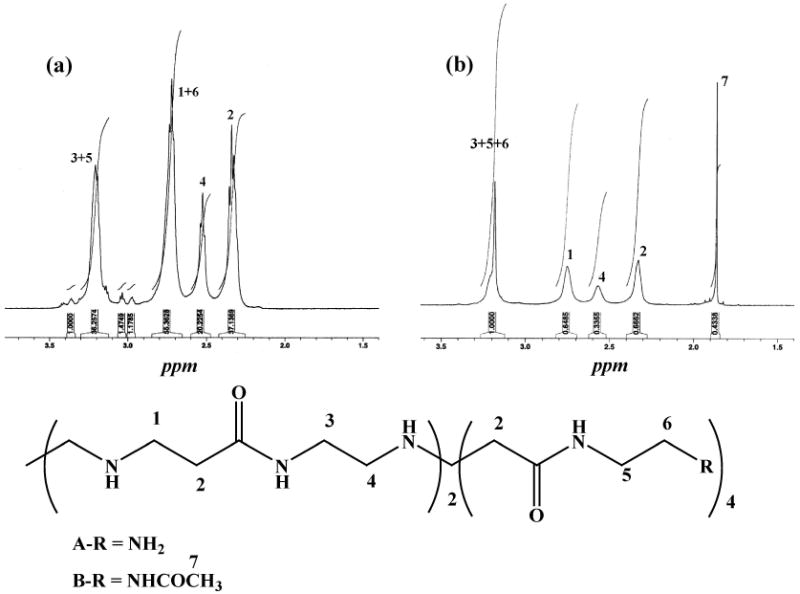
1H NMR spectra of {(Au3+)7-G5.NH2} complexes (a) and {(Au0)7-G5.NHAc} DSNPs (b). A schematic representation of the dendrimer structure used for NMR assignment is shown at the bottom.
For the {(Au0)7-G5.NGlyOH} DSNPs, it is clear that the 1H NMR signals related to protons 6, 4, and 8 shift downfield compared with the G5.NGlyOH dendrimers (Figure 3). Only a portion of the signal from protons 4 and 8 remain in the original positions. This implies that after the formation of {(Au0)7-G5.NGlyOH} DSNPs, the terminal secondary amines and tertiary amines of the G5.NGlyOH interact with the surface of Au NPs, while protons 9 and 10 representing the hydroxyl terminal groups are not significantly changed when compared with the signal in G5.NGlyOH dendrimers. However, the hydroxyl terminal groups help to stabilize the Au NPs since the terminal acetamide groups of G5.NHAc dendrimers induce the precipitation of Au NPs when forming Au DSNPs as described above (see also Figure S1 in the Supporting Information).
Figure 3.
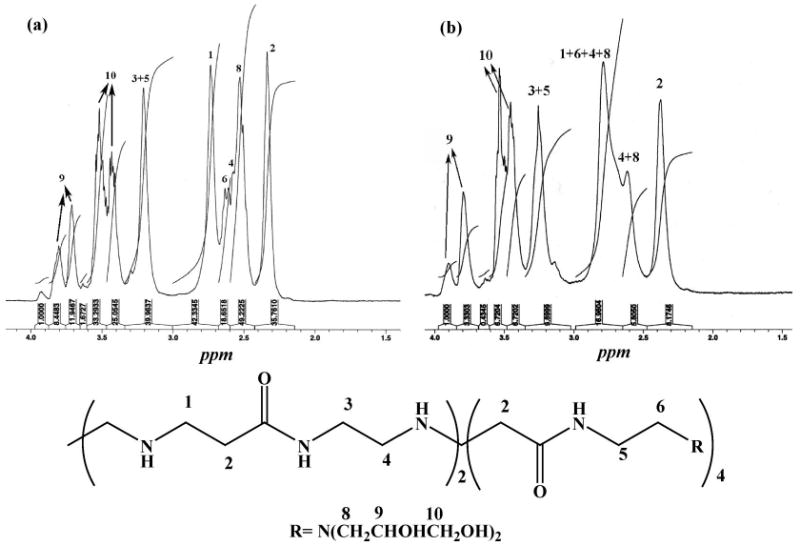
1H NMR spectra of G5.NGlyOH dendrimers (a) and {(Au0)7-G5.NGlyOH} DSNPs (b). A schematic representation of the dendrimer structure used for NMR assignment is shown at the bottom.
The morphology of the formed Au DSNPs was evaluated by TEM imaging. Parts a and b of Figure 4 show a bright-field TEM image of the {(Au0)7-G5.NHAc} DSNPs and the corresponding size distribution histogram, respectively. The average diameter of {(Au0)7-G5.NHAc} DSNPs is 13 ± 4.5 nm. The {(Au0)7-G5.NHAc} DSNPs are highly crystalline as confirmed by the selected area electron diffraction (SAED) pattern (Figure 4c). The indexed (111), (200), (220), and (311) rings in Figure 4c are indicative of the face-centered cubic (fcc) crystal structure of Au DSNPs. An EDS spectrum of the {(Au0)7-G5.NHAc} DSNPs confirms the existence of the Au element (Figure 4d). Figure 5 shows a bright-field TEM image (Figure 5a), a size distribution histogram (Figure 5b), and a high-resolution TEM image of the formed {(Au0)7-G5.NGlyOH} DSNPs (Figure 5c), respectively. It appears that the size of the {(Au0)7-G5.NGlyOH} DSNPs (8.5 ± 0.9 nm) is smaller and more monodisperse than that of {(Au0)7-G5.NHAc}, presumably due to the existence of the secondary amines and hydroxyl groups of the G5.NGlyOH dendrimers that more effectively prevent the aggregation of Au NPs during their formation process. A typical EDS spectrum (data not shown) verifies the existence of the Au element. A high-resolution TEM image (Figure 5c) of the {(Au0)7-G5.NGlyOH} DSNPs shows that most NPs are single crystals, further confirming the crystalline nature of the formed Au DSNPs. It is worthwhile to note that under the TEM conditions, dendrimers on the surfaces of Au NPs are invisible in the TEM images because of their low electron contrast when compared with metal Au.
Figure 4.
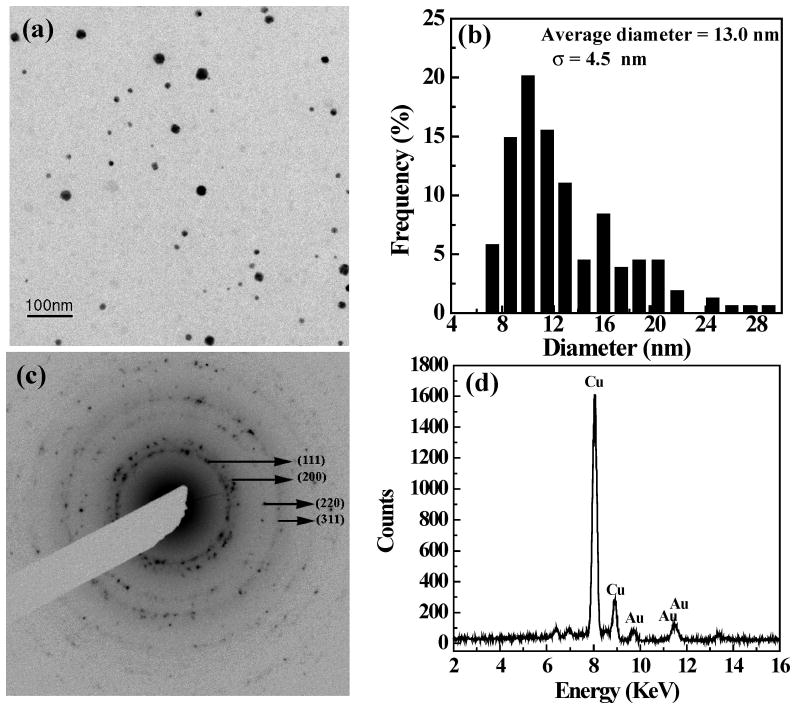
TEM image (a), size distribution histogram (b), selected area electron diffraction pattern (c), and EDS spectrum (d) of {(Au0)7-G5.NHAc} DSNPs.
Figure 5.
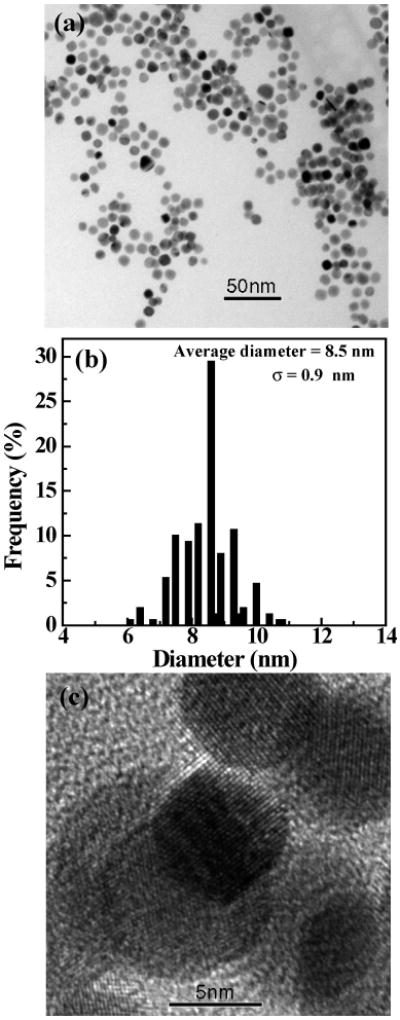
TEM image (a), size distribution histogram (b), and high-resolution TEM image (c) of {(Au0)7-G5.NGlyOH} DSNPs.
The formed {(Au0)7-G5.NHAc} and {(Au0)7-G5.NGlyOH} DSNPs are soluble in water and stable after at least 3 months of storage (photographs of the solution of {(Au0)7-G5.NHAc} and {(Au0)7-G5.NGlyOH} DSNPs are shown in the inset of Figure 1). The surface potential of the {(Au3+)7-G5.NH2} complexes (+39.7 mV) significantly decreased after the formation of the {(Au0)7-G5.NHAc} DSNPs (+9.8 mV), confirming the successful transformation of dendrimer terminal amines to acetamide groups. The slight positive charge is believed to result from the incomplete acetylation reaction as observed for dendrimers in the absence of metals.47 We note that the comparison of the surface potential of {(Au3+)7-G5.NH2} and {(Au0)7-G5.NHAc} is based only on the nature of the peripheral dendrimer groups, without taking into account that Au(III) has been converted to Au(0). However, this comparison gives us clear evidence to confirm the dendrimer surface modification. It is believed that the conversion of Au(III) to Au(0) would not induce significant changes of the surface potentials before and after acetylation reaction. For {(Au0)7-G5.NGlyOH} DSNPs, the surface potential was measured to be +26.8 mV. The more positive charge compared to the {(Au0)7-G5.NHAc} DSNPs is ascribed to the presence of the terminal secondary amines of the G5.NGlyOH dendrimers.45 This indicates that the Au DSNPs synthesized by using different approaches display different surface charges. It is worth noting that the stabilization of {(Au0)7-G5.NGlyOH} DSNPs may be mostly achieved by the interaction of the terminal secondary amines and hydroxyl groups of G5.NGlyOH dendrimers with Au NPs, while the higher surface potential of the particles would help them not aggregate and retain good colloidal stability through electrostatic repelling forces. However, it is difficult to make a comparison between dendrimer terminal groups and the higher surface potential of the particles in terms of the stabilization of the Au DSNPs. For the matter of surface potential effect, our earlier work shows that fully acetylated Au DENPs with a close to neutral surface potential still retain good colloidal stability.28
Dye-functionalized Au NPs are important for biological applications, especially in cellular markers and in in vivo biosensing and bioimaging.27,53 Since we had shown that acetamide-functionalized Au NPs can be formed by acetylation of G5.NH2 dendrimers complexed with AuCl4− ions, we attempted to synthesize surface-neutralized, dye-functionalized Au NPs by acetylation of the amine-terminated G5.NH2-AF594 conjugate complexed with a 10-molar equiv of HAuCl4. {(Au0)10-G5.NHAc-AF594} DSNPs were readily formed, and a TEM image (Figure 6) shows that the formed Au DSNPs are relatively monodisperse with a size of 16.0 ± 4.8 nm. Some of the particles do not show a spherical shape. The size distribution is more or less similar to that of {(Au0)7-G5.NHAc} DSNPs. High-resolution TEM images and EDS (data not shown) confirmed the high crystalline structure and the existence of the Au element, respectively. The 1H NMR spectrum of the {(Au0)10-G5.NHAc-AF594} DSNPs is similar to that of the control dendrimer G5.NHAc-AF594 in the absence of Au, indicating that the amine groups of AF594-functionalized G5 dendrimers were converted to acetamide groups (Figure S4 in the Supporting Information). The MALDI-TOF molar mass (Figure S5 in the Supporting Information) and 1H NMR characterization allow for the identification of the number of AF594 molecules conjugated onto each dendrimer. The practical number of AF594 molecules conjugated onto each dendrimer is 2.6, close to the initial molar ratio. This was also confirmed by UV–vis spectroscopy studies, using a free AF594 calibration curve. The dye-functionalized Au DSNPs are stable in aqueous solution for at least 6 months (a photograph of the aqueous solution of {(Au0)10-G5.NHAc-AF594} DSNPs and G5.NHAc-AF594 dendrimers is shown in Figure S6 in the Supporting Information). The surface plasmon band of the dye-functionalized Au NPs (approximately at 540 nm), however, is difficult to observe due to the overlap with the strong maximum absorbance of the dye AF594 at 594 nm in the UV–vis spectrum (Figure 7a). Fluorescence spectroscopy studies show that, when excited at 594 nm, the fluorescence intensity of {(Au0)10-G5.NHAc-AF594} DSNPs at 640 nm does not display appreciable changes as compared with the G5.NHAc-AF594 dendrimers (Figure 7b). In comparison with other dye-functionalized Au NPs,53 this approach allows for a straightforward functionalization of the Au NPs with a neutral surface, which can readily avoid the specific binding to cell surfaces when applied for biomedical applications.
Figure 6.
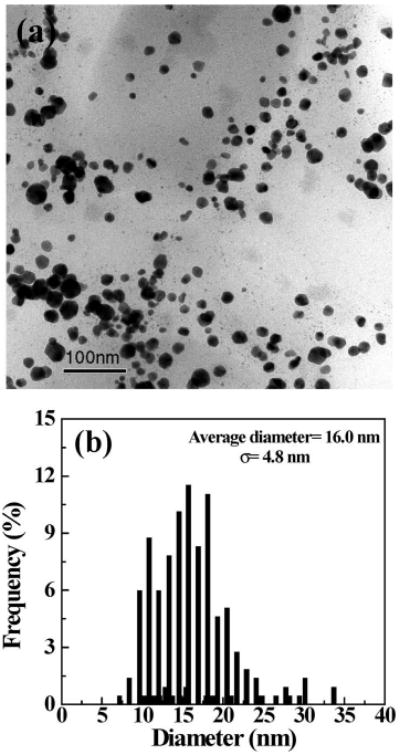
TEM image (a) and size distribution histogram (b) of {(Au0)10-G5.NHAc-AF594} DSNPs.
Figure 7.
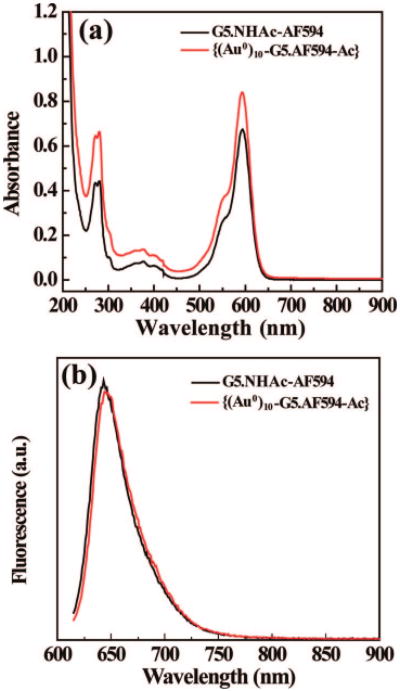
UV–vis (a) and fluorescence (b) spectra of G5.NHAc-AF594 dendrimers and {(Au0)10-G5.NHAc-AF594} DSNPs.
Conclusion
In summary, we have developed two simple approaches to spontaneously synthesizing Au DSNPs with acetamide and hydroxyl surfaces: by acetylation of dendrimer-Au (III) complexes and by mixing glycidol hydroxylated dendrimers with Au anions, respectively. Furthermore, AF594 dye-functionalized Au DSNPs were synthesized by acetylation of the G5.NH2-AF594 conjugate–Au(III) complexes. The formed functionalized Au DSNPs are water-soluble and stable for at least 3 months. It is possible that one could control the size of the formed Au DSNPs by changing the molar ratio between the dendrimers and Au atoms, or using dendrimers of different generations. Importantly, with the recent advances in dendrimer use for biomedical applications,54–57 we anticipate that prefunctionalized dendrimers with targeting, imaging, and drug molecules can be complexed with Au(III) ions for the spontaneous formation of Au DSNP nanodevices. This opens a new avenue for the fabrication of various metal DSNP nanodevices for a range of biomedical applications.
Supplementary Material
Acknowledgments
The authors thank Dr. Xiangdong Bi for his help with NMR experiments. This work was financially supported by the National Cancer Institute (NCI), National Institutes of Health (NIH), under contract No. NOI-CO-97111, 1 R01 CA119409, and 1 R01 EB002657. The 2010F microscope used in the study was funded by NSF through the Grant DMR-9871177.
Footnotes
Supporting Information Available: Photographs of the Au particle suspension prepared using preformed G5.NHAc dendrimers as stabilizers, TEM image, size distribution histogram, and photograph of the suspension of {(Au0)7-G5.NGlyOH} DSNPs prepared in the presence of free glycidol molecules, 1H NMR spectra of G5.NH2, G5.NHAc, and G5.NHAc-AF594 dendrimers, as well as {(Au0)10-G5.NHAc-AF594} DSNPs, MALDI-TOF spectrum of G5.NHAc-AF594 dendrimers, and photographs of aqueous suspension of G5.NHAc-AF594 dendrimers and {(Au0)10-G5.NHAc-AF594} DSNPs. This material is available free of charge via the Internet at http://pubs.acs.org.
References and Notes
- 1.Daniel MC, Astruc D. Chem Rev. 2004;104:293. doi: 10.1021/cr030698+. [DOI] [PubMed] [Google Scholar]
- 2.Rosi NL, Mirkin CA. Chem Rev. 2005;105:1547. doi: 10.1021/cr030067f. [DOI] [PubMed] [Google Scholar]
- 3.Parak WJ, Gerion D, Pellegrino T, Zanchet D, Micheel C, Williams SC, Boudreau R, Le Gros MA, Larabell CA, Alivisatos AP. Nanotechnology. 2003;14:R15. [Google Scholar]
- 4.Li D, Cui Y, Wang K, He Q, Yan X, Li J. Adv Funct Mater. 2007;17:3134. [Google Scholar]
- 5.Li D, He Q, Cui Y, Li J. Chem Mater. 2007;19:412. [Google Scholar]
- 6.Li D, He Q, Cui Y, Wang K, Zhang X, Li J. Chem Eur J. 2007;13:2224. doi: 10.1002/chem.200600839. [DOI] [PubMed] [Google Scholar]
- 7.Lu Y, Mei Y, Drechsler M, Ballauff M. Angew Chem, Int Ed. 2006;45:813. doi: 10.1002/anie.200502731. [DOI] [PubMed] [Google Scholar]
- 8.Sandhu KK, McIntosh CM, Simard JM, Smith SW, Rotello VM. Bioconj Chem. 2002;13:3. doi: 10.1021/bc015545c. [DOI] [PubMed] [Google Scholar]
- 9.Shenhar R, Rotello VM. Acc Chem Res. 2003;36:549. doi: 10.1021/ar020083j. [DOI] [PubMed] [Google Scholar]
- 10.Mayya KS, Caruso F. Langmuir. 2003;19:6987. [Google Scholar]
- 11.Goodman CM, McCusker CD, Yilmaz T, Rotello VM. Bioconj Chem. 2004;15:897. doi: 10.1021/bc049951i. [DOI] [PubMed] [Google Scholar]
- 12.Yuan JJ, Schmid A, Armes SP, Lewis AL. Langmuir. 2006;22:11022. doi: 10.1021/la0616350. [DOI] [PubMed] [Google Scholar]
- 13.Scott RWJ, Wilson OM, Crooks RM. J Phys Chem B. 2005;109:692. doi: 10.1021/jp0469665. [DOI] [PubMed] [Google Scholar]
- 14.Crooks RM, Zhao M, Sun L, Chechik V, Yeung LK. Acc Chem Res. 2001;34:181. doi: 10.1021/ar000110a. [DOI] [PubMed] [Google Scholar]
- 15.Garcia ME, Baker LA, Crooks RM. Anal Chem. 1999;71:256. doi: 10.1021/ac980588g. [DOI] [PubMed] [Google Scholar]
- 16.Kim YG, Oh SK, Crooks RM. Chem Mater. 2004;16:167. [Google Scholar]
- 17.Esumi K, Suzuki A, Aihara N, Usui K, Torigoe K. Langmuir. 1998;14:3157. [Google Scholar]
- 18.Esumi K, Suzuki A, Yamahira A, Torigoe K. Langmuir. 2000;16:2604. [Google Scholar]
- 19.Esumi K. Top Curr Chem. 2003;227:31. [Google Scholar]
- 20.Esumi K. Encycl Nanosci Nanotechnol. 2004;2:317. [Google Scholar]
- 21.Hayakawa K, Yoshimura T, Esumi K. Langmuir. 2003;19:5517. [Google Scholar]
- 22.Shi X, Ganser TR, Sun K, Balogh LP, Baker JR., Jr Nanotechnology. 2006;17:1072. [Google Scholar]
- 23.Balogh L, Valluzzi R, Laverdure KS, Gido SP, Hagnauer GL, Tomalia DA. J Nanopart Res. 1999;1:353. [Google Scholar]
- 24.Bielinska A, Eichman JD, Lee I, Baker JR, Jr, Balogh L. J Nanopart Res. 2002;4:395. [Google Scholar]
- 25.Zheng J, Petty JT, Dickson RM. J Am Chem Soc. 2003;125:7780. doi: 10.1021/ja035473v. [DOI] [PubMed] [Google Scholar]
- 26.Grohn F, Bauer BJ, Akpalu YA, Jackson CL, Amis EJ. Macromolecules. 2000;33:6042. [Google Scholar]
- 27.Shi X, Wang S, Meshinchi S, Van Antwerp M, Bi X, Lee I, Baker JR., Jr Small. 2007;3:1245. doi: 10.1002/smll.200700054. [DOI] [PubMed] [Google Scholar]
- 28.Shi X, Wang S, Sun H, Baker JR., Jr Soft Matter. 2007;3:71. doi: 10.1039/b612972b. [DOI] [PubMed] [Google Scholar]
- 29.Shi X, Sun K, Balogh LP, Baker JR., Jr Nanotechnology. 2006;17:4554. [Google Scholar]
- 30.Lesniak W, Bielinska AU, Sun K, Janczak KW, Shi X, Baker JR, Balogh LP. Nano Lett. 2005;5:2123. doi: 10.1021/nl051077u. [DOI] [PubMed] [Google Scholar]
- 31.Shi X, Lee I, Baker JR., Jr J Mater Chem. 2008;18:586. [Google Scholar]
- 32.Haba Y, Kojima C, Harada A, Ura T, Horinaka H, Kono K. Langmuir. 2007;23:5243. doi: 10.1021/la0700826. [DOI] [PubMed] [Google Scholar]
- 33.Knecht MR, Garcia-Martinez JC, Crooks RM. Langmuir. 2005;21:11981. doi: 10.1021/la051475c. [DOI] [PubMed] [Google Scholar]
- 34.Yu SH, Coelfen H, Mastai Y. J Nanosci Nanotechnol. 2004;4:291. doi: 10.1166/jnn.2004.030. [DOI] [PubMed] [Google Scholar]
- 35.Bronstein LM, Sidorov SN, Gourkova AY, Valetsky PM, Hartmann J, Breulmann M, Colfen H, Antonietti M. Inorg Chim Acta. 1998;280:348. [Google Scholar]
- 36.Sakai T, Alexandridis P. Langmuir. 2004;20:8426. doi: 10.1021/la049514s. [DOI] [PubMed] [Google Scholar]
- 37.Sakai T, Alexandridis P. J Phys Chem B. 2005;109:7766. doi: 10.1021/jp046221z. [DOI] [PubMed] [Google Scholar]
- 38.Sun X, Dong S, Wang E. Macromolecules. 2004;37:7105. [Google Scholar]
- 39.Sun X, Jiang X, Dong S, Wang E. Macromol Rapid Commun. 2003;24:1024. [Google Scholar]
- 40.Ishii T, Otsuka H, Kataoka K, Nagasaki Y. Langmuir. 2004;20:561. doi: 10.1021/la035653i. [DOI] [PubMed] [Google Scholar]
- 41.Sardar R, Park JW, Shumaker-Parry JS. Langmuir. 2007;23:11883. doi: 10.1021/la702359g. [DOI] [PubMed] [Google Scholar]
- 42.Khopade AJ, Caruso F. Nano Lett. 2002;2:415. [Google Scholar]
- 43.Kim BS, Lebedeva OV, Koynov K, Gong H, Caminade AM, Majoral JP, Vinogradova OI. Macromolecules. 2006;39:5479. [Google Scholar]
- 44.Lebedeva OV, Kim BS, Groehn F, Vinogradova OI. Polymer. 2007;48:5024. [Google Scholar]
- 45.Shi X, Lesniak W, Islam MT, Mu˜Niz MC, Balogh L, Baker JR., Jr Colloid Surf A. 2006;272:139. [Google Scholar]
- 46.Shi X, Bányai I, Rodriguez K, Islam MT, Lesniak W, Balogh P, Balogh L, Baker JR., Jr Electrophoresis. 2006;27:1758. doi: 10.1002/elps.200500818. [DOI] [PubMed] [Google Scholar]
- 47.Majoros IJ, Keszler B, Woehler S, Bull T, Baker JR., Jr Macromolecules. 2003;36:5526. [Google Scholar]
- 48.Thomas TP, Myaing MT, Ye JY, Candido K, Kotlyar A, Beals J, Cao P, Keszler B, Patri AK, Norris TB, Baker JR., Jr Biophys J. 2004;86:3959. doi: 10.1529/biophysj.103.034462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Alvarez MM, Khoury JT, Schaaff TG, Shafigullin MN, Vezmer I, Whetten RL. J Phys Chem B. 1997;101:3706. [Google Scholar]
- 50.Bergamini G, Ceroni P, Balzani V, Gingras M, Raimundo JM, Morandi V, Merli PG. Chem Commun. 2007:4167. doi: 10.1039/b708115d. [DOI] [PubMed] [Google Scholar]
- 51.Lee WI, Bae Y, Bard AJ. J Am Chem Soc. 2004;126:8358. doi: 10.1021/ja0475914. [DOI] [PubMed] [Google Scholar]
- 52.Garcia-Martinez JC, Crooks RM. J Am Chem Soc. 2004;126:16170. doi: 10.1021/ja046567n. [DOI] [PubMed] [Google Scholar]
- 53.Shukla R, Bansal V, Chaudhary M, Basu A, Bhonde RR, Sastry M. Langmuir. 2005;21:10644. doi: 10.1021/la0513712. [DOI] [PubMed] [Google Scholar]
- 54.Kukowska-Latallo JF, Candido KA, Cao Z, Nigavekar SS, Majoros IJ, Thomas TP, Balogh LP, Khan MK, Baker JR., Jr Cancer Res. 2005;65:5317. doi: 10.1158/0008-5472.CAN-04-3921. [DOI] [PubMed] [Google Scholar]
- 55.Majoros IJ, Thomas TP, Mehta CB, Baker JR., Jr J Med Chem. 2005;48:5892. doi: 10.1021/jm0401863. [DOI] [PubMed] [Google Scholar]
- 56.Thomas TP, Majoros IJ, Kotlyar A, Kukowska-Latallo JF, Bielinska A, Myc A, Baker JR., Jr J Med Chem. 2005;48:3729. doi: 10.1021/jm040187v. [DOI] [PubMed] [Google Scholar]
- 57.Patri AK, Kukowska-Latallo JF, Baker JR., Jr Adv Drug Delivery Rev. 2005;57:2203. doi: 10.1016/j.addr.2005.09.014. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.