

Abstract
Co-infection with Hepatitis C virus (HCV) is present in one-third of all Human Immunodeficiency Virus-infected (HIV) individuals in the United States and is associated with rapid progression of liver fibrosis and poor response to pegylated interferon (IFN) and ribavirin. In this study, we examined gene expression profiles in peripheral blood mononuclear cells (PBMCs) from different groups of individuals who are mono- or co-infected with HIV and HCV. Data showed that HIV and HCV viremia up-regulate genes associated with immune activation and immunoregulatory pathways. HCV viremia is also associated with abnormalities in all peripheral immune cells, suggesting a global effect of HCV on the immune system. Interferon-α-induced genes were expressed at a higher level in PBMCs from HIV-infected individuals. HCV and HIV infections leave distinct profiles or gene expression of immune activation in PBMCs. HIV viremia induces an immune activated state; by comparison, HCV infection induces immunoregulatory and pro-inflammatory pathways that may contribute to progression of liver fibrosis. An aberrant type-I IFN response seen exclusively in HIV-infected individuals could be responsible for the poor therapeutic response experienced by HIV/HCV co-infected individuals receiving interferon-α based current standard of care.
Keywords: immunity, immune activation, microarray, interferon-inducible genes, biomarker
Introduction
Chronic co-infection with hepatitis C virus (HCV) is documented in one-third of all HIV-infected persons in the United States,, and is associated with increased morbidity and mortality relative to mono-infection with either virus (1, 2). Since the advent of antiretroviral therapy (ART) for controlling HIV replication in vivo, AIDS-associated opportunistic infections have declined considerably (3). However, recent data suggest an increasing number of HIV-infected individuals are now dying from liver disease (3–5). Moreover, HCV/HIV co-infected individuals see a rapid progression of liver fibrosis to cirrhosis when compared to HCV mono-infected individuals (6). Several adverse effects associated with ART are exacerbated in HCV/HIV co-infected individuals, making it difficult to accomplish adequate virologic control of HIV infection among such individuals (7–12). Additionally, HCV/HIV co-infected individuals have a higher HCV RNA viral load than do HCV mono-infected individuals (13–17). Finally, co-infection with HIV decreases the rates of sustained virologic response (SVR) of HCV and increases the rate of relapses after discontinuation of anti-HCV therapy among those who have achieved an end-of treatment response (ETR) to combination therapy (14–17).
Chronicity of infection with HCV mono-infected individuals is associated with an inconspicuous immune response against the virus (18); in contrast, in HIV mono-infected individuals, the resultant immune response is readily detectable, but is unable to contain HIV replication, leading to establishment of chronic infection (19). The characterization of and relationships between (the immune responses against HCV and HIV in co-infected individuals are not completely understood.
To determine the differential host immune responses to each virus, we employed a DNA microarray study using PBMCs from five different groups of individuals. DNA microarrays has been used previously to study the pathogenesis of HCV (20) and HIV in mono-infected individuals (21). However, such studies failed to define gene expression imprints and/or adequately compare the differentiated gene profiles induced by each of these viruses alone and in co-infection because the studies did not involve direct comparison of the gene expression profiles of HCV and HIV co-infected individuals. In this study, we performed DNA microarray analysis on PBMCs from HIV negative, HIV viremic, HIV aviremic, HCV viremic, and HCV/HIV co-infected individuals to determine the differential gene expression among these groups. To our knowledge this study is the most comprehensive DNA microarray study that involves cross sectional analysis of all 5 control groups. These genetic imprints provide insights into the pathophysiology of chronic HCV infection in those subjects who are co-infected with HIV.
Methods
Study subjects
Peripheral Blood Mononuclear Cells (PBMCs) were obtained by venipuncture from thirty-three subjects belonging to the following clinical categories: HIV negative (n=7), HCV mono-infected viremic (n=7), HIV mono-infected viremic (n=8), HCV/HIV co-infected (n=5), and HIV aviremic (n=6) (Table 1A). All donors signed informed consents approved by the Institutional Review Board (IRB) of the National Institute of Allergy and Infectious Diseases (Bethesda, MD).
Table 1.
Table 1A. Demographics and immune profiles of Study Participants:Immune profiling was performed by flow cytometry for all individuals except HCV monoinfected individuals. HIV and HCV viral load had a lower limit of detection of 50 copies/mL and 615 IU/mL respectively. Group A consisted of healthy individuals seronegative for HIV and HCV; group B included HCV mono-infected viremic individuals; group C constituted HIV mono-infected viremic individuals; group D contained HCV/HIV co-infected individuals that were HCV viremic and HIV aviremic, and group E consisted of HIV mono-infected, but aviremic individuals. For Group E individuals, antiretroviral therapy (ART) consisted of at least one HIV protease inhibitor and/or one non-nucleoside reverse transcriptase inhibitor and two reverse transcriptase inhibitors.
Table 1B. Functional Characteristics of all the genes selected for validation in the study
| Table 1A | |||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| Group | Start Date |
Age | Gender | Race | Risk | HCV genotype |
TCD4 | CD4 (%) |
HIV VL |
HCV VL | HCV Treatment Response |
| A | 10/27/05 | 41 | F | White | N/A | N/A | 737 | 55 | N/A | N/A | N/A |
| A | 10/18/05 | 34 | M | White | N/A | N/A | 1225 | 43 | N/A | N/A | N/A |
| A | 10/26/05 | 56 | F | White | N/A | N/A | 606 | 59 | N/A | N/A | N/A |
| A | 10/26/05 | 37 | F | Black | N/A | N/A | 490 | 44 | N/A | N/A | N/A |
| A | 10/25/05 | 42 | M | White | N/A | N/A | 863 | 46 | N/A | N/A | N/A |
| A | 10/17/05 | 46 | F | White | N/A | N/A | 726 | 47 | N/A | N/A | N/A |
| A | 10/20/05 | 39 | M | Hispanic | N/A | N/A | 352 | 37 | N/A | N/A | N/A |
| B | 10/31/05 | 51 | M | White | IVDU | 1b | N/A | 2,500,000 | NR | ||
| B | 11/9/05 | 53 | F | White | IVDU | 1a | N/A | 473000 | RELAPSER | ||
| B | 10/24/05 | 51 | M | Black | IVDU | 1 | N/A | 441,000 | SVR | ||
| B | 10/26/05 | 45 | M | White | IVDU | 1a | N/A | 3,820,000 | RELAPSER | ||
| B | 11/9/05 | 42 | M | White | IVDU | 2 | N/A | 10,900,000 | SVR | ||
| B | 11/2/05 | 59 | F | Black | IVDU | 2b | N/A | 7,810,000 | SVR | ||
| B | 10/26/05 | 70 | M | White | IVDU | 1b | N/A | 3,830,000 | SVR | ||
| C | 12/13/05 | 41 | M | White | MSM | N/A | 290 | 22 | 29576 | N/A | N/A |
| C | 12/19/05 | 20 | M | Black | HETERO | N/A | 588 | 27 | 168629 | N/A | N/A |
| C | 12/2/05 | 42 | M | White | MSM | N/A | 337 | 36 | 32602 | N/A | N/A |
| C | 12/13/05 | 36 | F | Black | HETERO | N/A | 415 | 17 | 1964 | N/A | N/A |
| C | 11/10/05 | 34 | M | Hispanic | MSM | N/A | 356 | 21 | 23457 | N/A | N/A |
| C | 12/1/05 | 33 | M | Hispanic | MSM | N/A | 318 | 14 | 112697 | N/A | N/A |
| C | 12/1/05 | 59 | F | Black | HETERO | N/A | 507 | 27 | 12648 | N/A | N/A |
| C | 12/19/05 | 23 | M | Hispanic | HETERO | N/A | 15 | 2 | 108507 | N/A | N/A |
| D | 12/22/05 | 49 | M | Black | IVDU | 1a | 1233 | 45 | 121 | >7,692,310 | Naïve |
| D | 11/28/05 | 40 | M | Black | MSM | 1a | 1460 | 44 | 49 | 4,976,400 | Viral breakthrough |
| D | 11/16/05 | 51 | M | Black | IVDU | 1b | 727 | 30 | 49 | 3945420 | NR |
| D | 12/8/05 | 49 | F | Black | IVDU | 1a | 794 | 25 | 49 | 1054510 | Naïve |
| D | 12/8/05 | 55 | M | Black | MSM | 1b | 1008 | 43 | 49 | 9504730 | NR |
| E | 11/16/05 | 44 | M | White | MSM | N/A | 745 | 31 | 49 | N/A | N/A |
| E | 11/28/05 | 41 | M | Black | HETERO | N/A | 258 | 16 | 49 | N/A | N/A |
| E | 6/20/05 | 45 | F | Hispanic | HETERO | N/A | 202 | 10 | 49 | N/A | N/A |
| E | 11/16/05 | 36 | F | Hispanic | HETERO | N/A | 462 | 22 | 76 | N/A | N/A |
| E | 11/22/05 | 51 | M | White | MSM | N/A | 362 | 32 | 49 | N/A | N/A |
| E | 12/20/05 | 44 | M | Hispanic | MSM | N/A | 464 | 33 | 296 | N/A | N/A |
| Table 1B | |||
|---|---|---|---|
| Name of Gene |
Function | Relationship with Disease |
Relevance in this study |
| CD10 | Surface marker for immature B cells | Upregulated in HCV | Global effect of HCV replication on B cells |
| NKp30 | NK activating Receptor | Upregulated in HCV | Increased in chronic HCV and HCV/HIV |
| CD80 | Major T-Cell co-stimulatory factor | Upregulated in HCV | Global effect of HCV replication on T cells |
| CX3CL1 | Neutrophil chemo attractant involved in angiogenesis, inflammation and tissue healing | Upregulated in HCV | Increased in chronic HCV |
| IGF-1R | Cell surface receptor that increases insulin secretion upon stimulation | Upregulated in HCV | Increased in chronic HCV and HCV/HIV |
| CCL-7 | Monocyte chemotaxis | Upregulated in HCV | Increased in chronic HCV |
| CCL-20 | Lymphocyte chemotaxis | Upregulated in HCV | Increased in chronic HCV |
| APOBEC3A | mRNA editing enzyme | Upregulated in HIV | HIV-induced type-I IFN response |
| APOBEC3G | Interferes with replication of retroviruses | Upregulated in HIV | HIV-induced type-I IFN response |
| TRIM5 | Iinnate immune defense against retroviruses | Upregulated in HIV | HIV-induced type-I IFN response |
| EIF2AK2 | Viral defense, IFN-inducible gene | Upregulated in HIV | HIV-induced type-I IFN response |
| G1P3 | Regulation of apoptosis, IFN-inducible gene | Upregulated in HIV | HIV-induced type-I IFN response |
| IFI27 | Impacts cellular apoptosis, IFN-inducible gene | Upregulated in HIV | HIV-induced type-I IFN response |
| IFI44 | Impacts cellular apoptosis, IFN-inducible gene | Upregulated in HIV | HIV-induced type-I IFN response |
| IFIT1 | IFN-inducible gene | Upregulated in HIV | HIV-induced type-I IFN response |
| IFIT3 | IFN-inducible gene | Upregulated in HIV | HIV-induced type-I IFN response |
| IFITM1 | IFN-inducible gene | Upregulated in HIV | HIV-induced type-I IFN response |
| IFITM3 | IFN-inducible gene | Upregulated in HIV | HIV-induced type-I IFN response |
| IFNA2 | IFN-inducible gene | Upregulated in HIV | HIV-induced type-I IFN response |
| IFNB | IFN-inducible gene | Upregulated in HIV | HIV-induced type-I IFN response |
| IFNG | IFN-inducible gene | Upregulated in HIV | HIV-induced type-I IFN response |
| IRF7 | Transcriptional activation of virus-inducible cellular genes, IFN-inducible gene | Upregulated in HIV | HIV-induced type-I IFN response |
| ISG15 | Ubiquitin like modifier for innate defense, IFN-inducible gene | Upregulated in HIV | HIV-induced type-I IFN response |
| ISG20 | Ubiquitin like modifier for innate defense, IFN-inducible gene | Upregulated in HIV | HIV-induced type-I IFN response |
| LY6E | IFN-inducible gene | Upregulated in HIV | HIV-induced type-I IFN response |
| MX1 | Responsible for antiviral state against influenza virus infection, IFN-inducible gene | Upregulated in HIV | HIV-induced type-I IFN response |
| MX2 | Responsible for antiviral state against influenza virus infection, IFN-inducible gene | Upregulated in HIV | HIV-induced type-I IFN response |
| OAS1 | Viral RNA degradation and the inhibition of viral replication, IFN-inducible gene | Upregulated in HIV | HIV-induced type-I IFN response |
| OAS2 | Viral RNA degradation and the inhibition of viral replication, IFN-inducible gene | Upregulated in HIV | HIV-induced type-I IFN response |
| PLSCR1 | Responsible for the translocation of phospholipids, IFN-inducible gene | Upregulated in HIV | HIV-induced type-I IFN response |
| PPIA | Involved in T cell activation, IFN-inducible | Upregulated in HIV | HIV-induced type-I IFN response |
| SP110 | Transcriptional coactivator, Ifn-inducible gene | Upregulated in HIV | HIV-induced type-I IFN response |
| STAT1 | IFN-signaling | Upregulated in HIV | HIV-induced type-I IFN response |
Isolation of PBMCs and RNA
PBMCs were isolated from white blood cells by the standard Ficoll-Hypaque Plus (Amersham Biosciences, Uppsala, Sweden) density gradient separation technique. RNA was isolated using Qiagen mRNA isolation kits (Qiagen, Germantown, MD) following the manufacturer’ protocol.
Cell Cultures
Each patient’ PBMCs (2 ×106) were incubated in 12-wellplates with complete RPMI 1640 medium (Invitrogen, Carlsbad, CA) supplemented with 10%fetal calf serum (Hyclone Laboratories, Logan, UT), enicillin-streptomycin (Invitrogen), and L-glutamine (Invitrogen),. Cultures were incubated at 37° in a 5% CO2 incubatorfor up to 48-hours, and supernatants fromeach culture were collected.
DNA Microarray Analysis
PBMCs were analyzed using Affymetrix U133A 2.0 oligonucleotide arrays according to the protocols specified by the manufacturer (Affymetrix, Santa Clara, CA). A significant analysis of microarray (SAM) algorithm was used to determine the genes that were differentially expressed after an extensive filtering processes (22). Geneswith low variabilityor undetectable expression levels (for the majority of samples) were eliminated from analysisif the Guanosine-Cytosine Robust Multi Array values for these genes were within the interquartile range of<0.263 or a 75thpercentile of <5.
bDNA Multiplex Assay
Validation of DNA microarray data was performed using a novel customized bDNA multiplex assay capable of detecting the expression of 35 genes. The RNA transcripts are released from cells in the presence of Lysis Mixture and hybridized to the Probe Sets. The RNA-Probe Set complexes are captured to their respective Capture Beads through the cooperative hybridization of multiple CEs with the Capture Probes on the Capture Beads during an overnight incubation. Signal amplification is performed by sequential hybridization of the bDNA Amplifier and biotinylated Label Probe. The Streptavidin-conjugated R-Phycoerythrin (SAPE) binds to the biotinylated Label Probe. The Capture Beads are analyzed using a Luminex instrument. The amount of each target RNA present in a sample is quantified by determining the amount of SAPE fluorescence signal and the identity of the beads.
Flow Cytometry
For flow-cytometric analyses, the following combinations of fluorochrome-conjugated antibodies were used: CD3 (allophycocyanin-conjugated [APC]) with CD54 (phosphatidylethanolamine [PE]), CCR2 (PE), CCR7 (PE), CD10 (PE), CD80 (PE), CD86 (PE), CD274 (PE), CX3CR1 (PE), IGF-1R (PE), and NCR3 (PE). All antibodies and appropriate isotype controls were obtained from BD Biosciences, San Jose, CA. After washing, PBMCs were incubated with appropriate antibodies for 30 min at 4°C. The cells were washed, fixed, and suspended in 1% paraformaldehyde in PBS and flow-cytometric analysis was performed on a FACS Array (BD Biosciences). For subset analysis, a lymphocyte gate and gates uniquely identifying CD3+ cells were applied, ~100,000 events were collected, and the frequency of CD3+ and CD3− expressing each receptor was analyzed with FlowJo software (TreeStar, Ashland, OR).
ELISA
Culture supernatants from PBMCs at 48 hours were tested for levels of IL-23A, β2 microglobulin, TNF, CCL-7, CCL-20, IL-8, and CXCL1 by enzyme-linked immunosorbent assay (ELISA; R&D Systems, Minneapolis, MN).
Statistical Analysis
ANOVA with Tukey’s multiple comparison test was used to compare means of the independent groups. The paired t test with the Bonferroni adjustment for multiple testing was used to compare paired responses.
Results
Differential gene expression profiles in PBMCs of HCV and HIV infected individuals
In order to identify the gene expression profiles induced by both HCV and HIV, we performed DNA microarray analyses using total RNA isolated from fresh PBMCs from the aforementioned five patient groups. There were no significant differences in the 5 groups based on the major demographic characteristics such as sex, age, treatment responses, etc (Table 1A). Using Affymetrix human genome U133A oligonucleotide arrays consisting of probes encompassing over 22,000 genes and a SAM algorithm (22), we identified 1813 differentially expressed genes (Fig 1). The corresponding genes and samples from the individuals were grouped by using hierarchical clustering. Differences in relative levels of gene expression (Z-score) are indicated in color, where red indicates up-regulation and green indicates down-regulation relative to that of corresponding gene expression in controls (Fig 1). The hierarchical analyses classified the genes into six distinct clusters based on differential expression between the five groups. Of these, Cluster 1 consists of 174 genes down-regulated both in HCV and HIV infected individuals (group B and C). Cluster 2 consists of 369 genes up-regulated in HCV-infected individuals (groups B and D). Cluster 3 consists of genes up-regulated in HIV mono-infected individuals (group C and E). Cluster 4 consists of genes up-regulated in only HIV viremic individuals (group C). Cluster 5 consists of 302 genes down-regulated in only HCV mono-infected individuals (group B). Cluster 6 consists of 599 genes up-regulated in only HCV mono-infected individuals (group B). To identify HCV-induced changes in gene expression in PBMCs, we chose to focus our analyses on clusters 2, 5 and 6. In this regard, cluster 2 distinctively showed genes up-regulated by HCV viremia as observed in both HIV negative and positive individuals. Meanwhile, clusters 5 and 6 showed genes that are either down-regulated or up-regulated by HCV viremia alone as observed in HCV mono-infected individuals. Hierarchical clustering analyses indicated that there was a similarity in the transcriptional profile of genes that were differentially expressed in PBMCs of HCV-infected individuals (seen in cluster 2 genes) and also distinct gene expression profiles in HCV mono-infected and HCV/HIV co-infected individuals (seen in cluster 5 and 6 genes). Representative genes that belong to each cluster identified using rigorous literature-mining algorithms and statistical analyses are shown in Fig 1.
Figure 1. Clustering of differentially expressed genes in PBMCs from 5 groups (See Methods section).
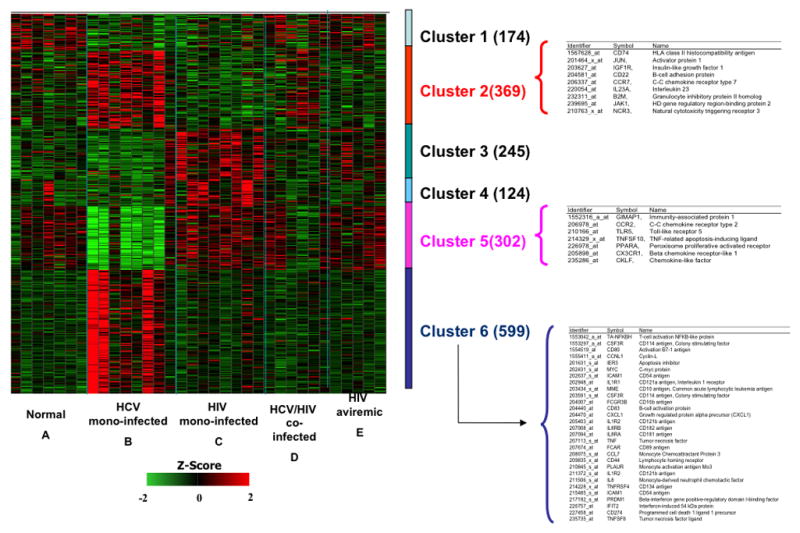
Levels of gene expression were assayed using Affymetrix Human Genome U133A chips. A total of 1,813 differentially expressed genes were identified. Genes were grouped using K-means clustering, and samples were grouped by hierarchical clustering. Differences in relative levels of gene expression (Z-score) are indicated in color, where red indicates up-regulation and green indicates down-regulation relative to that of corresponding gene expression in controls. The numbers in parentheses indicate the number of genes in each cluster. Some genes are listed twice because certain probes recognize multiple regions of a single gene. Cluster 1 consists of genes down-regulated both in group B and C individuals. Cluster 2 consists of genes up-regulated in groups B and D individuals. Cluster 3 consists of genes up-regulated in group C and E. Cluster 4 consists of genes up-regulated only in group C individuals. Cluster 5 consists of genes down-regulated only in group B individuals. Cluster 6 consists of genes up-regulated only in group B individuals. Select biologically relevant genes of Clusters 2, 5 and 6 are shown on the right.
Selection of genes for validation of DNA microarray by amplification
To validate our DNA microarray data, we selected genes based on an extensive literature-mining algorithm, significance of microarray analysis data and biology of the disease process (both HIV and HCV). This analytical approach following the biology of response rather than the mere expression levels of genes will help us ascertain our results are driven by the biology (disease process, infection status etc) rather than race, age, sex etc. Although this approach reduces the influence of these variables in interpretation of results, it will certainly not eliminate the influence completely, warranting validation of these results in a larger study. We performed gene amplification from RNA, estimated surface expression of receptors of freshly isolated PBMCs and the levels of secreted proteins in culture supernatants. Gene amplification performed by bDNA analysis of selected genes that were differentially regulated using a custom-designed bDNA array is shown in Fig 2. Two candidate genes OAS1 (2A & B) and MX1 (2 C & D) were selected for validation of gene expression and were consistently reproduced by bDNA assay.
Figure 2. Validation of DNA microarray data by bDNA analysis.

DNA microarray expression of IFIG genes was validated using a customized bDNA assay. The expression of two representative IFIGs by DNA microarray (left) and bDNA assay (right) for the two gene candidates OAS1 (A,B) and MX1 (C,D) indicate that HIV-infected individuals have a higher level of IFIG expression overall than the other groups of individuals. Fold change of gene expression is calculated as the change within a group compared to that observed in normal volunteers. The expression of both OAS1 and MX1 were significantly different between Group C and others by microarray (p<0.03, and p<0.02 respectively) and bDNA (p<0.04and p<0.01 respectively).
Selection of genes/gene products for validation by microarray flow cytometry and ELISA
For further validation of our DNA microarray analyses at the level of surface expression of proteins, we selected most biologically relevant gene products based on biological relevance (Fig. 3 and Table 1B). From cluster 2, we chose IGF-1R, a cell surface receptor that increases insulin secretion upon stimulation (23); CCR7, which is a chemokine homing receptor of immune cells (24) and NKp30, which is one of the NK activating receptors facilitating killing of infected targets (25). From cluster 5, we examined CCR2 and CX3CR1, two chemokine receptors involved in inflammatory response and lymphocyte activation (26). From cluster 6, we studied the expression of CD10, CD54, CD80, and CD274. CD10 is a cell surface marker of immature B cells (27); CD54 is involved with cell-cell adhesion and formation of immunological synapses (28); CD80 is a major T-cell co-stimulatory factor (29); and CD274 is a cell surface molecule with a T-cell regulatory function (30). To elucidate the effect of HIV and HCV on the secretory function of PBMCs, we tested the ability of PBMCs to secrete the following proteins, CCL-7, CCL-20, CX3CL1, IL-8 and TNF-α. CCL-7 (MCP-3) is a chemokine that attracts monocytes to sites of inflammation and regulates macrophage function (24). CCL-20, otherwise called liver activation regulated chemokine (LARC) or macrophage inflammatory protein-3 A (MIP-3α), is a chemokine that attracts lymphocytes, but is a weaker target for monocytes (31). CX3CL-1 also know as GRO-α or fractalkine, is a cytokine belonging to the CX3C chemokine family and is a neutrophil chemo attractant, which is also involved in angiogenesis, inflammation and tissue healing (32). TNF-α and IL-8 are pro-inflammatory cytokines secreted by T cells that mediate chemotaxis and inflammatory responses (33). All these genes formed part of cluster 6, which are up-regulated in HCV-mono-infected individuals.
Figure 3. Average differential gene expression profiles of the five groups of individuals.
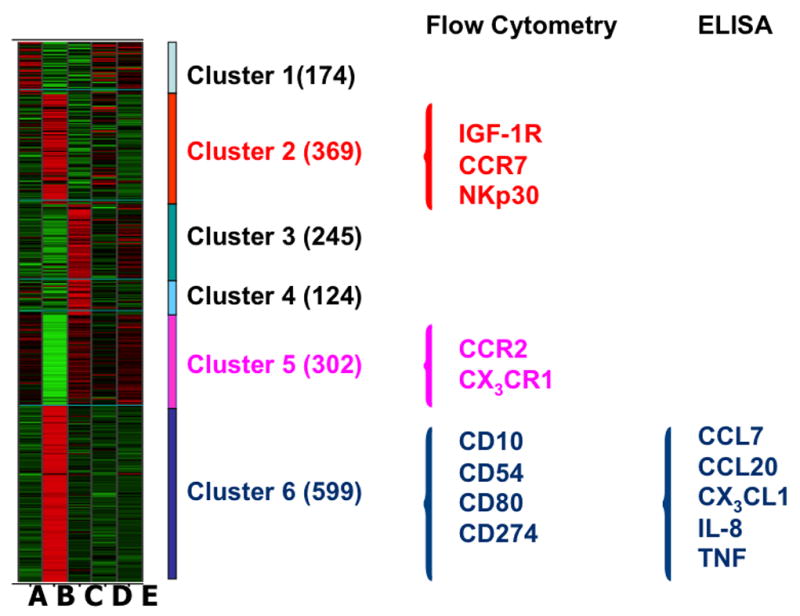
Using either flow cytometry (surface expression) or ELISA (secreted proteins) selected genes were validated based on their biological relevance (following literature-mining algorithms). Clusters 2, 5 and 6 were selected based on the identification of cell surface receptors and secreted proteins most relevant to HCV infection status.
HCV infection has a global effect on peripheral blood immune competent cells
To investigate the effect of ongoing HCV replication on various peripheral blood immune competent cells, we performed flow cytometry analysis for the expression of surface molecules whose genes were differentially regulated in the DNA microarray analysis. As show in Figure 4, HCV replication has profound effects on B cells, NK cells, and on antigen presenting cells. B cells from HCV-monoinfected individuals expressed a significantly higher percentage of CD10+ immature B cells in their peripheral blood when compared to that seen in the other groups (14.2 ± 0.8% (Group B) vs. 7.1 ± 0.3% (Group A), 8.2 ± 1.1% (Group C), 3.2 ± 0.1% (Group D), and 3.9 ± 0.3%; p<0.03 for Group B versus the others). A significantly higher proportion of NK cells from HCV monoinfected and HIV/HCV co-infected individuals expressed the natural cytotoxicity receptor 3 (NCR3 or NKp30) on their surface compared to the other 3 groups (3 ± 0.4% (Group A), 9.8 ± 0.3% (Group B), 2.6 ± 0.4% (Group C), 8.7 ± 0.7% (Group D), and 1.8 ±0.2% (Group E); p<0.04 for Groups B and D vs Groups A and C and p<0.03 for Groups B and D vs Group E). The percentage of cells expressing CD80 in the peripheral blood of HCV mono-infected subjects was significantly higher compared to that seen with the other 4 groups (5.1 ± 0.6% (Group A), 14.3 ± 0.9% (Group B), 4.8 ± 0.8% (Group C), 5.1± 0.7% (Group D), and 4.4 ± 0.5% Group E; p value <0.03 for Group B vs the others). The levels of expression of CD274 (PD-L1) were not significantly different among the groups (data not shown).
Figure 4. HCV Infection has a Global Effect on Immune Cells.
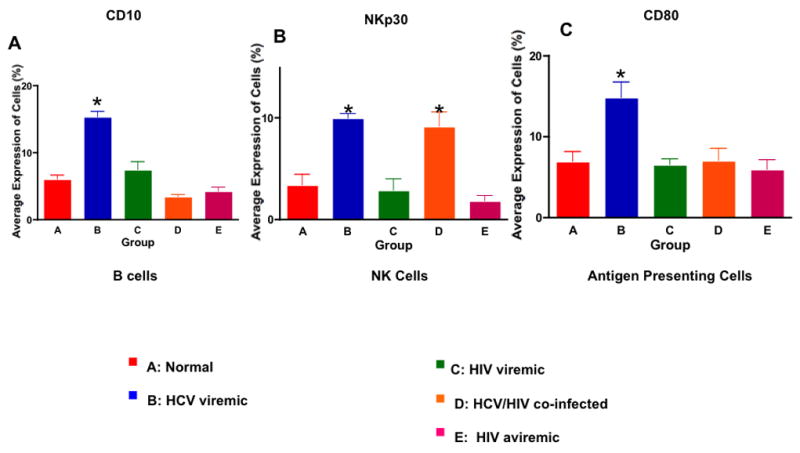
A. The percentage of CD10+ immature B cells in the peripheral blood measured by flow cytometry from 5 individuals in the HCV mono-infected group was significantly higher than that of the other 4 groups (p<0.03). B. The percentage of NKp30+ cells among NK cells in the peripheral blood was measured by flow cytometry from the 5 groups. HCV-infected (both mono and HIV co-infected) individuals had a significantly higher percentage of NKp30+ NK cells than that of the other groups (p<0.04). C. The percentage of CD80+ cells among CD3- in HCV mono-infected individuals was significantly higher than that of the other 4 groups (p<0.03).
Higher levels of markers of hepatic injury were secreted by PBMCs of individuals with chronic hepatitis C infection
Several non-invasive markers of liver fibrosis have been identified in individuals with chronic hepatitis B and C infection (34). We selected markers of liver fibrosis CX3CL1 and IGF-R1 from cluster 6 and 2 whose gene expressions were also up-regulated in groups B and D (HCV mono-infected and HCV/HIV co-infected subjects) (Fig 3). CX3CL1 and its receptor CX3CR1 have been found to be associated with liver injury and have been suggested to be a marker of liver disease (35). When we measured the levels of CX3CL1 expression in the supernatants of PBMCs, the cells from HCV mono-infected subjects produced significantly higher levels of CX3CL1 than did cells from other 4 groups (Fig 5A; 276 ± 18 pg/mL (Group A), 2870 ± 420 pg/mL (Group B), 1020±180 pg/mL (Group C), 950± 110 pg/mL (Group D), and 180 ±30 pg/mL (Group E); p <0.02 for Group B vs Groups C and D and p<0.01 for Group B vs Groups A and E). IGF-R1 expression (Mean Fluorescent Intensity) was significantly higher among HCV mono-infected and HCV/HIV co-infected subjects than that of the other groups (72 ± 3% (Group A), 198± 9% (Group B), 82 ± 8% (Group C), 202±12% (Group D), and 87± 5% (Group E); p value < 0.03 for Groups B and D vs others. These results also validate the respective gene expression pattern observed with DNA microarray analysis.
Figure 5. Chronic HCV Infection is Associated with Markers of Liver Injury.
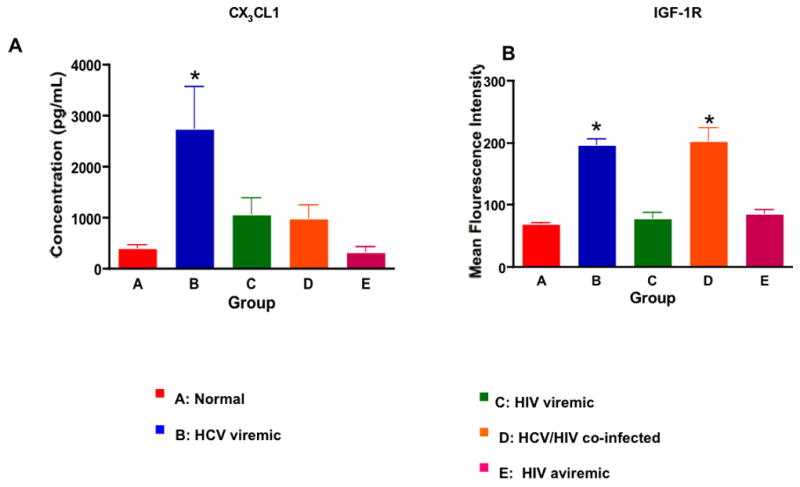
A. The levels of CX3CL1 present in the supernatants of PBMCs from the 5 groups were determined by ELISA. PBMCs from HCV mono-infected individuals secreted significantly higher levels of CX3CL1 than did the other 4 groups (p<0.02). B. Mean Fluorescent Intensity (MFI) of IGF-1R expression on PBMCs showed that HCV-infected (both mono and HIV co-infected) individuals had significantly higher expression of IGF-1R than did the other 3 groups (p<0.02).
PBMCs from HCV mono-infected subjects secrete higher levels of pro-inflammatory cytokines
Since both HCV and HIV are chronic viral infections that could result in immune activation and pro-inflammatory responses, we measured the levels of pro-inflammatory cytokines whose genes were differentially expressed among the groups in the DNA microarray analysis (Cluster 6, Fig 3). The levels of CCL-7 (MCP-3) in the culture supernatants of PBMCs from HCV mono-infected subjects were significantly increased compared to those of the other 4 groups (Fig 6A; 90 ± 9 pg/mL (Group A), 2190±650 pg/mL (Group B), 150 ± 15 pg/mL (Group C), 240 ± 22 pg/mL (Group D), 75 ± 5 pg/mL (Group E); p< 0.001 between Group B vs the others). The levels of CCL-20 (MIP-3α) were also significantly elevated in the culture supernatants of PBMCs from HCV mono-infected subjects compared to those of the other 4 groups (Fig 6B; 10 ± 1 pg/mL (Group A), 51 ± 8 pg/mL (Groups B), 2 ± 0.1 pg/mL (Group C), 48 ± 9 pg/mL (Group D), 1.8 ± 0.1 pg/mL (Group E); p <0.01 between Group B vs Groups A and D and p<0.005 for Group B vs Groups C and E). These results consistently validate our DNA microarray data.
Figure 6. Chronic HCV infection is associated with increased levels of pro-inflammatory chemokines.
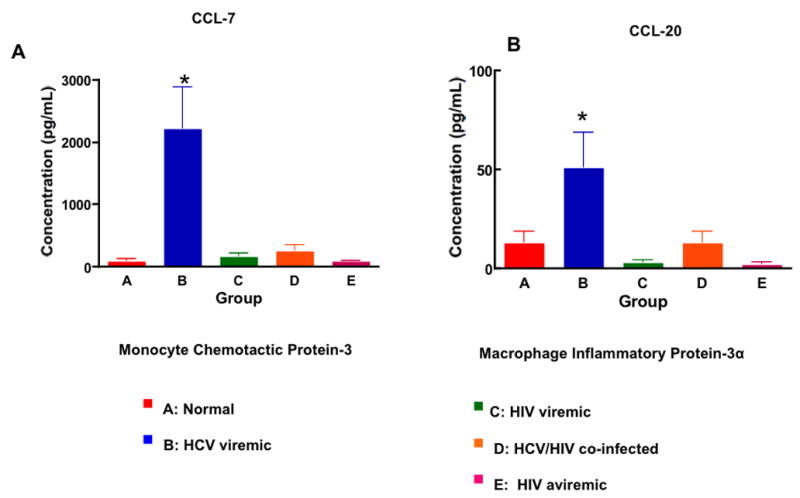
A. The levels of CCL-7 (Monocyte Chemotactic Protein -3) present in the supernatants of PBMCs from the 5 groups were quantified by ELISA. PBMCs from HCV mono-infected individuals secreted significantly higher levels of CCL-7 than did the other 4 groups (p<0.001). B. The levels of CCL-20 (Macrophage Inflammatory Protein-3 α) present in the supernatants of PBMCs from HCV mono-infected individuals were significantly higher than those of the other 4 groups (p<0.01).
HIV-infected subjects express higher levels of IFIG than do HCV mono-infected or normal subjects
We had observed previously that interferon inducible gene (IFIG) expression in PBMCs is significantly higher in HCV/HIV co-infected individuals who fail to respond to treatment (36). Furthermore, we demonstrated that non-responders to IFN-α therapy failed to induce IFIG expression even after exogenous IFN-α treatment (manuscript in preparation). It is unclear whether HCV/HIV co-infected individuals have a higher level of IFIG expression when compared to HCV mono-infected individuals. Therefore, we examined the microarray data for IFIG expression in all 5 groups (Fig 7, A through F) and measured the expression of 25 IFIG by bDNA multiplex. Our results showed that HIV mono-infected individuals had significantly higher expression of IFIG genes than did the other groups (p value <0.05).
Figure 7. HIV-infection results in increased expression of IFIG that is not seen in normal individuals and HCV-infected individuals.

The gene expression profile of several differentially regulated genes was measured using a customized bDNA assay. IFIG genes were found to be up-regulated in HIV viremic individuals compared to other groups of individuals. (p<0.05). Panel A: IFI27 gene expression was significantly higher in PBMCs from Group C compared to Group A (p<0.04), Group B (p<0.035), Group D (p<0.048), and Group E (p<0.034). Panel B: LY6E gene expression was significantly higher in PBMCs from Group C compared to Group A (p<0.02), Group B (p<0.01), Group D (p<0.04), and Group E (p<0.015). Panel C: IFI44 gene expression was significantly higher in PBMCs from Group C compared to Group A (p<0.038), Group B (p<0.02), Group D (p<0.04), and Group E (p<0.038). Panel D: OAS2 gene expression was significantly higher in PBMCs from Group C compared to Group A (p<0.02), Group B (p<0.005), Group D (p<0.03), and Group E (p<0.028). Panel E: IRF7 gene expression was significantly higher in PBMCs from Group C compared to Group A (p<0.042), Group B (p<0.039), Group D (p<0.04), and Group E (p<0.044). Panel F: MX2 gene expression was significantly higher in PBMCs from Group C compared to Group A (p<0.02), Group B (p<0.018), Group D (p<0.022), and Group E (p<0.02).
Discussion
In this study, we demonstrate that HCV and HIV infections induce distinct immunological profiles in PBMCs as determined by gene expression. While HCV infection leads to the expression of genes that reflect a pr-oinflammatory immune response, mainly on non-T cells. HIV infection induces an immune activation profile involving CD4+ and CD8+ T cells Liver activation-specific inflammatory markers were induced in PBMCs of both HCV mono- and HIV/HCV co-infected individuals.. HCV infection induces activation of all peripheral immune cells reflecting a global effect of chronic HCV replication on the immune system Moreover, the IFIG expression, which is highly predictive of the therapeutic response to IFN-α based therapy, is expressed at a higher level in HIV-infected individuals, re-iterating a mechanistic role for type-I IFN baseline hyper-expression in vivo in HCV/HIV co-infected individuals who fail to clear HCV with IFN-α therapy.
Several studies have demonstrated that HIV infection induces a state of immune activation, which is responsible in part for the immunopathogenesis of the disease (37). HIV viremia has been shown to induce cellular activation as demonstrated by serum markers (38), activation of B cells (39), NK cells (25), pDCs (40), CD4+ (41) and CD8+ T cells (42). Although analysis of HIV-induced gene expression profiles in PBMCs was not the primary objective in this study, our results largely confirmed the activation of peripheral T cells in HIV-infected viremic subjects, when compared to those who were aviremic or HIV seronegative. These changes were most remarkable among CD8+ T cells, which showed over expression of genes coding for surface markers, such as CD38, HLA DR, CD25, as well as granzyme and perforin genes, which represent an activated peripheral CD8 response to ongoing HIV replication (data not shown).
Unlike HIV, HCV primarily infects and replicates in human hepatocytes (43). Although several studies have shown that HCV can be detected in non-hepatic cells and tissues, maximal replication is sustained only in primary hepatocytes (44). Recent studies also have shown that chronic HCV infection does not lead to the extent of immune activation of T cells seen in HIV infected subjects (43). Our results also show increased expression of certain genes and expression of various receptors on NK cells (NKp30), B cells (CD10), and pDCs (CD80) in PBMCs from HCV-infected subjects, when compared with HIV-infected or HIV seronegative subjects. This increased expression of NKp30 on NK cells suggests a level of activation of NK cells in chronically viremic HCV mono-infected subjects. Future functional studies should confirm whether this increased expression of a natural cytotoxicity receptor may result in enhanced cytotoxic capacity of NK cells from HCV viremic individuals or whether the over expression of this receptor merely represents a state of aberrant activation or anergy of NK cells from HCV viremic subjects. HCV-infected subjects had a significantly increased proportion of CD10+ immature B cells when compared to normal controls. Expansion of CD10+ B cells have been described in other chronic persistent viral infections such as HIV and is thought to represent an accelerated release of immature B cells as a result of IL-7 that is responding to the CD4+ T cell lymphopenia (39). In this regard, it is possible that chronic HCV infection also results in B cell dysfunction and might explain to some extent the defective humoral immune responses previously described in chronic HCV-infected subjects (45). Finally, PBMCs from HCV mono-infected subjects express higher levels of the T cell co-stimulatory molecule, CD80 when compared to controls. The functional significance of the over expression of this molecule on antigen presenting cells is not completely understood, yet it may represent chronic activation of antigen presenting cells as a result of the persistence of HCV antigens.
Several inflammatory cytokines have been linked to liver damage and the recruitment of effector cells to the liver. There are several markers of liver injury and dysfunction that are elevated in peripheral blood. Expression of CX3CL1 was increased in individuals infected with HCV when compared to those who do not have underlying liver disease. Recent studies have suggested that fractalkine (CX3CL1) recruits CX3CR1-expressing monocytes to the liver that may participate in the process of hepatic inflammation, in agreement with the pro-inflammatory role of fractalkine in other conditions of inflammation (35). These findings reiterate that CX3CL1/CX3CR1 interactions do play a significant role in inducing the hepatic inflammation seen in chronic HCV infection. Our results indicate that fractalkine expression is elevated in the peripheral blood cells of individuals with chronic HCV infection and suggest that this parameter be useful as a marker of liver fibrosis/injury, when validated in larger studies. Furthermore, metabolic abnormalities, such as insulin resistance, diabetes, hyperlipidemia are much more common in HCV-infected individuals than in the other groups (37, 40). The expression of IGF-1R on cells from HCV-infected individuals was higher than that in the other patient groups. As demonstrated in our study, PBMCs of HCV mono-infected individuals produce increased levels of several proinflammatory cytokines. These results suggest a role for these cytokines in recruiting effector inflammatory cells to the liver and also serve as a non-invasive marker of liver fibrosis. Both HCV mono and HIV/HCV co-infected individuals have increased serum levels of markers of liver fibrosis compared to seronegative HIV individuals and HIV-infected individuals without liver disease. These findings, when validated in larger studies will definitively reiterate their role as possible non-invasive markers of liver fibrosis.
In summary, some immune markers such as NKp30 and IGF-R1 seem to be up-regulated among patients with HCV with or without HIV co-infection, some others seem to be selectively up-regulated in HCV mono-infected subjects (CD10, CD80, CCL-7 CCL20 and CX3CL1). It is plausible that HIV infection may have resulted in regulating the expression of some of these receptors. Additionally, it is also possible that the liver disease staging might have influenced the levels of some of these cytokines as well. Further studies are warranted to address whether HIV infection specifically interferes with the effects of chronic HCV infection on immune cells.
Our previous studies have described that high IFIG expression is the single most important predictor of therapeutic non-response to IFN-based treatment for HCV (36). This study demonstrates higher expression of IFIG expression among HIV-infected individuals than HCV mono-infected individuals; suggesting HIV infection is the major driving factor in turning on a type-I IFN signature gene expression. Although our study does not involve follow up analysis of treatment responses of all HCV-infected patients, this is the only study that has compared HCV mono-infected subjects to HIV/HCV co-infected subjects with respect to IFIG expression. The results are highly suggestive that HIV co-infection drives type-I IFN signature gene expression, which could result in blunting of responsiveness to exogenous IFN therapy. A recent study has suggested that increased IFIG expression was seen in the liver of HCV mono-infected patients who were IFN-non-responders than those who achieved SVR (46). However, this study did not examine the hepatic IFIG expression, nor did select out patient population based on treatment response. However, a comparative study to look at hepatic and peripheral IFIG expression among HCV-moni0fected patients is warranted to see if there is a strong correlation between the two compartments.
In summary, our study offers a comprehensive analysis of the differential regulation of host immune responses in HCV and HIV-infected subjects. The results show that both chronic viral infections have distinct immunological profiles that are consistent with the pathogenesis of the disease process. Future studies will be focused on identifying the specific mechanisms involved in the interactions of HIV and HCV that contribute to the establishment of chronicity and the accelerated progression of liver disease seen in co-infected individuals.
Acknowledgments
This research was supported in whole by the Intramural Research Program of the NIH, [National Institute of Allergy and Infectious Diseases and National Institute of Digestive Diseases and Kidney]
Abbreviations
- HCV
Hepatitis C virus
- ART
antiretroviral therapy
- RNA
ribonucleic acid
- HIV
human immunodeficiency virus
- ETR
end of treatment response
- AIDS
acquired immune disease syndrome
- PBMC
peripheral blood mononuclear cell
- PCR
polymerase chain reaction
- ELISA
Enzyme linked immunosorbent assay
- FACS
fluorescent-activated cell sorting
- IRB
Institutional Review Board
- SAM
significant analysis of microarray
- SAPE
Stretavidin-conjugated R-Phycoerythrin
- CCR
CC chemokine receptor
- PE
phosphatidylethanolamine
- APC
allophycocyanin-conjugated
- CCL
CC chemokine ligand
- IL
interleukin
- GRO
growth regulated oncogene
- MCP
macrophage inflammatory proteins
- MCP
monocyte chemotactic protein
- CXCR
CXC chemokine receptor
- TNF
tumor necrosis factor
- CXCL
CXC chemokine ligand
- IFN
interferon
- IFIG
interferon inducible genes
Footnotes
Publisher's Disclaimer: Disclaimer
The content of this publication does not necessarily reflect the views of policies of the Department of Health and Human Services, nor does mention of trade names, commercial products or organization imply endorsement by the U.S. Government.
Conflict of Interest Statement
None of the other authors have any conflicts of interest to report.
References
- 1.Alter MJ, Kruszon-Moran D, Nainan OV, McQuillan GM, Gao F, Moyer LA, Kaslow RA, et al. The prevalence of hepatitis C virus infection in the United States, 1988 through 1994. N Engl J Med. 1999;341:556–562. doi: 10.1056/NEJM199908193410802. [DOI] [PubMed] [Google Scholar]
- 2.Sherman KE, Rouster SD, Chung RT, Rajicic N. Hepatitis C Virus prevalence among patients infected with Human Immunodeficiency Virus: a cross-sectional analysis of the US adult AIDS Clinical Trials Group. Clin Infect Dis. 2002;34:831–837. doi: 10.1086/339042. [DOI] [PubMed] [Google Scholar]
- 3.Selik RM, Byers RH, Jr, Dworkin MS. Trends in diseases reported on U.S. death certificates that mentioned HIV infection, 1987–1999. J Acquir Immune Defic Syndr. 2002;29:378–387. doi: 10.1097/00126334-200204010-00009. [DOI] [PubMed] [Google Scholar]
- 4.Jain MK, Skiest DJ, Cloud JW, Jain CL, Burns D, Berggren RE. Changes in mortality related to human immunodeficiency virus infection: comparative analysis of inpatient deaths in 1995 and in 1999–2000. Clin Infect Dis. 2003;36:1030–1038. doi: 10.1086/368186. [DOI] [PubMed] [Google Scholar]
- 5.Selik RM, Lindegren ML. Changes in deaths reported with human immunodeficiency virus infection among United States children less than thirteen years old, 1987 through 1999. The Pediatric infectious disease journal. 2003;22:635–641. doi: 10.1097/01.inf.0000073241.01043.9c. [DOI] [PubMed] [Google Scholar]
- 6.Benhamou Y, Bochet M, Di Martino V, Charlotte F, Azria F, Coutellier A, Vidaud M, et al. Liver fibrosis progression in human immunodeficiency virus and hepatitis C virus coinfected patients. The Multivirc Group. Hepatology. 1999;30:1054–1058. doi: 10.1002/hep.510300409. [DOI] [PubMed] [Google Scholar]
- 7.Aceti A, Mangoni ML, Pasquazzi C, Fiocco D, Marangi M, Miele R, Zechini B, et al. Alpha-defensin increase in peripheral blood mononuclear cells from patients with hepatitis C virus chronic infection. J Viral Hepat. 2006;13:821–827. doi: 10.1111/j.1365-2893.2006.00762.x. [DOI] [PubMed] [Google Scholar]
- 8.Sulkowski M. HIV and hepatitis C virus co-infection. Hopkins HIV Rep. 1998;10:8, 12. [PubMed] [Google Scholar]
- 9.Sulkowski MS. Hepatitis C virus and HIV co-infection: a sleeping giant wakes. Hopkins HIV Rep. 1999;11:3, 10–12. [PubMed] [Google Scholar]
- 10.Sulkowski MS. Hepatitis C Virus Infection in HIV-infected Patients. Curr Infect Dis Rep. 2001;3:469–476. [PubMed] [Google Scholar]
- 11.Sulkowski MS, Mast EE, Seeff LB, Thomas DL. Hepatitis C virus infection as an opportunistic disease in persons infected with human immunodeficiency virus. Clin Infect Dis. 2000;30 (Suppl 1):S77–84. doi: 10.1086/313842. [DOI] [PubMed] [Google Scholar]
- 12.Sulkowski MS, Moore RD, Mehta SH, Chaisson RE, Thomas DL. Hepatitis C and progression of HIV disease. Jama. 2002;288:199–206. doi: 10.1001/jama.288.2.199. [DOI] [PubMed] [Google Scholar]
- 13.Eyster ME. Concurrent HIV and HCV infections hasten liver failure. Am Fam Physician. 1992;46:536. [PubMed] [Google Scholar]
- 14.Carrat F, Bani-Sadr F, Pol S, Rosenthal E, Lunel-Fabiani F, Benzekri A, Morand P, et al. Pegylated interferon alfa-2b vs standard interferon alfa-2b, plus ribavirin, for chronic hepatitis C in HIV-infected patients: a randomized controlled trial. Jama. 2004;292:2839–2848. doi: 10.1001/jama.292.23.2839. [DOI] [PubMed] [Google Scholar]
- 15.Laguno M, Murillas J, Blanco JL, Martinez E, Miquel R, Sanchez-Tapias JM, Bargallo X, et al. Peginterferon alfa-2b plus ribavirin compared with interferon alfa-2b plus ribavirin for treatment of HIV/HCV co-infected patients. Aids. 2004;18:F27–36. doi: 10.1097/00002030-200409030-00003. [DOI] [PubMed] [Google Scholar]
- 16.Soriano V, Perez-Olmeda M, Rios P, Nunez M, Garcia-Samaniego J, Gonzalez-Lahoz J. Hepatitis C virus (HCV) relapses after anti-HCV therapy are more frequent in HIV-infected patients. AIDS Res Hum Retroviruses. 2004;20:351–353. doi: 10.1089/088922204323048096. [DOI] [PubMed] [Google Scholar]
- 17.Torriani FJ, Ribeiro RM, Gilbert TL, Schrenk UM, Clauson M, Pacheco DM, Perelson AS. Hepatitis C virus (HCV) and human immunodeficiency virus (HIV) dynamics during HCV treatment in HCV/HIV coinfection. J Infect Dis. 2003;188:1498–1507. doi: 10.1086/379255. [DOI] [PubMed] [Google Scholar]
- 18.Rehermann B. Interaction between the hepatitis C virus and the immune system. Semin Liver Dis. 2000;20:127–141. doi: 10.1055/s-2000-9946. [DOI] [PubMed] [Google Scholar]
- 19.Fauci AS, Pantaleo G, Stanley S, Weissman D. Immunopathogenic mechanisms of HIV infection. Ann Intern Med. 1996;124:654–663. doi: 10.7326/0003-4819-124-7-199604010-00006. [DOI] [PubMed] [Google Scholar]
- 20.Bukh J, Miller RH, Purcell RH. Genetic heterogeneity of hepatitis C virus: quasispecies and genotypes. Semin Liver Dis. 1995;15:41–63. doi: 10.1055/s-2007-1007262. [DOI] [PubMed] [Google Scholar]
- 21.Chun TW, Justement JS, Lempicki RA, Yang J, Dennis G, Jr, Hallahan CW, Sanford C, et al. Gene expression and viral prodution in latently infected, resting CD4+ T cells in viremic versus aviremic HIV-infected individuals. Proc Natl Acad Sci U S A. 2003;100:1908–1913. doi: 10.1073/pnas.0437640100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Tibshirani R, Hastie T, Narasimhan B, Chu G. Diagnosis of multiple cancer types by shrunken centroids of gene expression. Proceedings of the National Academy of Sciences of the United States of America. 2002;99:6567–6572. doi: 10.1073/pnas.082099299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Kulkarni RN. Receptors for insulin and insulin-like growth factor-1 and insulin receptor substrate-1 mediate pathways that regulate islet function. Biochemical Society transactions. 2002;30:317–322. doi: 10.1042/bst0300317. [DOI] [PubMed] [Google Scholar]
- 24.Forster R, Davalos-Misslitz AC, Rot A. CCR7 and its ligands: balancing immunity and tolerance. Nature reviews. 2008;8:362–371. doi: 10.1038/nri2297. [DOI] [PubMed] [Google Scholar]
- 25.Byrd A, Hoffmann SC, Jarahian M, Momburg F, Watzl C. Expression analysis of the ligands for the Natural Killer cell receptors NKp30 and NKp44. PLoS ONE. 2007;2:e1339. doi: 10.1371/journal.pone.0001339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Saederup N, Chan L, Lira SA, Charo IF. Fractalkine deficiency markedly reduces macrophage accumulation and atherosclerotic lesion formation in CCR2−/−mice: evidence for independent chemokine functions in atherogenesis. Circulation. 2008;117:1642–1648. doi: 10.1161/CIRCULATIONAHA.107.743872. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Loken MR, Shah VO, Hollander Z, Civin CI. Flow cytometric analysis of normal B lymphoid development. Pathology and immunopathology research. 1988;7:357–370. doi: 10.1159/000157129. [DOI] [PubMed] [Google Scholar]
- 28.Lebedeva T, Dustin ML, Sykulev Y. ICAM-1 co-stimulates target cells to facilitate antigen presentation. Current opinion in immunology. 2005;17:251–258. doi: 10.1016/j.coi.2005.04.008. [DOI] [PubMed] [Google Scholar]
- 29.Slavik JM, Hutchcroft JE, Bierer BE. CD28/CTLA-4 and CD80/CD86 families: signaling and function. Immunologic research. 1999;19:1–24. doi: 10.1007/BF02786473. [DOI] [PubMed] [Google Scholar]
- 30.Dong H, Chen X. Immunoregulatory role of B7-H1 in chronicity of inflammatory responses. Cellular & molecular immunology. 2006;3:179–187. [PMC free article] [PubMed] [Google Scholar]
- 31.Zeremski M, Petrovic LM, Talal AH. The role of chemokines as inflammatory mediators in chronic hepatitis C virus infection. Journal of viral hepatitis. 2007;14:675–687. doi: 10.1111/j.1365-2893.2006.00838.x. [DOI] [PubMed] [Google Scholar]
- 32.Sager R, Haskill S, Anisowicz A, Trask D, Pike MC. GRO: a novel chemotactic cytokine. Advances in experimental medicine and biology. 1991;305:73–77. doi: 10.1007/978-1-4684-6009-4_9. [DOI] [PubMed] [Google Scholar]
- 33.Mahalingam S, Karupiah G. Chemokines and chemokine receptors in infectious diseases. Immunology and cell biology. 1999;77:469–475. doi: 10.1046/j.1440-1711.1999.00858.x. [DOI] [PubMed] [Google Scholar]
- 34.Rockey DC, Bissell DM. Noninvasive measures of liver fibrosis. Hepatology (Baltimore, Md. 2006;43:S113–120. doi: 10.1002/hep.21046. [DOI] [PubMed] [Google Scholar]
- 35.Wasmuth HE, Zaldivar MM, Berres ML, Werth A, Scholten D, Hillebrandt S, Tacke F, et al. The fractalkine receptor CX3CR1 is involved in liver fibrosis due to chronic hepatitis C infection. Journal of hepatology. 2008;48:208–215. doi: 10.1016/j.jhep.2007.09.008. [DOI] [PubMed] [Google Scholar]
- 36.Lempicki RA, Polis MA, Yang J, McLaughlin M, Koratich C, Huang DW, Fullmer B, et al. Gene expression profiles in hepatitis C virus (HCV) and HIV coinfection: class prediction analyses before treatment predict the outcome of anti-HCV therapy among HIV-coinfected persons. The Journal of infectious diseases. 2006;193:1172–1177. doi: 10.1086/501365. [DOI] [PubMed] [Google Scholar]
- 37.Appay V, Sauce D. Immune activation and inflammation in HIV-1 infection: causes and consequences. The Journal of pathology. 2008;214:231–241. doi: 10.1002/path.2276. [DOI] [PubMed] [Google Scholar]
- 38.Sipsas NV, Sfikakis PP, Touloumi G, Pantazis N, Choremi H, Kordossis T. Elevated serum levels of soluble immune activation markers are associated with increased risk for death in HAART-naive HIV-1-infected patients. AIDS patient care and STDs. 2003;17:147–153. doi: 10.1089/108729103321619755. [DOI] [PubMed] [Google Scholar]
- 39.Malaspina A, Moir S, Ho J, Wang W, Howell ML, O’Shea MA, Roby GA, et al. Appearance of immature/transitional B cells in HIV-infected individuals with advanced disease: correlation with increased IL-7. Proceedings of the National Academy of Sciences of the United States of America. 2006;103:2262–2267. doi: 10.1073/pnas.0511094103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Boasso A, Shearer GM. Chronic innate immune activation as a cause of HIV-1 immunopathogenesis. Clinical immunology (Orlando, Fla. 2008;126:23–242. doi: 10.1016/j.clim.2007.08.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Kestens L, Vanham G, Vereecken C, Vandenbruaene M, Vercauteren G, Colebunders RL, Gigase PL. Selective increase of activation antigens HLA-DR and CD38 on CD4+ CD45RO+ T lymphocytes during HIV-1 infection. Clinical and experimental immunology. 1994;95:436–441. doi: 10.1111/j.1365-2249.1994.tb07015.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Kestens L, Vanham G, Gigase P, Young G, Hannet I, Vanlangendonck F, Hulstaert F, et al. Expression of activation antigens, HLA-DR and CD38, on CD8 lymphocytes during HIV-1 infection. AIDS (London, England) 1992;6:793–797. doi: 10.1097/00002030-199208000-00004. [DOI] [PubMed] [Google Scholar]
- 43.Kanto T, Hayashi N. Immunopathogenesis of hepatitis C virus infection: multifaceted strategies subverting innate and adaptive immunity. Internal medicine (Tokyo, Japan) 2006;45:183–191. doi: 10.2169/internalmedicine.45.1530. [DOI] [PubMed] [Google Scholar]
- 44.Dahari H, Major M, Zhang X, Mihalik K, Rice CM, Perelson AS, Feinstone SM, et al. Mathematical modeling of primary hepatitis C infection: noncytolytic clearance and early blockage of virion production. Gastroenterology. 2005;128:1056–1066. doi: 10.1053/j.gastro.2005.01.049. [DOI] [PubMed] [Google Scholar]
- 45.Chen M, Sallberg M, Sonnerborg A, Weiland O, Mattsson L, Jin L, Birkett A, et al. Limited humoral immunity in hepatitis C virus infection. Gastroenterology. 1999;116:135–143. doi: 10.1016/s0016-5085(99)70237-4. [DOI] [PubMed] [Google Scholar]
- 46.Chen L, Borozan I, Feld J, Sun J, Tannis LL, Coltescu C, Heathcote J, Edwards AM, McGilvray ID. Hepatic gene expression discriminates responders and nonresponders in treatment of chronic hepatitis C viral infection. Gastroenterology. 2005;128(5):1437–44. doi: 10.1053/j.gastro.2005.01.059. [DOI] [PubMed] [Google Scholar]