

Abstract
The Pd/phosphine-catalyzed reaction of 1 with aryl bromides leads to the selective synthesis of either 6-aryl octahydrocyclopenta[b]pyrroles (3), the corresponding 5-aryl isomers 5, diarylamine 2, or hexahydrocyclopenta[b]pyrrole 4 depending on the structure of the phosphine ligand. These transformations are effective with a variety of different aryl bromides, and provide 3-5 with excellent levels of diastereoselectivity (dr ≥ 20:1). The changes in product distribution are believed to derive from the influence of Pd-catalyst structure on the relative rates of reductive elimination, β-hydride elimination, alkene insertion, and alkene dissociation processes in a mechanistically complex reaction. The effect of phosphine ligand structure on product distribution is described in detail, along with analysis of a proposed mechanism for these transformations.
Introduction
A significant challenge in the development of metal-catalyzed reactions is the suppression of competing mechanistic pathways without inhibiting desired steps in a catalytic cycle. In recent years several remarkable transformations have been effected through the use of palladium catalysts that minimize side reactions (e.g. β-hydride elimination) while still allowing reductive elimination or transmetalation processes to occur.1 Despite these achievements, the factors that affect the relative rates of competing mechanistic pathways in catalytic reactions (e.g. reductive elimination vs. olefin insertion, or C-C vs. C-N bond-forming reductive elimination) are not well understood. If these fundamental processes could be controlled, the selective construction of a diverse array of products from common starting materials could be achieved under similar reaction conditions by varying catalyst structure.
 |
(1) |
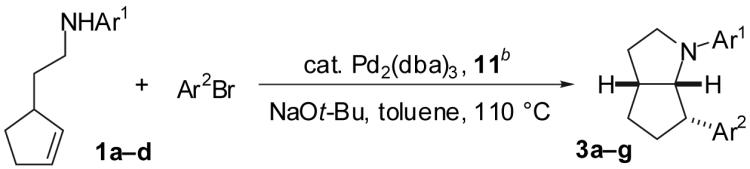
We recently described a new stereoselective synthesis of N-aryl pyrrolidines via Pd-catalyzed reactions of γ-(N-arylamino)alkenes with aryl bromides (eq 1).2,3,4 These transformations lead to the formation of a C-C and a C-N bond along with up to two stereocenters in a single process. During the course of these studies, we noted that the Pd/P(o-tol)3-catalyzed reaction of 1a with 4-bromobiphenyl provides an unexpected mixture of four products (Scheme 1, 2-5). We suggested that these products may derive from a common intermediate (6), which undergoes reductive elimination to afford 2,5 or intramolecular alkene insertion into the Pd-N bond6,7,8 followed by: a) reductive elimination to afford 3,9 b) β-hydride elimination and alkene displacement to afford 4, or c) β-hydride elimination, reinsertion, and reductive elimination to afford 5 (Scheme 1).2 Despite the apparently complex mechanism of this reaction, we felt that selective formation of 2-5 might be achieved by altering the catalyst structure and/or the nitrogen nucleophilicity to control the relative rates (k1-k7) of each possible step in the catalytic cycle. This would provide access to a variety of products with a 2-azabicyclo[3.3.0]octane core, which is found in a number of biologically active molecules.10,11 In this Article we present a detailed description of the effects of ligand structure, nitrogen nucleophilicity and aryl halide electronics on the ratios of products observed in reactions of 1 with aryl bromides. These studies have led to the selective synthesis of products 2-5 in good yields from a single precursor via the use of an appropriate catalyst for each transformation. The experiments described herein also provide further evidence to support the mechanistic hypothesis outlined in Scheme 1.
Scheme 1.

(a) Conditions: 1.0 equiv 1a, 1.2 equiv 4-Ph(C6H4)Br, 1.4 equiv NaOtBu, 1 mol % Pd2(dba)3, 4 mol % P(o-tol)3, toluene, 110 °C. Isolated yield: 2 (19%), 3 (32%), 4 (5%), 5 (12%) for Ar = 4-methoxyphenyl, Ar1 = 4-(phenyl)phenyl.
Results
Effect of Ligand Structure on Product Distribution
Prior to the start of our optimization studies, we sought to identify catalyst properties that might be expected to provide some degree of selectivity for each of the products 2-5. The mechanistic proposal described above (Scheme 1) suggests that if the rate of reductive elimination from 6 (k1) is fast relative to all other steps, then 2 should be obtained in high yield. Thus, we hypothesized that the selective synthesis of product 2 could be achieved in a straightforward manner by employing palladium catalysts supported by bulky, electron-rich phosphine ligands that are known to be highly effective at promoting C-N bond forming reductive elimination reactions.5
We reasoned that if the rates of all possible C-N and C-C bond forming reductive elimination steps (k1, k5, and k7) could be minimized, the reaction may be directed towards product 4 provided that the rate of alkene insertion into the Pd-N bond is not completely inhibited. The two main classes of phosphines that are well documented in their ability to decrease the rate of reductive elimination reactions are small, electron-rich ligands (e.g. PMe2Ph)12 and chelating ligands with small bite angles (e.g. dppe).12,13 Therefore, these types of ligands seemed to be potentially viable candidates for the selective formation of 4.14
The selective formation of products 3 and 5 was more difficult to envision, as the formation of these products requires conditions that minimize C-N bond-forming reductive elimination without completely suppressing C-C bond-forming reductive elimination. In the case of product 3, the rate of C-C bond forming reductive elimination of intermediate 7 would need to be faster than competing β-hydride elimination (k3>k4). We reasoned that it might be feasible to achieve selectivity for 3 by employing chelating ligands with moderate to large bite angles (e.g. dppf, BINAP, or dpe-phos) to minimize the rate of β-hydride elimination15 (k4) while still allowing C-C bond forming reductive elimination16 of 7 to occur at a reasonable rate (k3).17 If adequate control of the relative rates of C-N and C-C bond-forming reductive elimination could not be achieved through variation of phosphine ligand, we felt it should be possible to decrease the rate of C-N bond forming reductive elimination by employing an electron-deficient N-aryl substituent on the substrate 1.18
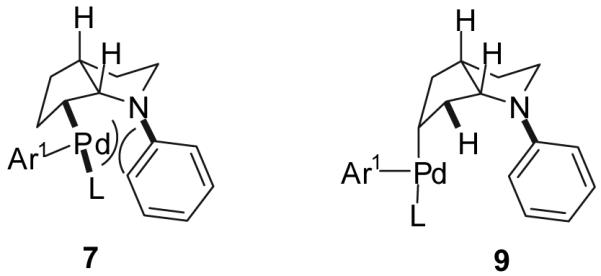
The selective synthesis of 5 would require a scenario in which the rate of reversible β-hydride elimination19 from 7 to afford 8 is fast relative to the rate of reductive elimination from 7 (k4>k3). In this scenario it seemed likely that the equilibrium between alkylpalladium complex 7 and π-complex 8 would lie far to the right, as examination of molecular models reveals a severe steric interaction between the N-aryl substituent and the metal fragment in 7 (Figure 1). In principle, 8 could undergo reversible reinsertion of the alkene into the Pd-H bond with opposite regiochemistry19 to afford 9; the position of the equilibrium between 8 and 9 is difficult to predict. However, if equilibration is rapid and the rate of reductive elimination from 9 is faster than the rate of the alkene displacement from 8 (k7>k5) then selective formation of 5 should occur. There was not sufficient literature precedent to allow us to predict how ligand variation would affect the relative rates of the required olefin insertion processes vs. reductive elimination reactions. However, the effects of ligand steric and electronic properties on the rates of reductive elimination reactions are well documented.20 Additionally, we expected that increasing the size and electron-donor ability of the ligand should slow the rate of alkene displacement from 4, which likely proceeds through an associative mechanism involving attack of an external nucleophile.19,21,22,23 Thus, it seemed feasible that the correct combination of ligand steric and electronic properties could potentially provide good selectivity for 5.
Figure 1.
In order to probe the hypotheses described above, we examined the Pd-catalyzed reaction of 1a with 4-bromotoluene in the presence of a variety of different phosphine ligands (eq 2). All reactions examined proceeded to completion with a catalyst loading of 1 mol % of Pd2(dba)3 except reactions conducted with PMe3 and PEt3, which both stopped at ca. 80% conversion. In all cases compounds 2-5 accounted for ≥95% of the products detected by GC and/or 1H NMR analysis of the crude reaction mixtures, and products derived from oxidation of 1a to the corresponding N-aryl imine via β-hydride elimination of 65 were not observed in most reactions.
As shown in Table 1, experiments with monodentate trialkylphosphine ligands24 demonstrated that selectivity is highly dependent on the steric bulk of the ligand, and that excellent selectivity for products 2, 4, and 5 can be achieved with the appropriate ligand. In general, the very bulky ligands (cone angle ≥178°) favored N-arylation, the medium-sized ligands (cone angle = 160-174°) favored the formation of 5, and the very small ligands (cone angle ≤132°) favored the formation of 4. Use of preformed L2Pd complexes1a,25 gave similar results to those obtained with Pd2(dba)3/phosphine mixtures.
 |
(2) |
Table 1. Trialkylphosphine Ligand Effects(a).
| Entry | Ligand | Cone Angle | 2 | 3a | 4 | 5a |
|---|---|---|---|---|---|---|
| 1(b),(c) | PMe3·HBF4 | 11826a | 0 | 0 | 98 | 2 |
| 2(b),(c) | PEt3·HBF4 | 13226a | 0 | 0 | 92 | 8 |
| 3 | P(t-Bu)2Me·HBF4 | 16126b | 0 | 0 | 0 | 100 |
| 4 | Pd[PMe(t-Bu)2]2 | 16126b | 0 | 0 | 0 | 100 |
| 5 | Pd[PCy3]2 | 17026a | 1 | 2 | 1 | 96 |
| 6 | PCy3 | 17026a | 2 | 2 | 6 | 90 |
| 7 | Pd[P(t-Bu)Cy2]2 | 17427 | 6 | 6 | 0 | 88 |
| 8 | Pd[P(t-Bu)2Cy]2 | 17827 | 81 | 5 | 0 | 14 |
| 9 | Pd[P(t-Bu)3]2 | 18226a | 100 | 0 | 0 | 0 |
| 10(b) | P(t-Bu)3·HBF4 | 18226a | 98 | 2 | 0 | 0 |
Conditions: 1.0 equiv 1a, 2.0 equiv 4-bromotoluene, 1.2 equiv NaOtBu, 1 mol % Pd2(dba)3, 4 mol % ligand, toluene (0.25 M), 110 °C. The product ratios refer to GC ratios of products 2-5 that have been corrected for GC response factor. All reactions proceed to completion unless otherwise noted.
The reaction was conducted with 1.4 equiv 4-bromotoluene.
The reaction proceeded to 80% conversion.
The selectivities observed in Pd-catalyzed reactions conducted with other monodentate phosphine ligands are shown in Table 2. In general, sterically bulky ligands favor the formation of 2 and/or 3, whereas smaller ligands provide 4 and/or 5 as the major products. Of the ligands examined in this series, bulky, electron-rich phosphines provided the highest selectivity for 2, and the selectivity for 4 increased with decreasing ligand size. Use of ligands that were extremely bulky (e.g tris[2,4,6-trimethoxyphenyl]phosphine), and/or extremely electron poor (e.g. P[C6F5]3, P[o-C6H4CF3]3) resulted in low reactivity.28
Table 2. Effect of Other Monodentate Phosphine Ligands(a).
| Entry | Ligand | Cone Angle | 2 | 3a | 4 | 5a |
|---|---|---|---|---|---|---|
| 1 | t-Bu2P(o-biphenyl) (10) | 94 | 6 | 0 | 0 | |
| 2 | 2-(Dicyclohexylphosphino)-2′-(N,N-dimethylamino)biphenyl | 76 | 24 | 0 | 0 | |
| 3 | Cy2P(o-biphenyl) | 52 | 39 | 1 | 8 | |
| 4 | 2-(Diphenylphosphino)-2′-(N,N-dimethylamino)biphenyl (11) | 56 | 40 | 2 | 2 | |
| 5 | P(o-tol)3 | 19426a | 18 | 42 | 9 | 31 |
| 6 | P[C6H4(m-CF3)]3 | 16526c | 4 | 31 | 32 | 33 |
| 7 | P[C6H4(p-CF3)]3 | 14526d | 3 | 28 | 26 | 43 |
| 8 | PPh3 | 14526a | 3 | 9 | 55 | 33 |
| 9 | P[C6H4(p-OMe)]3 | 14526c | 0 | 5 | 59 | 36 |
| 10 | P(2-furyl)3 | 0 | 8 | 82 | 10 | |
| 11 | Ph2PMe | 13626a | 0 | 0 | 86 | 14 |
| 12(b) | PhPMe2 | 12226a | 0 | 0 | 92 | 8 |
Conditions: 1.0 equiv 1a, 2.0 equiv 4-bromotoluene, 1.2 equiv NaOtBu, 1 mol % Pd2(dba)3, 4 mol % ligand, toluene (0.25 M), 110 °C. The product ratios refer to GC ratios of products 2-5 that have been corrected for GC response factor. All reactions proceed to completion unless otherwise noted.
The reaction was conducted with 1.4 equiv 4-bromotoluene.
To probe the effect of chelation and ligand bite angle on product distribution, a number of bidentate ligands were employed in the transformation shown in eq 1; the results are described in Table 3. Systematic variation of ligand bite angle did not result in clear trends of product distribution. However, electronic effects similar to those described above for reactions involving monodentate ligands were observed within series of ligands that possess identical backbone structures. For example, reactions catalyzed by mixtures of Pd2(dba)3 and 1,2-bis(phosphino)ethane derivatives (entries 2-4 and 6) afforded increasing amounts of 4 and 5a (relative to 2 and 3a) as the electron-donicity of the phosphines increased. Similarly, use of 1,2-bis(diphenylphosphino)ferrocene as ligand afforded predominantly 2 and 3a, whereas 1,2-bis(diisopropylphosphino)ferrocene provided 5a as the major product (entries 10 and 11).
Table 3. Effect of Chelating Phosphine Ligands.
| Entry | Ligand | Bite Angle(c) | 2 | 3a | 4 | 5a |
|---|---|---|---|---|---|---|
| 1 | dppm | 7329a | 12 | 11 | 51 | 26 |
| 2 | dppe-p-CF3 | 86(d) | 20 | 25 | 28 | 27 |
| 3(g) | dppe | 8629a | 5 | 7 | 63 | 25 |
| 4 | dppe-p-OMe | 86(d) | 3 | 4 | 69 | 24 |
| 5 | dpp-benzene | 87(e),29b | 87 | 3 | 7 | 4 |
| 6 | dcpe | 8729c | 11 | 4 | 19 | 66 |
| 7 | dppp | 9129a | 31 | 58 | 8 | 3 |
| 8 | BINAP | 9329d | 32 | 9 | 37 | 22 |
| 9(g) | dppb | 9429e | 4 | 10 | 56 | 30 |
| 10 | dppf | 9929f | 27 | 42 | 12 | 19 |
| 11 | dppf-i-Pr | 10329g | 13 | 12 | 0 | 75 |
| 12 | dpe-phos | 104(f),29h | 19 | 27 | 37 | 17 |
| 13 | xantphos | 108(f),29h | 54 | 46 | 0 | 0 |
Conditions: 1.0 equiv 1a, 2.0 equiv 4-bromotoluene, 1.2 equiv NaOtBu, 1 mol % Pd2(dba)3, 2 mol % ligand, toluene (0.25 M), 110 °C. The product ratios refer to GC ratios of products 2-5 that have been corrected for GC response factor. All reactions proceed to completion unless otherwise noted.
The reaction was conducted with 1.4 equiv 4-bromotoluene.
Bite angles were obtained from crystal structures of bis(phosphine)PdCl2 complexes.
The bite angle was estimated based on analogy to dppe.
The bite angle was obtained from a crystal structure of the bis(phosphine)PdBr2 complex.
The bite angle was obtained from a crystal structure of the bis(phosphine)Pd(1,1-dimethylallyl)tetrafluoroborate complex.
Approximately 3% of an imine side product derived from substrate oxidation was detected by GC and GC/MS analysis of the crude reaction mixture.
In summary, the best selectivity for 2 was obtained with the bulky, monodentate, electron-rich ligands P(t-Bu)3 and t-Bu2P(o-biphenyl) (10). Use of the small, monodentate electron-rich ligands PMe3, PEt3, and PhPMe2 provided the best selectivity for 4, whereas the medium-sized, electron-rich ligands P(t-Bu)2Me and PCy3 provided the highest selectivity for 5a. None of the ligands examined provided excellent selectivity for 3a, however dppp provided modest selectivity for 3a, with 2 formed as the major side product. The xantphos, Cy2P(o-biphenyl), and 2-(diphenylphosphino)-2′-(N,N-dimethylamino)biphenyl (11) ligands also provided close to 1:1 ratios of 2:3a with only small amounts of 4 and 5a generated. Although the selectivity for 3a was not ideal, we reasoned that we could further improve the ratio of 3a:2 by employing substrates bearing electron-withdrawing N-aryl substituents, which would slow the rate of competing C-N bond-forming reductive elimination (see below).5
Selective Synthesis of 5-Aryl Octahydrocyclopenta[b]pyrroles (3): Effect of Aryl Bromide Structure and N-Aryl Substituent
With optimized conditions in hand, we examined the reaction of several derivatives of 1 with a number of different aryl bromides; the results are summarized below. As shown in Table 4, the reaction of 4-bromotoluene with 1a afforded a modest 44% isolated yield of 3a in the presence of the Pd2(dba)3/dppp catalyst system (entry 1). Analysis of the crude reaction mixture showed that the four products (2, 3a, 4, and 5a) were formed in a 25:65:10:0 ratio; the Pd2(dba)3/11 catalyst provided a 48:48:2:2 ratio of 2:3a:4:5a (entry 2). As expected, significantly higher yields of isomer 3 were obtained with substrates bearing electron-deficient N-aryl substituents (entries 3-5, and 7). For example, the reaction of substrate 1d bearing a N-para-carbo-tert-butoxyphenyl group with 4-bromotoluene afforded a 75% yield of 3e (entry 7). Use of 11 as ligand provided superior results in reactions of substrates 1c and 1d, which failed to react in the presence of the Pd2(dba)3/dppp catalyst system. Aryl halide electronics also had a significant effect on the selectivity with which products 3a-g were formed. As noted above, the reaction of electron-deficient substrate 1d with 4-bromotoluene afforded 3e in 75% isolated yield as the sole detectable product (entry 7), whereas the reaction of 1d with the electron-rich 4-bromoanisole afforded a separable 9:1 mixture of 3f:4d (entry 8), and the reaction of 1d with an aryl bromide bearing an electron-withdrawing substituent led to the formation of significant amounts of N-arylated side products (entry 9). In all cases examined, the 2-azabicyclo[3.3.0]octane derivatives 3a-g were obtained with diastereomeric ratios of ≥20:1.
Table 4. Selective Synthesis of 3.
 | ||||
|---|---|---|---|---|
| Entry | Substrate(c) | Crude Ratio 2:3:4:5 | Product | Isolated Yield 3 |
| 1 | 1a | 25:65:10:0 |  |
44 %(d) |
| 2 | 1a | 48:48:2:2 | - | |
| 3 | 1b | 20:70:10:0 |  |
65 %(d) |
| 4 | 1b | 30:65:2:3 | - | |
| 5 | 1c | 10:80:10:0 | 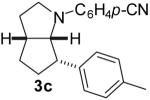 |
69 %(e) |
| 6 | 1c | 0:100:0:0 | 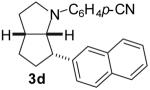 |
64 % |
| 7 | 1d | 0:100:0:0 | 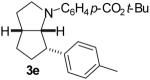 |
75 % |
| 8 | 1d | 0:90:10:0 | 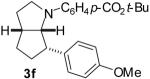 |
58 % |
| 9 | 1d | 30:60:10:0 | 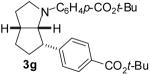 |
49 % |
Conditions: 1.0 equiv amine, 1.4 equiv Ar2Br, 1.2 equiv NaOt-Bu, 1 mol % Pd2(dba)3, 2 mol % 11, toluene (0.25 M), 110 °C.
11 = 2-diphenylphosphino-2′-(N,N-dimethylamino)biphenyl.
1a: Ar = p-(MeO)C6H4; 1b: Ar = p-(Cl)C6H4; 1c : Ar = p-(NC)C6H4; 1d: Ar = p-(t-BuO2C)C6H4.
Ligand = dppp.
This material contained 7 % of 4c as an inseparable impurity.
Selective Synthesis of 6-Aryl Octahydrocyclopenta[b]pyrroles (5): Effect of Aryl Bromide Structure and Nitrogen Electronics
As shown in Table 5, the selective synthesis of isomer 5 was achieved for a variety of different substrate combinations using the Pd2(dba)3/P(t-Bu)2Me·HBF4 catalyst system. Good to excellent selectivities and yields were obtained in transformations of substrates bearing electron-rich, -neutral, or -poor N-substituents. The reactions also proceeded in good yield with aryl bromides of varying electronic properties, although the ratios of 5:3 decreased when certain electron-deficient or o-substituted aryl bromides were employed as coupling partners (entries 3, 4, and 6). Products 2 and 4 were not observed except in the reaction of 1c with 3-bromopyridine (entry 5). In all cases the products 5a-g were obtained with ≥ 20:1 dr.
Table 5. Selective Synthesis of 5.
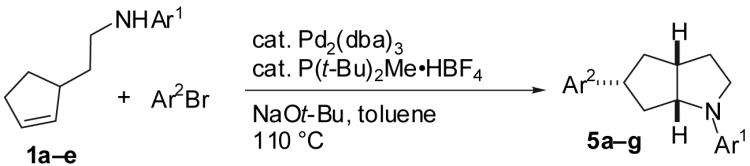 | ||||
|---|---|---|---|---|
| Entry | Substrate(b) | Crude Ratio 2:3:4:5 | Product | Isolated Yield 5 |
| 1 | 1a | 0:0:0:100 | 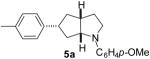 |
74 % |
| 2 | 1a | 0:0:0:100 | 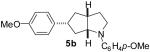 |
75 % |
| 3 | 1b | 0:15:0:85 | 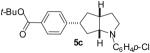 |
71 % |
| 4 | 1d | 0:10:0:90 | 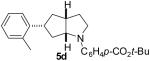 |
69 % |
| 5 | 1c | 5:10:10:75 | 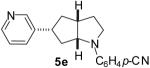 |
67 %(c) |
| 6 | 1e | 0:10:0:90 | 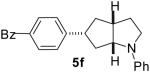 |
85 % |
| 7 | 1e | 0:0:0:100 | 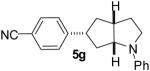 |
76 % |
Conditions: 1.0 equiv amine, 1.4 equiv Ar2Br, 1.2 equiv NaOt-Bu, 1 mol % Pd2(dba)3, 2 mol % P(t-Bu)2Me·HBF4, toluene (0.25 M), 110 °C.
1a : Ar = p-(MeO)C6H4; 1b: Ar = p-(Cl)C6H4; 1c: Ar = p-(NC)C6H4; 1d: Ar =p-(t-BuO2C)C6H4; 1e Ar = Ph.
This material contained 7 % of isomer 3 as an inseparable impurity.
Selective Synthesis of Oxidative Amination Product 4 and N-Arylated Product 2
The synthesis of oxidative amination30 product 4 was achieved in 60% yield as shown in eq 3; the best results were obtained with PMe3 as ligand. Although 4 is the sole product formed under these conditions, the reaction did not proceed beyond 80% consumption of starting material; use of 2 mol % Pd2(dba)3 led to only modest improvements in conversion and yield. Reactions in which PMePh2, PMe2Ph, or P(2-furyl)3 were employed as ligands all provided similar isolated yields (44-54%) despite differences in selectivity due to the difficult chromatographic separation of 4 from 5.31
 |
(3) |
The N-arylated product 2 was obtained in 88% yield using the Pd2(dba)3/t-Bu2P(o-biphenyl) (10) catalyst system (eq 4). The scope of this transformation was not explored due to the large body of existing literature on the effect of aryl bromide substitution on Pd-catalyzed N-arylation reactions.5
 |
(4) |
Discussion
Synthetic Scope and Limitations
The palladium-catalyzed reaction of N-aryl-2-(cyclopent-2-enyl)ethylamines 1 with aryl bromides provides 5- or 6-aryloctahydrocylopenta[b]pyrroles in moderate to good yields with excellent diastereoselectivity (>20:1 dr). The 5-aryl regioisomers 5a-g are selectively formed in reactions that employ the medium-sized electron-rich phosphine P(t-Bu)2Me as a ligand for palladium, whereas use of catalysts comprised of Pd2(dba)3 and either dppp or 11 leads to selective formation of the 6-aryl products 3a-g. The yields obtained in reactions that afford predominantly 5-aryl products are not greatly affected by the electronics of either the amine N-aryl substituent or the aryl bromide. In contrast, reactions that afford 6-aryl products provide the highest yields when substrates bearing electron-poor N-aryl substituents are employed in combination with electron-neutral or slightly electron-rich aryl bromides. Functional groups such as non-enolizable nitriles, ketones, and esters are tolerated in these transformations, and the synthesis of 5-aryl products bearing o-substituents or heterocylic groups is also feasible. The reaction of 1a with 4-bromotoluene in the presence of a catalytic amount of Pd2(dba)3/10 leads to clean N-arylation of the substrate to afford 2, whereas use of the Pd2(dba)3/PMe3 catalyst system provides oxidative amination product 4 in moderate yield.
The results described above (Table 4 entry 1, Table 5 entry 1, and eq 3-4) demonstrate that a single starting material can be converted to four different products (2-5) in moderate to excellent yield by changing only the nature of the metal catalyst employed in the reaction. For example, in separate experiments 1a was treated with 4-bromotoluene and an appropriate palladium catalyst to afford either 2 (88% yield), 3a (44% yield), 4 (60% yield), or 5a (74% yield). Although the selectivity for formation of 3a was modest (a 65:35 ratio of 3a: 2 + 4 + 5a was obtained under optimal conditions)32, products 2, 4, and 5a were each obtained with excellent selectivity (≥94:6). High selectivity for isomer 3 was achieved by changing the substrate’s N-aryl substituent to an electron-withdrawing p-C6H4CO2t-Bu group. The reaction of this modified substrate (1d) with 4-bromotoluene and the Pd2(dba)3/11 catalyst afforded 3e in 75% yield with 100:0 selectivity (Table 4, entry 7).
Mechanism and Ligand Effects
In addition to the hypothesis outlined in Scheme 1, a number of other mechanistic pathways could potentially lead to the formation of products 3-5 (Scheme 2).33 The ligand effects, stereoselectivity, and product stereochemistry described above can be used to evaluate the viability of the various possible mechanisms for this transformation. For example, product 3 could potentially derive from intermolecular carbopalladation of 1a followed by C-N bond-forming reductive elimination (Scheme 2, Path A). However, this pathway is unlikely due to the high stereoselectivity for formation of products in which the aryl group is installed syn to the C-3 methylene. If the reactions were to occur via intermolecular carbopalladation without chelation of the amino group or formation of an amido complex it is likely that steric repulsion between the Pd(Ar)(X) complex and the alkyl chain would favor formation of the anti-diastereomer 12 (or at least would likely provide mixtures of diastereomers).34 The formation of 3 via Wacker-type aminopalladation (Path B) can be ruled out, as this pathway would result in anti-addition of the heteroatom and the metal across the carbon-carbon double bond to provide 12 rather than the observed products 3a-g.35 The reductive Heck-arylation of 4 (Path C) can also be discounted as a possible mechanism for the generation of 3a and/or 5a, as the transformation of 4 to 3a and/or 5a is not observed under the conditions employed for the conversion of 1a to 3a or 5a.36
Scheme 2.

The data described above also argue against insertion of the alkene into the Pd-C bond of 6 or a related mechanism involving amino chelate-directed carbopalladation from intermediate 6′. As shown below (Scheme 3) alkene insertion into the Pd-C bond of 6 or 6′ would afford 13 or 13′. In principle 13 or 13′ could be converted to 3a via sp3C-N bond-forming reductive elimination. However, C-C bond-forming alkene insertions are generally irreversible,37 and this pathway could not provide either 4 or 5a from intermediates 13 or 13′. In addition, ligands that are known to slow the rate of C-C and sp2C-N bond-forming reductive elimination (e.g. dppe or PMe3)12 would also be likely to slow the rate of sp3C-N bond-forming reductive elimination. If the reductive elimination from 13 or 13′ were slowed, it is likely that β-hydride elimination of 13 or 13′ would afford product 14. However, use of dppe or PMe3 in the catalytic reactions leads instead to the formation of 4 as the major product. Thus, a mechanistic pathway involving intermediate 13 or 13′ cannot account for all products that are obtained and is also not consistent with the observed ligand effects.
Scheme 3.

In contrast, the data described above in Tables 1-5 can be explained using the mechanistic model outlined in Scheme 1. For example, the trends observed with trialkylphosphines show that as the steric bulk of the ligand is decreased, the product ratio shifts from predominantly 2 (P[t-Bu]2Cy, P[t-Bu]3) to 5 (PMe[t-Bu]2, PCy3, P[t-Bu]Cy2) to 4 (PMe3, PEt3). The decrease in ligand size would be expected to slow the rate of reductive elimination from 6→2,5,20 which may lead to the formation of 7. When medium-sized electron-rich ligands are employed it appears that β-hydride elimination from 7→8 occurs more rapidly than reductive elimination from 7, but the conversion of 8→9→5 is fast relative to the rate of alkene displacement from 8. This is presumably due to a combination of ligand steric and electronic properties that slow the associative ligand substitution process required to covert 8→4.19,21 In contrast, with the smaller ligands the metal becomes more sterically accessible for attack by an external nucleophile, and the rate of associative alkene displacement from 8 to afford 4 likely becomes fast relative to the rate of olefin reinsertion and reductive elimination. These arguments could also be used to explain similar trends observed in reactions that involve other monodentate ligands; the smaller ligands favor the formation of 4 (Table 2, entries 10-12) and larger ligands favor the formation of 2 and/or 3 (Table 2, entries 1-5). A comparison of reactions involving isosteric triphenylphosphine derivatives (Table 2, entries 7-9) reveals that increased amounts of products 2 and 3 are generated as phosphine basicity is decreased. This trend is also consistent with the mechanism described in Scheme 1, as ligands that increase the rate of C-N and/or C-C bond-forming reductive elimination20 should favor the formation of 2 and 3 over products 4 and 5, which are proposed to derive from β-hydride elimination processes.
Examination of the data shown in Table 3 does not reveal a clear relationship between bite angle of chelating ligands and product distribution. This may be due in part to the fact that the coordination chemistry of many of these ligands is complicated, and it is difficult to say whether or not some of the ligands are coordinated solely in a cis-bidentate fashion. For example, trans-chelated Pd-complexes of xantphos have been described,38 and dppb is known to form bridging complexes with two palladium atoms.39 However, ligands that are known to decrease the rate of reductive elimination (e.g. dppe)12 disfavor the formation of 2 and 3, which is consistent with our mechanistic hypothesis shown in Scheme 1. In addition, the electronic effects20 observed within a structurally similar series of ligands are also consistent with the hypothesis described above. For example, use of the electron-poor dppe-p-CF3 ligand resulted in the increased formation of 2 and 3 relative to dppe and, use of the electron-rich dppe-p-OMe led to a slight increase in the formation of 4.
At the outset of these studies we hoped to be able to facilitate the selective formation of 3 by employing chelating ligands to minimize β-hydride elimination of 7. Although we found several ligands (e.g dppp, dppf, xantphos, and dpp-benzene) that afforded high ratios of 2 + 3a: 4 + 5a, which suggests that chelation does affect the relative rates of reductive elimination vs. β-hydride elimination from 6 and 7, we were unable to completely control the selectivity for formation of 3 over 2 by varying phosphine ligand structure. However, substitution of the nitrogen with an electron-deficient aryl group resulted in good to excellent selectivities for 3c-3g with the pseudo-chelating ligand 11,40 which is consistent with an expected decrease in the rate of reductive elimination from 6 when the nitrogen nucleophilicity is decreased.18
The observed effects of aryl bromide substitution on product distribution are also consistent with the mechanistic hypothesis outlined in Scheme 1. The rates of stoichiometric C-N and C-C bond-forming reductive elimination reactions from LnPd(Ar)(NR2) and LnPd(Ar)(R) complexes are known to decrease with increasing π-electron density on the aryl group.18a,41 When 1d is treated with the electron deficient t-butyl-4-bromobenzoate under conditions that favor formation of 3g, increased amounts of N-arylated products 2 are formed compared to the amounts generated by reaction of 1d with 4-bromotoluene (Table 4, entries 7 and 9). This presumably derives from an increase in the rate of reductive elimination of intermediate 6. In contrast, reaction of 1d with the electron-rich 4-bromoanisole leads to increased formation of 4d (Table 4, entry 8), which is consistent with a decrease in the rate of reductive elimination from intermediates 6, 7, and 9. Similarly, reactions of 1b or 1e with aryl bromides bearing para-carbonyl functionality under conditions that favor formation of 5 also provided small amounts of regioisomeric side product 3, which may derive from an increase in the rate of reductive elimination from 7 (Table 5, entries 3 and 6). This effect was not observed in the reaction of 1e with of 4-bromobenzonitrile, which provided 5g with essentially complete selectivity. Although this latter result is somewhat surprising, previous studies have shown that the rates of reductive elimination reactions often do not follow simple linear free energy relationships with the σ-values of arene substituents.18a,41
In addition to providing further support for our mechanistic hypothesis shown in Scheme 1, the results described above illustrate several important concepts that are potentially relevant to the future development of other new Pd-catalyzed reactions. First of all, these results demonstrate that the rate of both C-N and C-C bond-forming reductive elimination can be minimized without completely suppressing alkene insertion processes, which are required to transform 1 into 3-5. Furthermore, since C-N bond forming reductive elimination reactions require larger ligands than are needed to effect C-C bond forming reductive elimination,5 it is possible to minimize the rate of C-N bond-forming reductive elimination without completely suppressing C-C bond-forming reductive elimination. This allows for the selective synthesis of 3 and 5. These results also reaffirm the notion that phosphines have a large impact on the relative rates of reductive elimination and β-hydride elimination, and that the appropriate choice of ligand can be used to favor either transformation over the other. Finally, our data also suggests that steric and electronic properties of phosphine ligands may have a large impact on the rate of alkene displacement from Pd(II) complexes. Medium-sized electron-rich ligands appear to slow the rate of alkene displacement from a Pd(II)-alkene complex to a greater degree than they decrease the rate of C-C bond-forming reductive elimination, which facilitates the selective preparation of 5 and minimizes the formation of side product 4. However, as the size of the ligand decreases, a near complete reversal in selectivity is observed such that the formation of 4 is favored over 5 (Table 1, entries 1-7).
Conclusion
In conclusion, we have demonstrated that the selective conversion of 1 to four different products can be achieved by using ligand effects to control the relative rates of C-N and C-C bond-forming reductive elimination, β-hydride elimination, alkene insertion, and alkene displacement in a mechanistically complex process. These transformations allow for synthesis of a variety of 5- or 6-aryl octahydrocyclopenta[b]pyrroles (5 and 3, respectively) in good yield with high regioselectivity and diastereoselectivity. The observed effects of ligand structure on product ratio provide further evidence to support our mechanistic hypothesis and suggest that palladium(aryl)amido complex 6 is a plausible common intermediate in the conversion of 1 to 2, 3, 4, and 5.
Supplementary Material
Acknowledgment
The authors thank the University of Michigan and the National Institutes of Health-National Institute of General Medical Sciences (GM-071650) for financial support of this work. JPW thanks the Camille and Henry Dreyfus Foundation for a new faculty award, and Research Corporation for an innovation award. JEN acknowledges the University of Michigan for a Regents Fellowship and Pfizer for a Graduate Research Fellowship. Additional unrestricted funding was provided by 3M, Amgen, and Eli Lilly.
References and Notes
- (1).For recent examples, see:Kirchhoff JH, Netherton MR, Hills ID, Fu GC. J. Am. Chem. Soc. 2002;124:13662–13663. doi: 10.1021/ja0283899.Torraca KE, Huang X, Parrish CA, Buchwald SL. J. Am. Chem. Soc. 2001;123:10770–10771. doi: 10.1021/ja016863p.Kawatsura M, Hartwig JF. J. Am. Chem. Soc. 1999;121:1473–1478.Lee C-W, Oh KS, Kim KS, Ahn KH. Org. Lett. 2000;2:1213–1216. doi: 10.1021/ol0056426.Aggarwal VK, Davies PW, Moss WO. J. Chem. Soc., Chem. Commun. 2002:972–973. doi: 10.1039/b201311h.Coperet C, Negishi E. -i. Org. Lett. 1999;1:165–168. doi: 10.1021/ol9900663.
- (2).A portion of this work has been previously communicated. See:Ney JE, Wolfe JP. Angew. Chem. Int. Ed. 2004;43:3605–3608. doi: 10.1002/anie.200460060.
- (3).For related transformations that provide indolines or tetrahydrofurans see:Lira R, Wolfe JP. J. Am. Chem. Soc. 2004;126:13906–13907. doi: 10.1021/ja0460920.Wolfe JP, Rossi MA. J. Am. Chem. Soc. 2004;126:1620–1621. doi: 10.1021/ja0394838.Hay MB, Hardin AR, Wolfe JP. J. Org. Chem. 2005;70:3099–3107. doi: 10.1021/jo050022+.
- (4).For recent, complementary methods involving late transition metal-catalyzed synthesis of pyrrolidines from γ-aminoalkenes, see:Bender CF, Widenhoefer RA. J. Am. Chem. Soc. 2005;127:1070–1071. doi: 10.1021/ja043278q.Manzoni MR, Zabawa TP, Kasi D, Chemler SR. Organometallics. 2004;23:5618–5621.
- (5).(a) Muci AR, Buchwald SL. Top. Curr. Chem. 2002;219:131–209. [Google Scholar]; (b) Hartwig JF. In: Modern Arene Chemistry. Astruc D, editor. Wiley-VCH; Weinheim: 2002. p. 107. [Google Scholar]
- (6).(a) Boncella JM, Villanueva LA. J. Organomet. Chem. 1994;465:297–304. [Google Scholar]; (b) Villanueva LA, Abboud KA, Boncella JM. Organometallics. 1992;11:2963–2965. [Google Scholar]; (c) Helaja J, Göttlich R. J. Chem. Soc., Chem. Commun. 2002:720–721. doi: 10.1039/b201209j. [DOI] [PubMed] [Google Scholar]; (d) Tsutsui H, Narasaka K. Chem. Lett. 1999:45–46. [Google Scholar]; (e) Brice JL, Harang JE, Timokhin VI, Anastasi NR, Stahl SS. J. Am. Chem. Soc. 2005;127:2868–2869. doi: 10.1021/ja0433020. [DOI] [PubMed] [Google Scholar]
- (7).For examples of alkene insertion into Pt-N bonds see:Cowan RL, Trogler WC. Organometallics. 1987;6:2451–2453.Cowan RL, Trogler WC. J. Am. Chem. Soc. 1989;111:4750–4761.
- (8).For an example of a catalytic reaction that likely proceeds via insertion of norbornene into an Ir-N bond see:Casalnuovo AL, Calabrese JC, Milstein D. J. Am. Chem. Soc. 1988;110:6738–6744.
- (9).Milstein D, Stille JK. J. Am. Chem. Soc. 1979;101:4981–4991. [Google Scholar]
- (10).(a) Schultz AG, Dai M. Tetrahedron Lett. 1999;40:645–648. [Google Scholar]; (b) Borioni A, Del Guidice MR, Mustazza C, Gatta F. J. Heterocycl. Chem. 2000;37:799–810. [Google Scholar]
- (11).For recent synthetic approaches to the 2-azabicyclo[3.3.0]octane ring system, see:Aron ZD, Overman LE. Org. Lett. 2005;7:913–916. doi: 10.1021/ol047330z.Pecanha EP, Verli H, Rodrigues CR, Barreiro EJ, Fraga CAM. Tetrahedron Lett. 2002;43:1607–1611.Gansauer A, Pierobon M, Bluhm H. Synthesis. 2001:2500–2520.Denmark SE, Senanayake CBW. Tetrahedron. 1996;52:11579–11600.Larock RC, Yang H, Weinreb SM, Herr RJ. J. Org. Chem. 1994;59:4172–4178.
- (12).(a) Gillie A, Stille JK. J. Am. Chem. Soc. 1980;102:4933–4943. [Google Scholar]; (b) Driver MS, Hartwig JF. J. Am. Chem. Soc. 1997;119:8232–8245. [Google Scholar]
- (13).Dppe = 1,2-bis(diphenylphosphino)ethane, dppp = 1,3-bis(diphenylphosphino)propane, dppb = 1,4-bis(diphenylphosphino)butane, dppm = 1,1-bis(diphenylphosphino)methane, dpp-benzene = 1,2-bis(diphenylphosphino)benzene, dcpe = 1,2-bis(dicyclohexylphosphino)ethane, BINAP = (±)-2,2′-bis(diphenylphosphino)-1,1′-binaphthyl, dpe-phos = bis(2-diphenylphosphinophenyl)ether, dppf = 1,1′-bis(diphenylphosphinoferrocene), xantphos = 9,9-dimethyl-4,5-bis(diphenylphosphino)xanthene.
- (14).Our previous studies showed that use of certain electron-rich phosphines (e.g 2-dicyclohexylphosphinobiphenyl) provided acceptable yields in the Pd-catalyzed carboamination reactions of γ-aminoalkenes. Thus, it seemed possible that the alkene insertion into the Pd-N bond may also potentially proceed with other electron-rich phosphines. See reference 2.
- (15).Jensen DR, Schultz MJ, Mueller JA, Sigman MS. Angew. Chem. Int. Ed. 2003;42:3810–3813. doi: 10.1002/anie.200351997.Steinhoff BA, Stahl SS. Org. Lett. 2002;4:4179–4181. doi: 10.1021/ol026988e.and references cited therein.
- (16).Culkin DA, Hartwig JF. Organometallics. 2004;23:3398–3416.and references cited therein.
- (17).van Leeuwen PWNM, Kamer PCJ, Reek JNH, Dierkes P. Chem. Rev. 2000;100:2741–2770. doi: 10.1021/cr9902704. [DOI] [PubMed] [Google Scholar]
- (18).The rate of C-N bond-forming reductive elimination decreases as the nitrogen becomes less nucleophilic. See: (a) Reference 12b.Yin J, Buchwald SL. J. Am. Chem. Soc. 2002;124:6043–6048. doi: 10.1021/ja012610k.Yamashita M, Cuevas Vicario JV, Hartwig JF. J. Am. Chem. Soc. 2003;125:16347–16360. doi: 10.1021/ja037425g.
- (19).Qian H, Widenhoefer RA. J. Am. Chem. Soc. 2003;125:2056–2057. doi: 10.1021/ja0293002. [DOI] [PubMed] [Google Scholar]
- (20).The rate of reductive elimination generally increases as ligand size increases, and decreases as ligand basicity increases. Steric effects can outweigh electronic effects, as electron-rich ligands that are sterically bulky are known to promote reductive elimination. For reviews, see:Christmann U, Vilar R. Angew. Chem. Int. Ed. 2005;44:366–374. doi: 10.1002/anie.200461189.Brown JM, Cooley NA. Chem. Rev. 1988;88:1031–1046.
- (21).Ligand substitution reactions at d8-Pd(II) complexes generally occur via an associative mechanism. See:Shultz LH, Tempel DJ, Brookhart M. J. Am. Chem. Soc. 2001;123:11539–11555. doi: 10.1021/ja011055j.and references cited therein. For rare exceptions see:Bartolome C, Espinet P, Martin-Alvarez JM, Villafane F. Eur. J. Inorg. Chem. 2004:2326–2337.Louie J, Hartwig JF. J. Am. Chem. Soc. 1995;117:11598–11599.
- (22).Rates of associative ligand substitution should decrease as the electron density on the metal center increases due to electron-electron repulsion between the metal and the incoming nucleophile.
- (23).Increased π-backbonding from the electron-rich metal to the alkene may also play a role in decreasing the rate of alkene dissociation. For a discussion of πbackbonding in electron-rich Pd(II)-alkene complexes, see:Miki K, Shiotani O, Kai Y, Kasai N, Kanatani H, Kurosawa H. Organometallics. 1983;2:585–593.
- (24).Most air sensitive trialkylphosphine ligands were employed in the form of their air-stable, crystalline tetrafluoroborate salts. See:Netherton MR, Fu GC. Org. Lett. 2001;3:4295–4298. doi: 10.1021/ol016971g.
- (25).Yoshida T, Otsuka S. Inorg. Synth. 1990;28:113–119. [Google Scholar]
- (26).Tolman CA. Chem. Rev. 1977;77:313–348.Lee KJ, Brown TL. Inorg. Chem. 1992;31:289–294.Rahman MM, Liu HY, Prock A, Giering WP. Organometallics. 1987;6:650–658.(d) This value was estimated based on the cone angle of the closely related ligands PPh3 and P[C6H4(p-OMe)]3.
- (27).Approximate value calculated using the method of Tolman. See:Tolman CA, Seidel WC, Gosser LW. J. Am. Chem. Soc. 1974;96:53–60.
- (28).Use of trimesitylphosphine provided a very complex mixture of unidentified products.
- (29).(a) Steffen WL, Palenik GJ. Inorg. Chem. 1976;15:2432–2439. [Google Scholar]; (b) Estevan F, Garcia-Bernabe A, Lahuerta P, Sanau M, Ubeda MA, Galan-Mascaros JR. J. Organomet. Chem. 2000;596:248–251. [Google Scholar]; (c) Ganguly S, Mague JT, Roundhill DM. Acta. Cryst. Sect. C. 1994;C50:217–219. [Google Scholar]; (d) Ozawa F, Kubo A, Matsumoto Y, Hayashi T, Nishioka E, Yanagi K, Moriguchi K. Organometallics. 1993;12:4188–4196. [Google Scholar]; (e) Makhaev VD, Dzhabieva ZM, Konovalikhin SV, D’yachenko OA, Belov GP. Russ. J. Coord. Chem. 1996;22:563–567. [Google Scholar]; (f) Hayashi T, Konishi M, Kobori Y, Kumada M, Higuchi T, Hirotsu K. J. Am. Chem. Soc. 1984;106:158–163. [Google Scholar]; (g) Boyes AL, Butler IR, Quayle SC. Tetrahedron Lett. 1998;39:7763–7766. [Google Scholar]; (h) van Haaren RJ, Goubitz K, Fraanje J, van Strijdonck GPF, Oevering H, Coussens B, Reek JNH, Kamer PCJ, van Leeuwen PWNM. Inorg. Chem. 2001;40:3363–3372. doi: 10.1021/ic0009167. [DOI] [PubMed] [Google Scholar]
- (30).For other examples of Pd-catalyzed oxidative amination reactions of olefins, see: (a) reference 6e.Hegedus LS. Tetrahedron. 1984;40:2415–2434.Fix SR, Brice JL, Stahl SS. Angew. Chem. Int. Ed. 2002;41:164–166. doi: 10.1002/1521-3773(20020104)41:1<164::aid-anie164>3.0.co;2-b.Trend RM, Ramtohul YK, Ferreira EM, Stoltz BM. Angew. Chem. Int. Ed. 2003;42:2892–2895. doi: 10.1002/anie.200351196.Larock RC, Hightower TR, Hasvold LA, Peterson KP. J. Org. Chem. 1996;61:3584–3585. doi: 10.1021/jo952088i.
- (31).The use of PMe2Ph, PMePh2, or P(2-furyl)3 as ligands afforded isolated yields of 54%, 44%, and 53%, respectively.
- (32).The 65:35 ratio of products was determined by 1H NMR analysis of the crude reaction mixture and differs slightly from the 58:42 ratio measured by GC analysis of the crude reaction mixture (Table 3, entry 7). Similarly small differences were observed for the product ratios described in Table 2, entry 4 (measured by GC) as compared to Table 4, entry 2 (measured by 1H NMR). The small differences in the product ratios for these are likely due to experimental error associated with the measurement techniques.
- (33).The mechanism for Pd-catalyzed N-arylation reactions of amines is well documented and is believed to proceed via intermediate palladium(aryl)(amido) complexes such as 6. For lead references, see: (a) reference 5.Alcazar-Roman LM, Hartwig JF. J. Am. Chem. Soc. 2001;123:12905–12906. doi: 10.1021/ja016491k.Singh UK, Streiter ER, Blackmond DG, Buchwald SL. J. Am. Chem. Soc. 2002;124:14104–14114. doi: 10.1021/ja026885r.Guari Y, van Strijdonck GPF, Boele MDK, Reek JNH, Kamer PCJ, van Leeuwen PWNM. Chem. Eur. J. 2001;7:475–482. doi: 10.1002/1521-3765(20010119)7:2<475::aid-chem475>3.0.co;2-6.Hartwig JF. Synlett. 1997:329–340.
- (34).Intermolecular Heck arylations of substituted cyclic, bicyclic, or heterocyclic alkenes generally provide products resulting from arylation on the less hindered face of the double bond. See:Tietze LF, Petersen S. Eur. J. Org. Chem. 2000:1827–1830.Haberli A, Leumann CJ. Org. Lett. 2001;3:489–492. doi: 10.1021/ol007029s.Desmazeau P, Legros J-Y, Fiaud J-C. Tetrahedron Lett. 1998;39:6707–6710.Oliveira DF, Severino EA, Correia CRD. Tetrahedron Lett. 1999;40:2083–2086.
- (35).(a) Lei A, Lu X, Liu G. Tetrahedron Lett. 2004;45:1785–1788. [Google Scholar]; (b) Bäckvall JE, Björkman EE. J. Org. Chem. 1980;45:2893–2898. [Google Scholar]; (c) Akermark B, Bäckvall JE, Siirala-Hansen K, Sjoberg K, Zetterberg K. Tetrahedron Lett. 1974;15:1363–1636. [Google Scholar]
- (36).Treatment of 4 with 4-bromotoluene in the presence of NaOtBu and a catalytic amount of Pd2(dba)3 and either P(t-Bu)2Me·HBF4 or dppp at 110 °C in toluene did not result in any detectable reaction.
- (37).Beletskaya IP, Cheprakov AV. Chem. Rev. 2000;100:3009–3066. doi: 10.1021/cr9903048. [DOI] [PubMed] [Google Scholar]
- (38).(a) Zuideveld MA, Swennenhuis BHG, Boele MDK, Guari Y, van Strijdonck GPF, Reek JNH, Kamer PCJ, Goubitz K, Fraanje J, Lutz M, Spek AL, van Leeuwen PWNM. J. Chem. Soc., Dalton Trans. 2002:2308–2317. [Google Scholar]; b) Yin J, Buchwald SL. J. Am. Chem. Soc. 2002;124:6043–6048. doi: 10.1021/ja012610k. [DOI] [PubMed] [Google Scholar]
- (39).Sanger AR. J. Chem. Soc., Dalton Trans. 1977:1971–1976. [Google Scholar]
- (40).Ligand 11 is believed to act as a bidentate ligand via π-complexation of the aromatic ring bearing the dimethylamino group. See:Yin J, Rainka MP, Zhang X-X, Buchwald SL. J. Am. Chem. Soc. 2002;124:1162–1163. doi: 10.1021/ja017082r.and references cited therein.
- (41).Ozawa F, Kurihara K, Fujimori M, Hidaka T, Toyoshima T, Yamamoto A. Organometallics. 1989;8:180–188. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.