

SUMMARY
Central to the replication checkpoint are two protein kinases, ATR, and its downstream target kinase, Chk1. Signaling pathways leading to activation of ATR-Chk1 have been extensively investigated; however, events that mediate checkpoint termination and replication fork restart are less well understood. Here, we define a coupled activation-destruction mechanism of Chk1 that regulates checkpoint termination and cellular sensitivity to replicative stress. DNA damage-induced phosphorylation or mutation of a conserved motif of Chk1 both activates Chk1 and exposes a degron-like region at the carboxyl-terminus of Chk1 to a Fbx6-containing SCF (Skp1-Cul1-F-box) E3 ligase, which mediates the ubiquitination and degradation of Chk1, and, in turn, terminates the checkpoint. The expression levels of Chk1 and Fbx6 proteins showed an inverse correlation in both cultured cancer cell lines and in a small cohort of human breast tumor tissues. Further, we show that low levels of Fbx6 and consequent impairment of replication stress-induced Chk1 degradation are associated with cancer cell resistance to killing by the chemotherapeutic agent, camptothecin (CPT). We propose that Fbx6-dependent Chk1 degradation contributes to S-phase checkpoint termination, and that a defect in this mechanism might increase tumor cell resistance to certain anticancer drugs.
INTRODUCTION
Mammalian cells respond to DNA damage by activating the ATM-Chk2- and/or ATR-Chk1-regulated checkpoints (Abraham, 2001; Shiloh, 2006). ATM-Chk2 mainly regulates DNA damage networks activated by DNA double strand breaks (DSBs). In contrast, ATR-Chk1 primarily responds to single strand DNA (ssDNA) with DSB junction structures generated during replication fork stalling, or through processing of DSBs, (Abraham, 2001; Cuadrado et al., 2006; Jazayeri et al., 2006; MacDougall et al., 2007; Myers and Cortez, 2006). In response to replication stress, ATR phosphorylates the Chk1 kinase at S317 and S345 (Liu et al., 2000; Zhao and Piwnica-Worms, 2001). These phosphorylation events trigger Chk1 activation, which, in turn, phosphorylates downstream substrates that orchestrate cell-cycle arrest, replication fork stabilization, and activate DNA repair responses (Bartek et al., 2004).
Significant progress has been made in elucidating the molecular circuitry underlying DNA damage checkpoint initiation (Abraham, 2001; Kastan and Bartek, 2004; Shiloh, 2006). However, checkpoint termination, which is essential for resumption of cell proliferation after DNA damage repair, is less well understood. Dephosphorylation of human γ-H2AX by phosphatase PP2A and human ATM by PP2A or PP2C type phosphatase PPM1D/Wip1 not only terminates the DNA damage checkpoint, but also facilitates DNA repair (Chowdhury et al., 2005; Goodarzi et al., 2004; Keogh et al., 2006; Shreeram et al., 2006). Similarly, Chk1 itself is a component of the checkpoint termination machinery, in that dephosphorylation of the activated Chk1 mediated by PP1, PP2A, or PPM1D/Wip1 phosphatases promotes recovery from cell-cycle arrest (den Elzen and O'Connell, 2004; Leung-Pineda et al., 2006; Lu et al., 2005; Yoda et al., 2006). In addition to phosphorylation and dephosphorylation, protein ubiquitination has emerged as an important mechanism that contributes to termination of DNA damage responses. For example, the adaptor protein, claspin, which is required for efficient Chk1 phosphorylation by ATR, is ubiquitinated by the β-Trcp1-containing SCF E3 ligase. The resultant proteasomal degradation of Claspin contributes to replication checkpoint termination by preventing phosphorylation of Chk1 by ATR (Bennett and Clarke, 2006; Mailand et al., 2006; Mamely et al., 2006; Peschiaroli et al., 2006). Recent studies identified a parallel process that reduces the pool of activated Chk1, particularly in cells exposed to persistent replication stress (Collis et al., 2007; Feng et al., 2008; Jurvansuu et al., 2007; Leung-Pineda et al., 2009; Zhang et al., 2005). These studies demonstrate that phosphorylation at S345 not only promotes full activation of Chk1, but also marks this protein for degradation via the ubiquitin-proteasome pathway (Collis et al., 2007; Feng et al., 2008; Zhang et al., 2005; Leung-Pineda et al., 2009).
The SCF E3 ligase complex mediates the ubiquitination of specific target proteins through the serial transfer of ubiquitin from the E1 ubiquitin activating enzyme to an E2 ubiquitin conjugating enzyme, and ultimately to a lysine residue(s) in the acceptor substrate, which is selected by the F-box protein component of the SCF E3 ligase (Cardozo and Pagano, 2004). The mammalian genome contains approximately 68 F-box proteins, which interact with distinct sets of ubiquitin acceptor proteins through specific interaction motifs, termed degrons (Cardozo and Pagano, 2004; Jin et al., 2004). In this study, we identified the F-box protein, Fbx6, as the targeting subunit of a Skp1-Cul1-Fbx6 E3 ligase that ubiquitinates activated Chk1. In addition, our findings indicate that the Fbx6 expression level might be a relevant determinant of Chk1 turnover and replication stress responses in human cancer cells
RESULTS
Fbx6 targets Chk1 for Ubiquitination and Degradation
We previously reported that Cul1- and Cul4A-containing SCF E3 ligases regulate Chk1 ubiquitination and degradation (Zhang et al., 2005). In this study, we focused our attention on the Cul1-containing SCF complex due to the more consistent effects of Cul1 knockdown in Chk1 levels observed in a panel of cancer cell lines (data not shown). To identify the F-box protein component of this SCF E3 ligase, we transfected A549 lung carcinoma cells with a validated SMARTpool™ siRNA library (Dharmacon) targeting all known human F-box genes. Immunoblot analysis of A549 cell extracts revealed 60–90% reduction in the level of several representative F-box proteins at 48 h after siRNA transfection (Figure 1A and data not shown). We reasoned that loss of the F-box protein that targets Chk1 for degradation would increase the basal level of Chk1, as observed in Cul1-depleted cells (Supplementary Figure S1A). After repetitive rounds of screening, we found that depletion of the F-box protein, Fbx6, but not other F-box proteins, including Skp2, consistently increased the basal expression level of Chk1 (Figure 1A). An independent shRNA lentiviral vector targeting the FBX6-encoded mRNA also increased the level of endogenous Chk1 (Figure 1B).
Figure 1. Fbx6 regulates Chk1 stability.

(A) A549 cells were transfected with 100 nM indicated siRNAs for 48 h, and blotted with indicated antibodies. (B) A549 cells were infected with control or Fbx6 lentivirus vectors for 48 h, and the indicated proteins were blotted. Cells were transfected with the indicated siRNAs and treated after 48 h with 160 µM CHX (C), or with 0.5 µM CPT (D), and representative Chk1 expression result is shown in the upper panels. Lower panels, quantitation of the Chk1 blots; data represent mean and standard deviation from 2–4 independent experiments.
Depletion of Cul1 or Fbx6, but not Skp2, caused an increase in the stability of Chk1 protein in cells treated with the protein synthesis inhibitor, cycloheximide (CHX) (Figure 1C). Several reports indicate that prolonged treatment of human cancer cells with certain cytotoxic agents, such as CPT (a topoisomerase-I inhibitor), induced Chk1 degradation (Collis et al., 2007; Feng et al., 2008; Jurvansuu et al., 2007; Leung-Pineda et al., 2009; Zhang et al., 2005). Transfection of A549 cells with siRNAs directed against either Cul1 or Fbx6, but not Skp2, suppressed the downregulation of Chk1 protein induced by CPT exposure (Figure 1D). Chk1 expression is regulated during the cell cycle (Kaneko et al., 1999); however, cell cycle analyses showed that siRNA transfection had only minor effect on the cell cycle profile of A549 cells (Supplementary Figure 1B). Hence, we concluded that the effect of Fbx6 on Chk1 expression was not a secondary consequence of altered cell cycle progression.
We then examined the interaction between Chk1 and Fbx6 using tagged proteins exogenously expressed in human embryonic kidney (HEK) 293T cells. Immunoprecipitates of Flag-tagged Fbx6, but not Skp2, contained co-precipitated HA-tagged Chk1 (Figure 2A). The absence of Chk1 in anti-Skp2 immunoprecipitates was not due to the failure of the antibody to pull down the intact SCF complex, because endogenous Cul1, another component of the SCF complex, was present in both immunoprecipitates (Figure 2A). To further define the Fbx6-Chk1 interaction, we generated several polypeptides corresponding to different regions of human Fbx6 (Figure 2B). The results show that the F-box associated (FBA) domain mediates the binding of Fbx6 to Chk1 (Figure 2C). The Fbx6-Chk1 interaction was only detected in the cytoplasmic compartment where Fbx6 is mainly localized (Supplementary Figure S2A, Figure 7C, and data not shown). Moreover, depletion of Fbx6 with the lentivirus shRNA vector partially blocked the downregulation of phospho-Chk1 in the cytoplasm after 6 h of CPT exposure (Supplementary Figure S2B). Finally, endogenous Fbx6 was co-immunoprecipitated with Chk1 in extracts from non-transfected cells (Figure 2D).
Figure 2. Fbx6 regulates Chk1 ubiquitination and degradation.
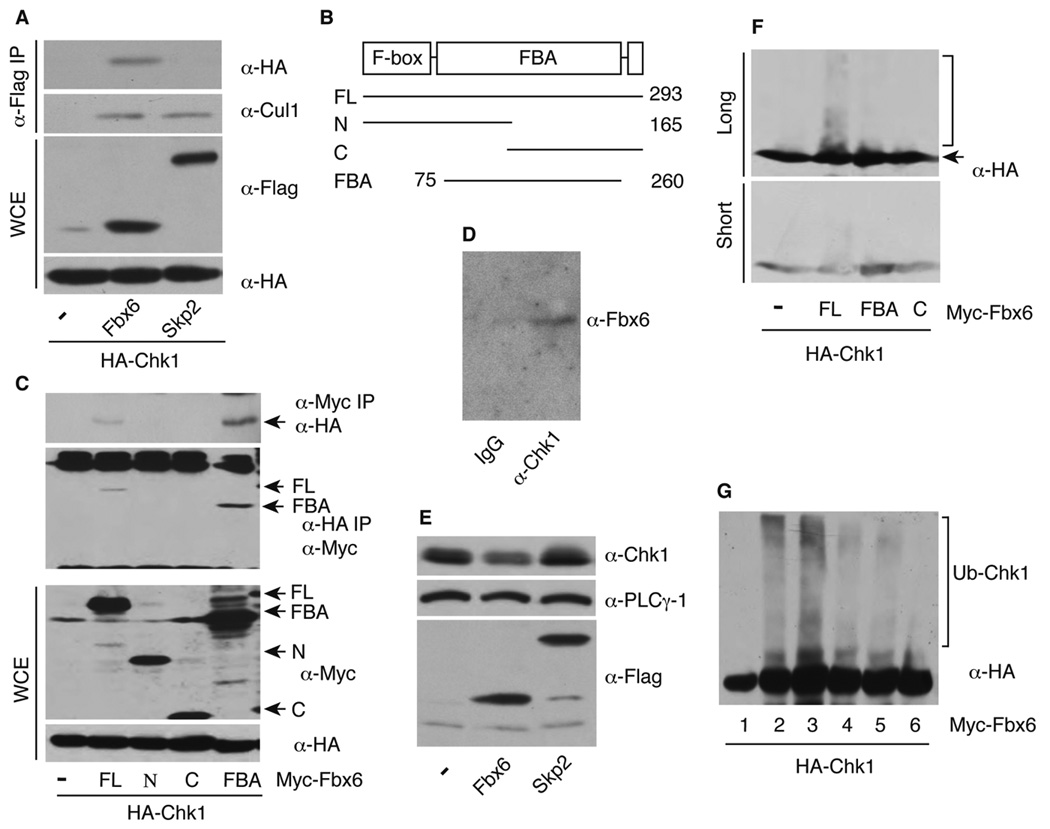
(A) Flag-tagged F-box proteins and HA-Chk1 were expressed in 293T cells, immunoprecipitated with anti-Flag antibodies and blotted with anti-HA antibodies. The same membrane was stripped and reblotted with anti-Cul1 antibodies. Whole cell extracts (WCE) were blotted for total protein expression. (B) Mutation of human Fbx6. Numbers represent the amino acid residues in human Fbx6. FL, N, C, and FBA represent the full-length, amino-terminus, carboxyl-terminus, and the FBA domain only of Fbx6, respectively. (C) Myc-tagged Fbx6 and HA-Chk1 were co-transfected into 293T cells, cell extract was divided into two aliquots for immunoprecipitation with anti-Myc or anti-HA antibodies, followed by immunoblotting with anti-HA and anti-Myc antibodies, respectively. Protein expression was also determined in WCE. (D) WCE from untreated A549 cells were immunoprecipitated with mouse IgG or anti-Chk1 antibodies, and blotted with anti-Fbx6 antibodies. (E) Expression of endogenous Chk1 from 293T cells stably transfected with pcDNA3-Flag-F-box proteins. (F) HEK 293T cells were transfected with HA-Chk1, Myc-tagged Fbx6 FL or mutants with His-ubiquitin for 48 h. Cells were lysed and blotted with anti-HA antibodies. (G) In vitro ubiquitination assay. 293T cells were transfected with HA-Chk1 with (lane 2) or without His-ubiquitin (lanes 1 and 3–6) for 48 h, cell extracts were immunoprecipitated with anti-HA (12CA5) antibodies, and the immobilized HA-Chk1 was used as the substrate in reactions containing soluble Myc-Fbx6 FL or mutant proteins as described in Experimental Procedures. Reaction products were immunoblotted with rat anti-HA antibodies. Lanes 1 and 2: no addition of either Fbx6 or the in vitro ubiquitination reaction reagents (lane 2 serves as a positive control for ubiquitinated Chk1); lane 3–5: in vitro ubiquitination with addition of Fbx6 FL, FBA, or the C-terminal mutant C, respectively; lane 6: same as lane 3 except no Fbx6 protein.
Figure 7. Inverse correlation between Chk1 and Fbx6 expression.

Asynchronously growing cells derived from (A) various tumor types and (B) non-small cell lung cancer were lysed and blotted with anti-Chk1 and anti-Fbx6 antibodies, and the same membranes were stripped and re-blotted with anti-tubulin and anti-PCNA antibodies, respectively. (C) Representative staining of Chk1 and Fbx6 in breast tumor sections. (D) The breast tumor staining data were analyzed and quantitated as described in Experimental Procedures. (E) Model for Chk1-regulated replication checkpoint activation and termination. The distal C-terminus of Chk1 interacts with the kinase domain likely through adaptor proteins (marked with ‘X’). After the phosphorylation-induced conformational change, the CM (conserved motif) and part of the regulatory domain form the degron-like region of Chk1. For simplicity, the spatial regulation of Chk1 is omitted in this model. See text for details.
We next determined whether overexpression of Fbx6 decreases the steady-state Chk1 expression. HEK 293T cells stably transfected with Flag-tagged Fbx6, but not Skp2, expressed lower levels of Chk1 than control cells (Figure 2E). Again, the effect of exogenously expressed Fbx6 on Chk1 levels was not attributable to an alteration in the cell-cycle distribution of the transfected cell population (Supplementary Figure S2C). To examine the role of Fbx6 in Chk1 ubiquitination, we transfected 293T cells with HA-Chk1 and His-ubiquitin with or without Fbx6 FL, FBA, or the F-box deleted C-terminal mutant (C represents the C-terminus of Fbx6), and blotted the whole cell extracts with a rat monoclonal anti-HA antibody that does not cross react with other non-HA-tagged proteins. Interestingly, a diffuse smear of anti-HA immunoreactivity, consistent with poly-ubiquitination of HA-Chk1, was readily observed in the Fbx6 FL co-transfected cells, but was substantially less noticeable in cells co-transfected with the FBx6-derived FBA or C constructs (Figure 2F). Moreover, only the Fbx6 FL protein supported ubiquitination of Chk1 in vitro (Figure 2G, lanes 3–5). Collectively, these results suggest that Fbx6 regulates ubiquitination of Chk1 in both untreated and CPT-treated cells.
A Degron-like Region in the Carboxyl-terminal Chk1
To define the region of Chk1 involved in the regulation of its stability, we generated a series of C-terminally truncated Myc-tagged Chk1 fragments (Figure 3A) and monitored protein stabilities after inhibiting new protein translation with CHX. The full-length (FL) Chk1 was relatively stable under the conditions tested; however, deletion of the distal C-terminal 55 amino acids significantly reduced the basal expression level of Chk1 and increased the rate of protein degradation (Figure 3B, fragment 2). These results are consistent with those reported in a previous study involving Xenopus Chk1 (Oe et al., 2001). Hence, the distal C-terminus stabilizes the Chk1 polypeptide. Strikingly, further deletion from the C-terminus partially or completely restored the stability of Chk1 (Figure 3B, fragments 3–5). Transient expression of these Chk1 fragments in Fbx6-expressing 293T cells revealed strong interactions between Fbx6 and Chk1 fragment 1 or 2, but not the shorter fragments (Figure 3C). Using the intrinsically unstable Chk1 fragment 2 as a surrogate for Fbx6-dependent Chk1 degradation, we observed that transfection of cells with the FBA domain only of Fbx6 increased the expression of the unstable Chk1 fragment 2 (Supplemental Figure S3A), presumably due to dominant-interference with the endogenous Fbx6-containing SCF E3 ligase. This Fbx6 construct had no effect on the cell cycle distribution of the transfected cell populations (Supplemental Figure S3B). We conclude from these studies that the regulatory region of Chk1 is involved in the interaction with Fbx6 that leads to Chk1 ubiquitination and degradation.
Figure 3. A degron-like region in Chk1.

(A) Schematic diagram of human Chk1. SQ, the Ser/Gln phosphorylation site cluster that includes S317 and S345; AIR, auto-inhibitory region. Letters on the left side identify the Chk1 fragment, and numbers on the right indicate the amino acid residue site based on the FL polypeptide. (B) 293T cells were transfected with Myc-tagged Chk1 fragments 1–5 for 48 h, treated with 160 µM CHX, with or without 10 µM MG132, for the indicated times, and expression of proteins was examined. (C) Myc-Chk1 fragments 1–5 were transfected into Fbx6-expressing stable 293T cells, immunoprecipitated with anti-Flag antibody and blotted with anti-Myc antibodies. The same membrane was stripped and re-blotted with anti-Flag antibodies. WCEs were blotted with anti-Myc antibodies. (D) EGFP, EGFP-D1, -D2, or – H2B were transfected into 293T cells for 48 h, cells were treated for 6 h with 12 µM MG132, and expression of fusion proteins was analyzed. (E) EGFP-D1 or –D2 were transfected into Fbx6 stably expressing 293T cells, immunoprecipitated with anti-Flag antibodies, and blotted with anti-GFP antibodies. (F) HEK293T cells were infected for 24 h with control or Fbx6 lentivirus shRNA vector, and then transfected with EGFP-D1 or –D2 plasmids. After another 48 h, cells were lysed and blotted with indicated antibodies. The asterisk denotes a non-specific band serving as the loading control. (G) HA-Chk1 WT, S34E, or S345A were transfected into Flag-Fbx6 expressing stable 293T cells, immunoprecipitated with anti-Flag antibodies, and blotted with rat anti-HA antibodies. WCE was blotted to determine total protein expression.
To further examine contributions of the regulatory domain, as well as the SQ cluster, to Chk1 protein stability, we generated two additional constructs, EGFP-D1 (330–421) and EGFP-D2 (368–421) (Figure 3A), and examined the expression of fusion proteins in the absence or presence of the proteasome inhibitor MG132. The control cells expressed a large amount of GFP protein, which was not increased by treatment with MG132. In contrast, EGFP-D1 and EGFP-D2 were expressed at much lower levels than the EGFP alone, and expression of both fusion proteins was strongly enhanced by MG132 (Figure 3D). MG132 did not alter the expression level of the control fusion protein EGFP-H2B (Figure 3D). Although a minimal degron region of Chk1 has not yet been defined, these data suggest that the regulatory domain (368–421) functions as a degron-like region that marks this protein for ubiquitination and degradation.
We next determined whether the degron-like region binds directly to Fbx6. Indeed, an interaction between Fbx6 and EGFP-D1 or EGFP-D2 was readily detected in transfected cells (Figure 3E). Depletion of Fbx6 with lentivirus shRNA increased the expression of both fusion proteins (Figure 3F). The D2 domain of Chk1 lacks the SQ cluster containing the S345 site required for damage-induced Chk1 degradation (Collis et al., 2007; Zhang et al., 2005), indicating that this ATR phosphorylation site was not directly involved in the Chk1-Fbx6 interaction. To confirm this prediction, we examined interactions between Chk1 wild type (WT), S345E, or S345A proteins and Fbx6. Fbx6 interacted with both WT and mutated forms of Chk1; however, the level of co-precipitated S345A Chk1 with Fbx6 was noticeably lower than that of the WT Chk1 (Figure 3G).
Lysine 436 Is a Candidate Ubiquitination Site in Chk1
Mass spectrometric analysis identified ubiquitin conjugated to K436 of Chk1 in MG132-treated MCF-7 cells (Denis et al., 2007). Therefore, we asked whether K436 is involved in regulating Chk1 protein turnover by comparing the stability of Chk1 WT and the K436R mutant in the presence of CHX. Chk1 K436R was noticeably more stable than WT Chk1 (Figures 4A and B), and this mutation substantially reduced Chk1 ubiquitination (Figure 4C). These data strongly suggest that K436 is a biologically relevant Chk1 ubiquitination site, but do not exclude the possibility that additional lysines in Chk1 are involved in its ubiquitination and degradation.
Figure 4. Regulation of Chk1 protein stability.
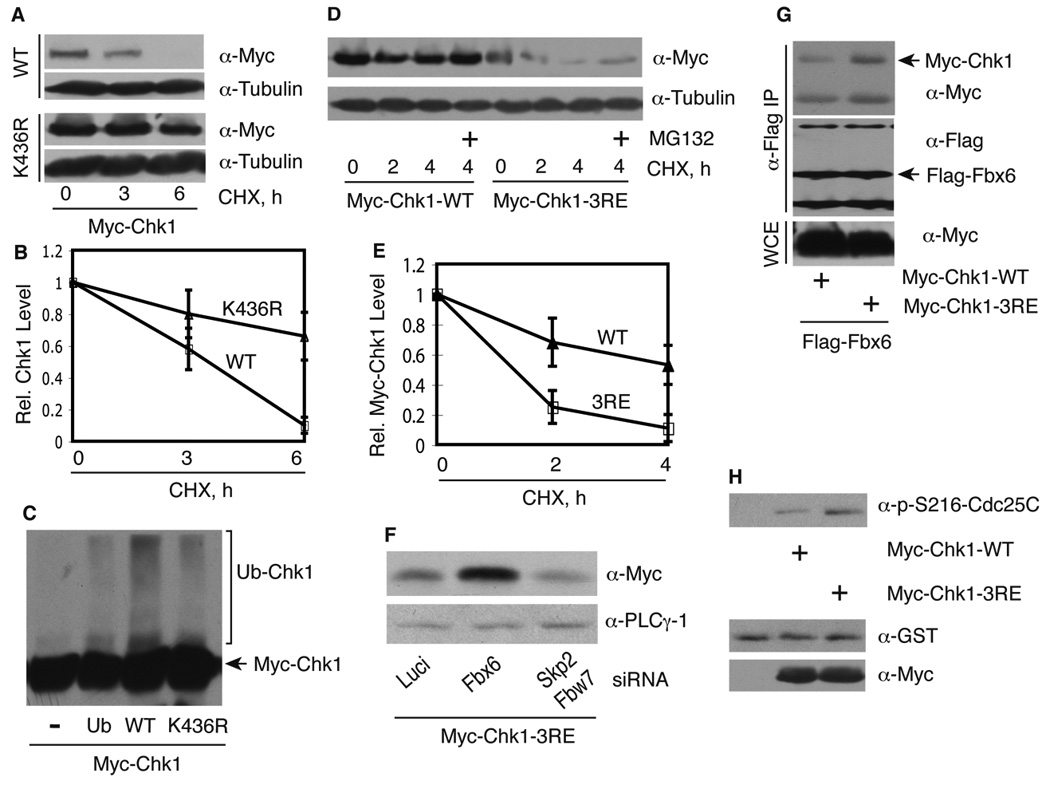
(A) 293T cells were transfected with Myc-Chk1 WT or K436R for 48 h, treated with 320 µM CHX, and blotted with indicated antibodies. (B) Anti-Myc blots as shown in panel A were quantitated from 2–4 independent experiments and plotted. Data represent mean and standard deviation. (C) 293T cells were transfected with Myc-Chk1 WT or K436R with or without His-ubiquitin for 48 h, lysed in 6 M guanidinium hydrochloride, and His-tagged ubiquitinated proteins were recovered on Co2+ beads, and eluates were blotted with anti-Myc antibodies. (D) Myc-Chk1 WT or 3RE mutant was expressed in 293T cells for 48 h, treated as described in Figure 3B, and protein expression was examined. (E) The anti-Myc blots in B from 2–3 independent experiments were quantitated and normalized to the control sample. Data represent mean and standard deviation. (F) HEK 293T cells were transfected with the indicated siRNAs for 24 h, then transfected with Myc-Chk1 3RE mutant, and protein expression was examined after an additional 48 h. (G) Myc-Chk1 WT or 3RE was transfected into Fbx6 expressing 293T cells for 48 h, treated for 4 h with 12.5 µM MG132 to allow accumulation of Chk1 3RE, cell extracts were immunoprecipitated with anti-Flag antibodies, and then blotted with anti-Myc antibodies. The same membrane was stripped and re-blotted with anti-Flag antibodies. Total protein levels were blotted in WCE. (H) In vitro kinase assay. Equal amount of immunoprecipitated Chk1 WT or 3RE mutant was added into the kinase reaction, and immunoblotted with a specific phospho-S216-Cdc25C antibody. Parallel samples without ATP are shown in the lower panel.
Coupled Activation and Degradation of Chk1
Recent studies suggest that the C-terminus of Chk1 is not only required for Chk1 activation, but may also function as an auto-inhibitory region (AIR) in cells, possibly through forming an intra-molecular interaction with the catalytic domain (Chen et al., 2000; Chen et al., 2009; Katsuragi and Sagata, 2004; Kosoy and O'Connell, 2008; Oe et al., 2001). In agreement with this model, GST-tagged polypeptides covering the AIR region of the C-terminus of Chk1 purified from bacteria precipitated the Myc-tagged kinase domain of Chk1 (fragment 5 in Figure 3A) from transfected 293T cell extracts (Supplementary Figures S4A and B) (Katsuragi and Sagata, 2004; Shann and Hsu, 2001). However, we cannot rule out the possibility that this intra-molecular interaction is facilitated by endogenous adaptor proteins, such as Rad9/Claspin or PCNA (Chen et al., 2009; Scorah et al., 2008).
If this intra-molecular interaction model is correct, mutation of the Chk1 C-terminus might disrupt this interaction, thereby generating a constitutively “open” configuration, characterized by increased kinase activity and simultaneous protein destabilization. To test this, we mutated three highly conserved Arg residues in a conserved motif (CM) within the regulatory region of Chk1 to Glu to create a 3RE Chk1 mutant (Supplementary Figure S4C). We observed that the basal expression level of Chk1 3RE was much lower than that of WT Chk1 due to an increased rate of proteasomal degradation (Figures 4D and E). These results are consistent with a recent report showing that deletion of three amino acids within the CM caused reduced expression of Saccharomyces cerevisiae Chk1 (Chen et al., 2009). Depletion of Fbx6, but not Skp2 or Fbw7, markedly increased the expression level of Chk1 3RE (Figure 4F). Moreover, when the level of Chk1 3RE was increased to that of WT Chk1 by MG132 treatment, Chk1 3RE bound more strongly to Fbx6 than WT Chk1 (Figure 4G), suggesting that Fbx6 mediates targeting of the 3RE mutant for ubiquitination and degradation. The immunoprecipitated 3RE mutant also displayed an elevated level of protein kinase activity in vitro toward a GST-Cdc25C substrate containing a Chk1 phosphorylation site at S216 (Figure 4H), consistent with the prediction that Chk1 3RE possesses an active conformation. Mutation of other residues in the CM of Xenopus Chk1 also increased its catalytic activity (Wang and Dunphy, 2000). A kinase-inactive version of Chk1 3RE was even more unstable than the Chk1 3RE containing a WT kinase domain (Supplementary Figure S5A), indicating that the instability of Chk1 3RE was not causally related to the increased basal protein kinase activity displayed by this mutant. In addition, the sub-cellular localization of the Chk1 3RE mutant was similar to that of Chk1 WT (Supplementary Figure S5B, best observed if one compares the short exposure for the WT with the long exposure for the 3RE mutant to compensate for differences in basal expression of the two proteins). Together, these data suggest that the 3RE mutation may disrupt the conformation and/or folding of Chk1, allowing increased accessibility or association of Fbx6 with Chk1.
Coupled Chk1 Degradation and Replication Arrest Recovery
We observed that moderate overexpression of Chk1 (approximately two-fold over the endogenous level) led to a G1/S-phase arrest, as indicated by reduced BrdU incorporation and decreased expression of the Chk1 target protein, Cdc25A (Supplementary Figure S6A and B, and data not shown). Interestingly, Schizoaccharomyces pombe Chk1 protein bearing a R to A mutation in the CM (corresponding to the third R in our 3RE Chk1 mutant) behaved as a constitutively active protein, and provoked a G2/M phase arrest in the absence of exogenous DNA damage (Kosoy and O'Connell, 2008).
Based on the elevated turnover rate of the Chk1 3RE mutant, we hypothesized that cells expressing this Chk1 variant would show a more rapid recovery from this G1/S-phase arrest than their counterparts expressing the Chk1 WT protein. For these studies, we transiently transfected U2-OS Tet/On cells with Tet-inducible Flag-Chk1 WT or 3RE constructs, and treated these cells with doxycycline for 16 h to induce FLAG-Chk1 protein expression (Supplementary Figures S6C and E). The cells were then released into doxycycline-free medium to terminate the exogenous CHK1 mRNA transcription, and recovery from the G1/S-phase arrest was measured by labelling the cells with BrdU (Supplementary Figure S6D). Due to the increased turnover rate of the Chk1 3RE mutant, the overall protein level of Chk1 3RE after doxycycline induction was much lower than that of the WT (Supplementary Figure S6E, 0 h time points). However, we were able to observe individual cells with similar levels of expression of Chk1 WT and 3RE by immunostaining for the Flag-Chk1 protein (data not shown), and in those cells, the WT and 3RE Chk1 proteins reduced the level of BrdU incorporation to similar extents at the time of doxycycline removal from the culture medium (Figure 5B, 0 h release). As expected, the expression of the 3RE mutant protein dropped more precipitously than that of Chk1 WT, and was almost undetectable at 24 h after release by both immunostaining and immunoblotting (Figure 5A and Supplementary Figure S6E). In parallel with the more rapid decline in Chk1 3RE protein expression, the Chk1 3RE-expressing cells resumed DNA replication in an accelerated fashion relative to cells expressing the Chk1 WT protein (Figure 5B). At 24 h after release, BrdU incorporation in 3RE-expressing cells returned to the level observed before doxycycline induction (Figure 5B, 0–24 h), as did the expression of the Cdc25A protein, which is degraded during checkpoint activation in a Chk1-dependent fashion (data not shown). Thus, the increased turnover rate of Chk1 3RE allows more rapid recovery from checkpoint-induced cell cycle arrest in the 3RE- versus the Chk1 WT-expressing cells.
Figure 5. Chk1 degradation and checkpoint recovery.

U2OS Tet/On cells transiently expressing Tet-inducible Flag-Chk1 WT or 3RE mutants were monitored for expression of Flag-Chk1 and cumulative BrdU incorporation as described. The fluorescence intensities (pixels) of Flag- and BrdU-positive cells were quantified with the Photoshop Histogram analysis program, and normalized to the 0 h and pre-induction samples for Flag and BrdU, respectively. (A) Relative Flag-Chk1 WT and 3RE expression levels. (B) Relative BrdU incorporation. Data represent mean and standard deviation from 50–100 cells. (C) In vitro trypsin digestion assay was performed as described in Experimental Procedures. (D) Equal amounts of phosphorylated and non-phosphorylated Myc-Chk1 beads were used to perform in vitro kinase assays as described in Figure 4F legend. Indicated samples were pre-treated for 15 min with 500 nM Chk1 kinase inhibitor PF-003946901 (Pfizer) on ice.
Phosphorylation Increases Chk1 Protease Sensitivity
Previous findings indicated that the ATR-dependent phosphorylation of Chk1 simultaneously activates and destabilizes this protein (Feng et al., 2008; Zhang et al., 2005; Leung-Pineda et al., 2009). Consequently, we asked whether phosphorylation of Chk1 triggers a transition from the inactive closed conformation to an open activated, more unstable conformation. We used a protease sensitivity assay to determine the relative sensitivities of the phosphorylated and non-phosphorylated Chk1 proteins (purified as described in Supplementary Information) to trypsin digestion. A similar approach was recently used to examine the effect of phosphorylation on the conformation and protein stability of the E3 ubiquitin ligase Itch (Gallagher et al., 2006). Phosphorylated Chk1 protein underwent significantly faster proteolysis than non-phosphorylated Chk1, with a half-life of around 0.75 min for phosphorylated Chk1 versus about 2.5 min for non-phosphorylated Chk1 (Figure 5C and Supplementary Figure S7B). We also confirmed that phosphorylated Chk1 displayed an elevated level of protein kinase activity that was abolished by the Chk1 kinase inhibitor PF-003946901 (Figure 5D) (Blasina et al., 2008). In support of the notion that phosphorylation at S345 is required for Chk1 degradation in vivo, the S345E mutant was more sensitive to trypsin digestion than either the Chk1 WT or S317E mutant (Supplementary Figure 7C).
Relationship between Chk1 Downregulation and CPT Sensitivity
Previous studies suggested that downregulation of Chk1 by prolonged exposure to replication stress correlated with cell killing presumably due to irreversible fork collapse and programmed cell death (Feijoo et al., 2001; Zhang et al., 2005). The clinical antitumor activity of CPT is limited in part by the relatively rapid emergence of drug-resistant clones of malignant cells, through mechanisms that are only partially understood (Rasheed and Rubin, 2003). We hypothesized that defects in the Chk1 destruction machinery might allow CPT-treated cells to maintain the viability of stalled replication forks during prolonged CPT exposure, thereby promoting drug resistance. To test this hypothesis, we searched the National Cancer Institute’s Developmental Therapeutics Program (NCI-DTP) database (http://dtp.nih.gov/docs/Static_Pages/Tables/609699gitb.html) for members of the NCI 60-cell line panel that displayed high-level (>100-fold) resistance to the CPT analog, topotecan, relative to the drug-sensitive A549 cell line. Three such topotecan-resistant cell lines (TK-10, HS578T, and MDA-MB-231) were tested for Chk1 downregulation after CPT exposure. Remarkably, two of these cell lines (MDA-MB-231 and TK-10) displayed profound defects in Chk1 degradation after CPT treatment (Figure 6A and Supplemental Figure S8A). Both cell lines were more resistant to the cytotoxic effect of CPT than was the A549 cell line (Figure 6B and Supplemental Figure S8B). The MDA-MB-231 cell line was subsequently chosen for more detailed analyses. Knockdown of Chk1 by siRNA dramatically increased cell killing of the resistant MDA-MB-231 cells by CPT (Figure 6C). Conversely, overexpression of WT Chk1 rendered sensitive A549 cells partially resistant to CPT-induced cell death (data not shown).
Figure 6. Chk1 degradation and CPT sensitivity.
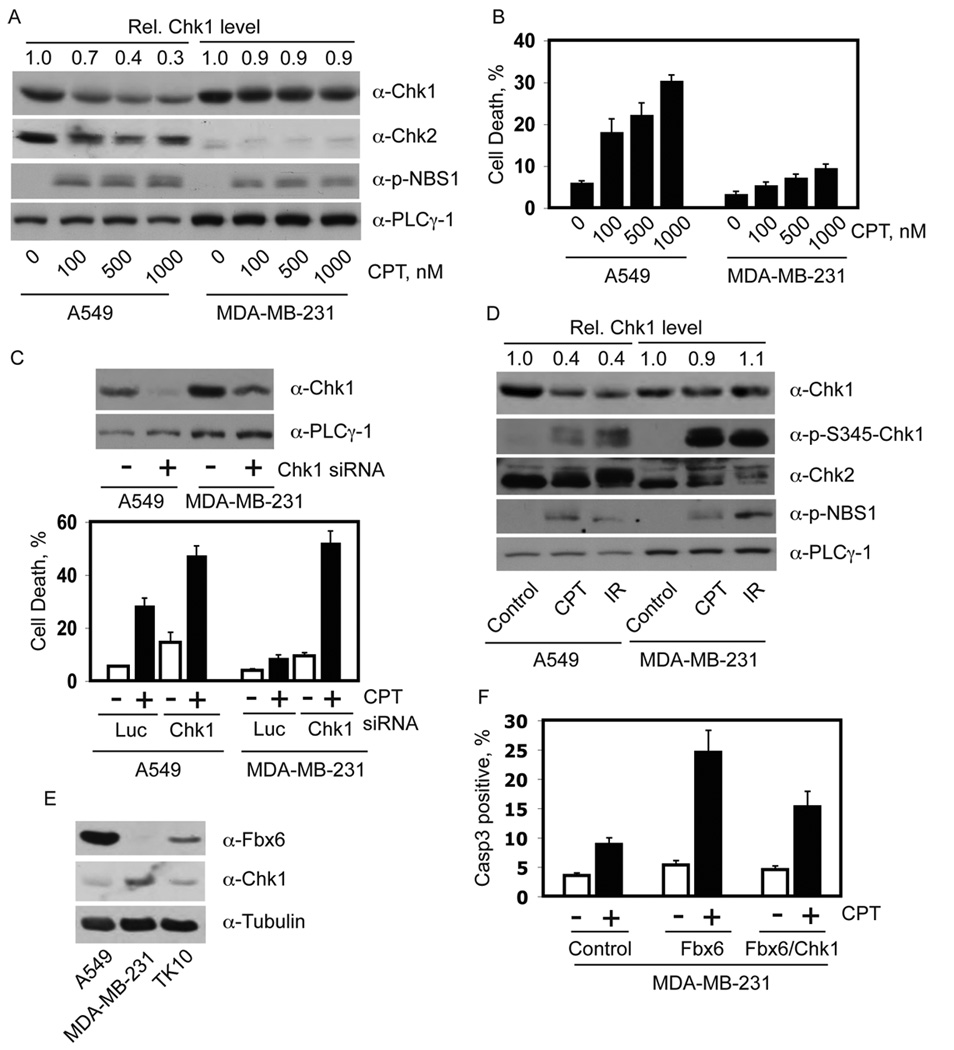
(A) Asynchronous A549 and MDA-MB-231 cells were treated for 8 h with the indicated concentrations of CPT. Protein expression was determined by immunoblotting with the indicated antibodies. Numbers at the top of each sample lane represent the relative Chk1 protein level, normalized to that obtained in the no-drug control. (B) Cells treated as described in panel A were cultured for 48 h in fresh medium. Cell death was determined by staining with trypan blue. The data are plotted as mean +/− standard deviation from 3 independent trials. (C) A549 and MDA-MB-231 cells were transfected for 48 h with the indicated siRNAs and treated with CPT. Upper panel, expression level of Chk1. Lower panel, Cell death after 36 h of release from CPT treatment. The data are presented as mean +/− standard deviation from three independent experiments. (D) Asynchronous A549 and MDA-MB-231 cells were treated with 500 nM CPT or 15 Gy IR. After 8 h, cell extracts were prepared and immunoblotted with the indicated antibodies. Numbers at the top of each sample lane represent relative Chk1 protein levels, normalized to that obtained in the non-treated control sample for each cell type. (E) Equal amounts of total proteins from exponentially growing A549, MDAMB-231, and TK-10 cells were blotted with purified rabbit anti-Fbx6 antibody first, then stripped and sequentially re-blotted with mouse anti-Chk1 and anti-tubulin antibodies. (F) MDAMB-231 cells grown on glass cover slides were transfected with control vector, Flag-Fbx6 only, or Flag-Fbx6 plus Myc-Chk1 expression vectors. After 48 h, cells were treated for 8 h with 500 nM CPT, and were released into drug-free medium for another 12 h; cells were then fixed and stained with anti-Flag and anti-Chk1 or anti-activated caspase 3 antibodies. The caspase 3 positive cells in control or Flag-Fbx6-expressing cells were counted; data represent mean +/− standard deviation from two independent experiments. At least 50 positive cells were counted in each experimental setting.
The Chk1 degradation defect was not specific to CPT, as MDA-MB-231 cells also failed to downregulate Chk1 after IR exposure (Figure 6D), which triggers DNA damage through distinct mechanisms from CPT. The resistant MDA-MB-231 cells exhibited a strong Chk1 phosphorylation response to CPT treatment (Figure 6D), suggesting that the Chk1 destruction defect in the drug-resistant cell line is not due to reduced coupling of ATR activation to Chk1 phosphorylation during replication stress.
To understand the mechanisms underlying the Chk1 degradation defect in the drug-resistant cancer cells, we examined the expression level of Fbx6, the E3 ligase that regulates Chk1 protein level, in these cell lines. The level of Fbx6 in the drug-resistant MDA-MB-231 and TK-10 cells was significantly lower than in the drug-sensitive A549 cells (Figure 6E and Supplementary Figure S8C). Conversely, the basal level of Chk1 was noticeably lower in A549 cells than in MDA-MB-231 and TK-10 cells, suggesting that the variable basal expression of Fbx6 might be one determinant of the steady-state Chk1 expression. In contrast, the expression of Cul1 and Skp1, two additional components of the Fbx6-containing SCF E3 ligase, was similar in these cell lines (data not shown).
If our hypothesis is correct, then forced expression of Fbx6 in the CPT-resistant cells should render these cancer cells more sensitive to CPT. Expression of Flag-Fbx6 in MDA-MB-231 cells reduced the endogenous level of Chk1, and these cells exhibited strong staining for the caspase 3 cleavage product after CPT treatment (Figure 6F and Supplementary Figures S9A and B), consistent with an increased sensitivity to CPT-induced cell killing. Co-expression of Chk1 partially reversed the drug-sensitive phenotype of the Fbx6-transfected MDA-MB-231 cells (Figure 6F). In contrast, depletion of Fbx6 decreased the CPT sensitivity in A549 cells, which was completely restored by depletion of Chk1 (Supplementary Figure S9C). The decrease in CPT sensitivity in these Fbx6-deficient cells was partially reversed by re-introduction of WT Fbx6. These data support the hypothesis that variations in Fbx6 expression influences the stability of Chk1, and the ability of cancer cells to recover from replication stress induced by CPT, and probably other replication stresses.
Inverse Relationship between Chk1 and Fbx6 expression
To gain further insights into the regulation of Chk1 by Fbx6, we examined the expression of Chk1 and Fbx6 proteins in multiple human cancer cell lines exemplifying three cancer sub-types (non-small cell lung cancer, glioblastoma, and breast cancer). Fbx6 and Chk1 proteins were variably expressed in these cell lines, even those derived from the same tissue of origin. Nonetheless, many of these cell lines (in our assessment, 15 out of 19 cell lines tested) exhibited a noticeable inverse relationship between the expression levels of Fbx6 and Chk1 (Figures 7A–B, and Supplementary Figures S11A–B). A semi-quantitative RT-PCR showed no correlation between the levels of FBX6 and CHK1 mRNA transcripts (Supplemental Figure S10), consistent with the idea that a post-transcriptional mechanism, i.e., Fbx6-dependent Chk1 protein turnover, underlies the inverse relationship between these two proteins.
We further examined the levels of Chk1 and Fbx6 proteins in sections of a panel of 16 breast tumor samples. This quantitative immunohistochemistry analysis was performed by Histo-Rx, Inc. (New Haven, CT) using the AQUA™ technology platform that allows reproducible measurements of proteins of interest in both the cytoplasmic and nuclear compartments of cells in fixed tissues. Our affinity-purified polyclonal anti-Fbx6 antibodies were used for Fbx6 immunohistochemistry after rigorous preliminary validation (Figure 7C). These tumor samples had not been categorized for cancer sub-type or stage at the time of excision. Remarkably, Fbx6 and Chk1 protein expression revealed a significant inverse correlation in these tumor tissues, as indicated by the trend line (Figure 7D). The linear regression is well fitted, with an R-square of 0.351. The trend line is the result of linear regression between Fbx6 and Chk1 AQUA scores with a Pearson correlation coefficient of R= −0.59 (p=0.016). Rank order correlation analysis of the data indicates a Spearman rho= −0.556 (p=0.025) (Figure 7D). These results suggest that the steady-state level of Chk1 is determined, at least in part, by the level of the Fbx6 in these tumors.
DISCUSSION
In this study, we identified an F-box protein, Fbx6, and a degron-like region at the C-terminus of Chk1 that regulate Chk1 ubiquitination and degradation in both normally cycling cells and during replication stress. We propose that the phosphorylation of Chk1 by ATR sets in motion of sequence of events that simultaneously initiates the replication checkpoint, and limits the duration of checkpoint signaling via the targeted degradation of Chk1 (Figure 7E). In the absence of replication stress, the Chk1 protein adopts a ‘closed’ conformation that is likely stabilized through interactions of the AIR region with the N-terminal kinase domain and heterologous adaptor proteins (marked as X), such as Rad9/Claspin or PCNA (Chen et al., 2009; Scorah et al., 2008). In this state, Chk1 kinase activity is suppressed, and the degron-like region, located in the regulatory domain, is not accessible to the Fbx6-containing SCF E3 ligase, and possibly a second E3 ligase containing Cul4A/DDB1 as well (Leung-Pineda et al., 2009). During replication stress, ATR-dependent phosphorylation of Chk1 promotes the transition to an open conformation, which leads to protein kinase activation, and concomitantly exposes the degron-like region, rendering Chk1 accessible to E3 ubiquitin ligases that ultimately destabilize the activated protein kinase. This coupled activation-destruction mechanism of Chk1 limits the duration of replication checkpoint activation, and helps to insure that replication checkpoint efficiently protects active replication forks, without inappropriately interfering with S phase progression. A definitive confirmation of this model will require a full structural analysis of the inactive and S345-phosphorylated versions of full-length Chk1.
Fbx6 is mainly detected in the cytoplasm in both cultured cells and in tumor tissues. We previously observed an interaction between Chk1 and Cul1 in the nucleoplasm (Zhang et al., 2005), which seems to contradict the current findings that Fbx6-dependent Chk1 degradation occurs in the cytoplasm. We noted that Cul1 is primarily found in the nuclear compartment, with only low level detected in the cytoplasm (Zhang et al., 2005). Hence, the sensitivity of our co-immunoprecipitation assay may have precluded detection of a cytoplasmic pool of Cul1-associated Chk1. Alternatively, Fbx6 may promote the shuttling of phosphorylated Chk1 protein from nucleoplasm to cytoplasm. Fbx6 might bind to both Cul1 and phosphorylated Chk1 in the nucleoplasm, and the resulting protein complex might be exported from the nucleus to the cytoplasm, where Chk1 is fully ubiquitinated and degraded. A recent precedent for this model involves the Fbx6 paralog protein NFB42 (or Fbx44), which was shown to promote the nuclear export and cytoplasmic ubiquitination of viral protein UL9 (Eom et al., 2004).
Recent studies suggest that at least three mechanisms inactivate Chk1 in vertebrate cells: (1) SCFβ-Trcp1-mediated degradation of Claspin, an essential mediator during the ATR-induced Chk1 phosphorylation (Bennett and Clarke, 2006; Mailand et al., 2006; Mamely et al., 2006; Peschiaroli et al., 2006); (2) dephosphorylation of activated Chk1 by PP1, PP2A, or PP2C type phosphatases (den Elzen and O'Connell, 2004; Leung-Pineda et al., 2006; Lu et al., 2005); and (3) Cul1 or Cul4A-dependent degradation of Chk1, as described in this paper and elsewhere (Zhang et al., 2005; Leung-Pineda et al., 2009). The rapid degradation of Claspin (see Supplementary Figure S11C) blocks further phosphorylation of Chk1 by ATR, whereas dephosphorylation reverses Chk1 activation without eliminating the protein. We propose that the ubiquitination pathways targeting Chk1 insure that activated Chk1 does not accumulate as cells progress through S phase, or when replication forks encounter transient impediments during normal DNA replication. Although additional work is needed to understand the factors that determine whether Chk1 is dephosphorylated and potentially available for another round of activation versus being ubiquitinated and irreversibly removed from the replication checkpoint machinery.
Our findings suggest that Chk1 expression may serve as an important determinant of tumor sensitivity to certain anticancer therapies that provoke high-intensity replication stress. These drugs strongly activate the ATR–Chk1 pathway, and, with prolonged exposure, cause the depletion of intracellular Chk1, presumably due to the inability of Chk1 protein synthesis to maintain the steady state pool of Chk1 in the setting of continuous Chk1 activation and degradation. The progressive loss of Chk1 eventually surpasses a threshold below which the replication checkpoint machinery can no longer maintain the viability of stalled replication forks, which leads to irreversible replication arrest and/or DNA strand breakage, and ultimately cell death. If this model is correct, we would expect that tumor cells bearing defects in the mechanisms responsible for the irreversible removal of Chk1 will manifest resistance to chronic replication stress induced by chemotherapy and possibly the tumor microenvironment itself. Indeed, we found that two of the three most CPT-resistant cancer cell lines in NCI 60-cell line panel displayed significant defects in CPT-induced Chk1 degradation, which appeared to be causally related to very low levels of Fbx6 expression. Similarly, an earlier study reported that a trinuclear platinum complex induced downregulation of Chk1 in the parent ovarian carcinoma cell line, but not in a drug resistant sub-clone (Perego et al., 2003). These findings suggest that alteration of Chk1 expression regulated by ubiquitination and degradation represents a common mechanism for anticancer therapy resistance.
In agreement with this hypothesis, we observed an overall inverse correlation between Fbx6 and Chk1 expression in both cultured cancer cell lines and in a small cohort of breast tumor tissues. Considering Chk1 expression is influenced by multiple parameters, both transcriptional and post-transcriptional, the observed inverse correlation between Chk1 and Fbx6 levels in cancer cell lines and breast tumor tissues clearly suggests that variations in Fbx6 expression have important implications for Chk1 expression and function in tumor cells. A critical next step will be to evaluate a much larger cohort of breast cancers and other tumor types to understand the true significance and broad importance of the Fbx6-Chk1 relationship in human cancers. Our findings suggest that an outcome-based study that correlates Fbx6 and Chk1 expression in primary tumors with patient responsiveness to drugs such as irinotecan, platinum compounds, and gemcitabine might reveal that Fbx6 expression is a predictive biomarker of tumor responsiveness to these important anticancer drugs.
EXPERIMENTAL PROCEDURES
siRNA Library Screening
The SMARTpool ™ siRNA library targeting all known human F-box genes was obtained from Dharmacon. An optimized protocol for siRNA screening involves the usage of 549 cells and 100 nM siRNA with the DharmaFECT 1 reagent. Expression levels of Chk1 and PLCγ-1 were analyzed by immunoblotting and quantitated by the NIH Image Software. Positive hits from the first round of screening were re-screened three times to identify the final set of candidate F-box proteins.
Cell Culture and Antibodies
Standard culture was used for cell lines in this study. MCF10A cells were grown in DMEM/F12 (1:1) with 5% equine serum, 0.5 µg/ml hydrocortisone, 5 µg/ml insulin, and 20 ng/ml EGF. Anti-Chk1 (G4, Santa Cruz Biotechnologies), anti-Flag (M2, Sigma), rat anti-HA (3F10, Roche), anti-Cul1 (Zymed), anti-tubulin (Sigma), anti-p-NBS1 (Santa Cruz), anti-BrdU (Immunological Direct), and anti-Cdc25A (Ab-3, Lab Vision) were used in this study. Anti-p-S317-Chk1, anti-p-S345-Chk1, anti-cleaved Caspase 3 and anti-p-S216-Cdc25C were from Cell Signaling. Anti-Myc ascites fluid was generated with the anti-Myc hybridoma line 9E10 from ATCC. Anti-PLCγ-1, anti-GST, and anti-GFP antibodies were generated in this laboratory. Anti-Fbx6 antibodies were generated against KLH-coupled synthetic peptide corresponding to amino acids 262 to 284 of human Fbx6, which lie very close to the C-terminus (see Figure 2B). Antiserum from peptide-injected rabbits was affinity purified using the AminoLink Plus Immobilization Kit (Pierce). Chemicals and immunoblot analyses were as described previously (Zhang et al., 2005).
Immunofluorescence Staining
Cells grown on glass cover slips were fixed in 3.7% formaldehyde for 15 min. For BrdU staining, cells were additionally treated with 2 N HCl/0.5% NP-40 for 30 min and washed extensively with 0.1 M Na2B4O7 (pH 8.5). Samples were blocked for 30 min in 10% goat serum/PBS, stained with rat anti-BrdU (Immunological Direct), mouse anti-Flag (Sigma), rabbit anti-Chk1 (Cell Signaling), or rabbit anti-caspase 3 cleavage product (Cell Signaling), followed by anti-rat (red), anti-mouse (green), anti-rabbit (red) secondary antibodies (Molecular Probes) according to each individual experimental setting. The samples were then incubated with DAPI for 5 min, mounted, and visualized using a laser microscope. The TIFF files of images were opened in Photoshop, and the average fluorescence intensity (pixels) of BrdU (red) or Flag (green) was quantitated using the Histogram analysis. More than 50 cells were quantitated for each sample. The average fluorescence intensities of Flag or BrdU were normalized to before or after doxcycline induction, respectively.
Tumor Tissue Staining
Tissue microarray slides consisting of primary breast tumor samples were deparaffinized at 60°C, hydrated in water, and then put in a PT Module™ (Lab Vision Corp, Fremont CA) with a 1X Tris EDTA Buffer, pH 9.0) for antigen retrieval. Staining was carried out on a LabVision 720 Autostainer (Labvision, Fremont, CA) using 1X TBS-Tween rinse and wash solution. Sections were incubated in Peroxidazed blocking reagent (Biocare Medical, Concord, CA) for 5 min followed by Background Sniper (Biocare Medical, Concord, CA) for 10 min and then rinsed three times for 5 min in 1×TBS Tween. Samples were incubated with the primary antibodies (anti-Chk1, Santa Cruz, sc-56291, mouse monoclonal, 1:100, or anti-Fbx6, generated in this study, rabbit polyclonal, 1:300) diluted in DaVinci Green (Biocare Medical, Concord, CA) for 16 h at 4°C. Each primary antibody was included in a cocktail with mouse anti-pan-cytokeratin (Dako, M3515012, Carpinteria, CA, 1:50) for Fbx6, or rabbit anti-pan-cytokeratin (Dako, Z0622, Carpinteria, CA, 1:200) for Chk1. After rinsing in 1X TBS-Tween, slides were incubated with a cocktail containing: Rabbit EnvisionPlus (Dako, Carpinteria, CA) and Alexa555-conjugated anti-mouse (Invitrogen, A21422, Carlsbad CA, 1:200) for Chk1; or mouse EnvisionPlus (Dako, Carpinteria, CA) and Alexa555-conjugated anti-rabbit (Invitrogen, A21428, Carlsbad CA, 1:200) for Fbx6, for 30 min in the dark. Samples were then incubated with the Cy5 tyramide amplification system (Perkin Elmer, SAT705A, 1:50 dilution in amplification buffer) for 10 min, mounted with Prolong anti-fade with DAPI (Invitrogen, P36931, Carlsbad CA) and allowed to dry overnight.
Image Acquisition, AQUA® Analysis and Validation
Images of tissue microarrays were obtained using a PM-2000™ instrument (HistoRx, New Haven, CT). High-resolution (20X magnification) images were taken for each spot in each relevant fluorescent channel. The images were subjected to quantitative analysis with the AQUA® system. Briefly, pan-cytokeratin was utilized to molecularly identify the epithelial regions and non-nuclear epithelial regions within the tumor tissue microarray samples, and DAPI was utilized to mark cell nuclei. Each spot was assessed for signal integrity, tissue spot integrity, and image integrity. Fbx6 and Chk1 were measured in the non-nuclear (cytoplasmic) epithelial compartment.
Statistical Analysis
All analyses were conducted using SPSS V15 (SPSS Inc., Chicago, Illinois). R-squared is a statistical measure of how well the regression line approximates the real data points. An R-square of 1.0 indicates that the regression line perfectly fits the data. Correlation between data sets was evaluated using both Pearson (R) and Spearman (r) coefficients. The Pearson coefficient examines correlation between AQUA scores in a linear regression, while the Spearman correlation coefficient value indicates the strength of rank correlation. A positive value indicates a direct correlation, and a negative value indicates an inverse correlation. For this study, the absolute value of Spearman correlation was stratified based on the following scale: 0.0<0.2 (no correlation), 0.2 to <0.4 (weak, low correlation), 0.4 to <0.7 (significant correlation), and >0.7 (strong, high correlation), with the associated p-value indicating the significance of the correlation.
Supplementary Material
ACKNOWLEDGEMENTS
We thank Histo-RX, Inc for immunohistochemistry of the breast tumor tissues. We also thank J. Meisenhelder for synthesizing peptides and Michele Pagano (NYU) and Kenna Anderes (Pfizer Global Research & Development, San Diego) for providing reagents. Y.W.Z. was a Susan G. Komen Postdoctoral Fellow (PDF0503489), and is currently funded by an NCI K99/R00 Career Development Award (5 K99 CA126173). T.H is a Frank and Else Schilling American Cancer Society Research Professor. This work was funded by USPHS grants (CA14195 and CA116402) from NCI to T.H.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
SUPPLEMENTAL DATA
Supplemental data including methods and eleven figures accompany this paper.
REFERENCES
- Abraham RT. Cell cycle checkpoint signaling through the ATM and ATR kinases. Genes Dev. 2001;15:2177–2196. doi: 10.1101/gad.914401. [DOI] [PubMed] [Google Scholar]
- Bartek J, Lukas C, Lukas J. Checking on DNA damage in S phase. Nat Rev Mol Cell Biol. 2004;5:792–804. doi: 10.1038/nrm1493. [DOI] [PubMed] [Google Scholar]
- Bennett LN, Clarke PR. Regulation of Claspin degradation by the ubiquitin-proteosome pathway during the cell cycle and in response to ATR-dependent checkpoint activation. FEBS Lett. 2006;580:4176–4181. doi: 10.1016/j.febslet.2006.06.071. [DOI] [PubMed] [Google Scholar]
- Blasina A, Hallin J, Chen E, Arango ME, Kraynov E, Register J, Grant S, Ninkovic S, Chen P, Nichols T, et al. Breaching the DNA damage checkpoint via PF-00477736, a novel small-molecule inhibitor of checkpoint kinase 1. Mol. Cancer. Ther. 2008;7:2394–2404. doi: 10.1158/1535-7163.MCT-07-2391. [DOI] [PubMed] [Google Scholar]
- Cardozo T, Pagano M. The SCF ubiquitin ligase: insights into a molecular machine. Nat. Rev. Mol. Cell. Biol. 2004;5:739–751. doi: 10.1038/nrm1471. [DOI] [PubMed] [Google Scholar]
- Chen P, Luo C, Deng Y, Ryan K, Register J, Margosiak S, Tempczyk-Russell A, Nguyen B, Myers P, Lundgren K, et al. The 1.7 A crystal structure of human cell cycle checkpoint kinase Chk1: implications for Chk1 regulation. Cell. 2000;100:681–692. doi: 10.1016/s0092-8674(00)80704-7. [DOI] [PubMed] [Google Scholar]
- Chowdhury D, Keogh MC, Ishii H, Peterson CL, Buratowski S, Lieberman J. gamma-H2AX dephosphorylation by protein phosphatase 2A facilitates DNA double-strand break repair. Mol. Cell. 2005;20:801–809. doi: 10.1016/j.molcel.2005.10.003. [DOI] [PubMed] [Google Scholar]
- Collis SJ, Barber LJ, Clark AJ, Martin JS, Ward JD, Boulton SJ. HCLK2 is essential for the mammalian S-phase checkpoint and impacts on Chk1 stability. Nat. Cell. Biol. 2007;9:391–401. doi: 10.1038/ncb1555. [DOI] [PubMed] [Google Scholar]
- Cuadrado M, Martinez-Pastor B, Murga M, Toledo LI, Gutierrez-Martinez P, Lopez E, Fernandez-Capetillo O. ATM regulates ATR chromatin loading in response to DNA double-strand breaks. J. Exp. Med. 2006;203:297–303. doi: 10.1084/jem.20051923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- den Elzen NR, O'Connell MJ. Recovery from DNA damage checkpoint arrest by PP1-mediated inhibition of Chk1. EMBO J. 2004;23:908–918. doi: 10.1038/sj.emboj.7600105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Denis NJ, Vasilescu J, Lambert JP, Smith JC, Figeys D. Tryptic digestion of ubiquitin standards reveals an improved strategy for identifying ubiquitinated proteins by mass spectrometry. Proteomics. 2007;7:868–874. doi: 10.1002/pmic.200600410. [DOI] [PubMed] [Google Scholar]
- Eom CY, Heo WD, Craske ML, Meyer T, Lehman IR. The neural F-box protein NFB42 mediates the nuclear export of the herpes simplex virus type 1 replication initiator protein (UL9 protein) after viral infection. Proc. Natl. Acad. Sci. USA. 2004;101:4036–4040. doi: 10.1073/pnas.0400738101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feijoo C, Hall-Jackson C, Wu R, Jenkins D, Leitch J, Gilbert DM, Smythe C. Activation of mammalian Chk1 during DNA replication arrest: a role for Chk1 in the intra-S phase checkpoint monitoring replication origin firing. J. Cell Biol. 2001;154:913–923. doi: 10.1083/jcb.200104099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feng JM, Zhu H, Zhang XW, Ding J, Miao ZH. Proteasome-dependent degradation of Chk1 kinase induced by the topoisomerase II inhibitor R16 contributes to its anticancer activity. Cancer Biol. Ther. 2008;7:1726–1731. doi: 10.4161/cbt.7.11.6728. [DOI] [PubMed] [Google Scholar]
- Gallagher E, Gao M, Liu YC, Karin M. Activation of the E3 ubiquitin ligase Itch through a phosphorylation-induced conformational change. Proc. Natl. Acad. Sci. USA. 2006;103:1717–1722. doi: 10.1073/pnas.0510664103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goodarzi AA, Jonnalagadda JC, Douglas P, Young D, Ye R, Moorhead GB, Lees-Miller SP, Khanna KK. Autophosphorylation of ataxia-telangiectasia mutated is regulated by protein phosphatase 2A. EMBO J. 2004;23:4451–4461. doi: 10.1038/sj.emboj.7600455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jazayeri A, Falck J, Lukas C, Bartek J, Smith GC, Lukas J, Jackson SP. ATM- and cell cycle-dependent regulation of ATR in response to DNA double-strand breaks. Nat. Cell. Biol. 2006;8:37–45. doi: 10.1038/ncb1337. [DOI] [PubMed] [Google Scholar]
- Jin J, Cardozo T, Lovering RC, Elledge SJ, Pagano M, Harper JW. Systematic analysis and nomenclature of mammalian F-box proteins. Genes Dev. 2004;18:2573–2580. doi: 10.1101/gad.1255304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jurvansuu J, Fragkos M, Ingemarsdotter C, Beard P. Chk1 instability is coupled to mitotic cell death of p53-deficient cells in response to virus-induced DNA damage signaling. J. Mol. Biol. 2007;372:397–406. doi: 10.1016/j.jmb.2007.06.077. [DOI] [PubMed] [Google Scholar]
- Kaneko YS, Watanabe N, Morisaki H, Akita H, Fujimoto A, Tominaga K, Terasawa M, Tachibana A, Ikeda K, Nakanishi M. Cell-cycle-dependent and ATM-independent expression of human Chk1 kinase. Oncogene. 1999;18:3673–3681. doi: 10.1038/sj.onc.1202706. [DOI] [PubMed] [Google Scholar]
- Kastan MB, Bartek J. Cell-cycle checkpoints and cancer. Nature. 2004;432:316–323. doi: 10.1038/nature03097. [DOI] [PubMed] [Google Scholar]
- Katsuragi Y, Sagata N. Regulation of Chk1 kinase by autoinhibition and ATR-mediated phosphorylation. Mol. Biol. Cell. 2004;15:1680–1689. doi: 10.1091/mbc.E03-12-0874. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Keogh MC, Kim JA, Downey M, Fillingham J, Chowdhury D, Harrison JC, Onishi M, Datta N, Galicia S, Emili A, et al. A phosphatase complex that dephosphorylates gammaH2AX regulates DNA damage checkpoint recovery. Nature. 2006;439:497–501. doi: 10.1038/nature04384. [DOI] [PubMed] [Google Scholar]
- Kosoy A, O'Connell MJ. Regulation of Chk1 by its C-terminal domain. Mol. Biol. Cell. 2008;19:4546–4553. doi: 10.1091/mbc.E08-04-0444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leung-Pineda V, Huh J, Piwnica-Worms H. DDB1 targets Chk1 to the Cul4 E3 ligase complex in normal cycling cells and in cells experiencing replication stress. Cancer Res. 2009;69:2630–2637. doi: 10.1158/0008-5472.CAN-08-3382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leung-Pineda V, Ryan CE, Piwnica-Worms H. Phosphorylation of Chk1 by ATR is antagonized by a Chk1-regulated protein phosphatase 2A circuit. Mol. Cell. Biol. 2006;26:7529–7538. doi: 10.1128/MCB.00447-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu Q, Guntuku S, Cui XS, Matsuoka S, Cortez D, Tamai K, Luo G, Carattini-Rivera S, DeMayo F, Bradley A, et al. Chk1 is an essential kinase that is regulated by Atr and required for the G(2)/M DNA damage checkpoint. Genes Dev. 2000;14:1448–1459. [PMC free article] [PubMed] [Google Scholar]
- Lu X, Nannenga B, Donehower LA. PPM1D dephosphorylates Chk1 and p53 and abrogates cell cycle checkpoints. Genes Dev. 2005;19:1162–1174. doi: 10.1101/gad.1291305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- MacDougall CA, Byun TS, Van C, Yee MC, Cimprich KA. The structural determinants of checkpoint activation. Genes Dev. 2007;21:898–903. doi: 10.1101/gad.1522607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mailand N, Bekker-Jensen S, Bartek J, Lukas J. Destruction of Claspin by SCFbetaTrCP restrains Chk1 activation and facilitates recovery from genotoxic stress. Mol. Cell. 2006;23:307–318. doi: 10.1016/j.molcel.2006.06.016. [DOI] [PubMed] [Google Scholar]
- Mamely I, van Vugt MA, Smits VA, Semple JI, Lemmens B, Perrakis A, Medema RH, Freire R. Polo-like kinase-1 controls proteasome-dependent degradation of Claspin during checkpoint recovery. Curr. Biol. 2006;16:1950–1955. doi: 10.1016/j.cub.2006.08.026. [DOI] [PubMed] [Google Scholar]
- Myers JS, Cortez D. Rapid activation of ATR by ionizing radiation requires ATM and Mre11. J. Biol. Chem. 2006;281:9346–9350. doi: 10.1074/jbc.M513265200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oe T, Nakajo N, Katsuragi Y, Okazaki K, Sagata N. Cytoplasmic occurrence of the Chk1/Cdc25 pathway and regulation of Chk1 in Xenopus oocytes. Dev. Biol. 2001;229:250–261. doi: 10.1006/dbio.2000.9968. [DOI] [PubMed] [Google Scholar]
- Perego P, Gatti L, Righetti SC, Beretta GL, Carenini N, Corna E, Dal Bo L, Tinelli S, Colangelo D, Leone R, et al. Development of resistance to a trinuclear platinum complex in ovarian carcinoma cells. Int. J. Cancer. 2003;105:617–624. doi: 10.1002/ijc.11140. [DOI] [PubMed] [Google Scholar]
- Peschiaroli A, Dorrello NV, Guardavaccaro D, Venere M, Halazonetis T, Sherman NE, Pagano M. SCFbetaTrCP-mediated degradation of Claspin regulates recovery from the DNA replication checkpoint response. Mol. Cell. 2006;23:319–329. doi: 10.1016/j.molcel.2006.06.013. [DOI] [PubMed] [Google Scholar]
- Rasheed ZA, Rubin EH. Mechanisms of resistance to topoisomerase I-targeting drugs. Oncogene. 2003;22:7296–7304. doi: 10.1038/sj.onc.1206935. [DOI] [PubMed] [Google Scholar]
- Scorah J, Dong MQ, Yates JR, 3rd, Scott M, Gillespie D, McGowan CH. A conserved proliferating cell nuclear antigen-interacting protein sequence in Chk1 is required for checkpoint function. J. Biol. Chem. 2008;283:17250–17259. doi: 10.1074/jbc.M800369200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shann YJ, Hsu MT. Cloning and characterization of liver-specific isoform of Chk1 gene from rat. J. Biol. Chem. 2001;276:48863–48870. doi: 10.1074/jbc.M108253200. [DOI] [PubMed] [Google Scholar]
- Shiloh Y. The ATM-mediated DNA-damage response: taking shape. Trends Biochem Sci. 2006;31:402–410. doi: 10.1016/j.tibs.2006.05.004. [DOI] [PubMed] [Google Scholar]
- Shreeram S, Demidov ON, Hee WK, Yamaguchi H, Onishi N, Kek C, Timofeev ON, Dudgeon C, Fornace AJ, Anderson CW, et al. Wip1 phosphatase modulates ATM-dependent signaling pathways. Mol. Cell. 2006;23:757–764. doi: 10.1016/j.molcel.2006.07.010. [DOI] [PubMed] [Google Scholar]
- Wang SX, Dunphy WG. Activation of Xenopus Chk1 by mutagenesis of threonine-377. FEBS Lett. 2000;487:277–281. doi: 10.1016/s0014-5793(00)02370-x. [DOI] [PubMed] [Google Scholar]
- Yoda A, Xu XZ, Onishi N, Toyoshima K, Fujimoto H, Kato N, Oishi I, Kondo T, Minami Y. Intrinsic kinase activity and SQ/TQ domain of Chk2 kinase as well as N-terminal domain of Wip1 phosphatase are required for regulation of Chk2 by Wip1. J. Biol. Chem. 2006;281:24847–24862. doi: 10.1074/jbc.M600403200. [DOI] [PubMed] [Google Scholar]
- Zhang YW, Otterness DM, Chiang GG, Xie W, Liu YC, Mercurio F, Abraham RT. Genotoxic stress targets human Chk1 for degradation by the ubiquitin-proteasome pathway. Mol. Cell. 2005;19:607–618. doi: 10.1016/j.molcel.2005.07.019. [DOI] [PubMed] [Google Scholar]
- Zhao H, Piwnica-Worms H. ATR-mediated checkpoint pathways regulate phosphorylation and activation of human Chk1. Mol. Cell. Biol. 2001;21:4129–4139. doi: 10.1128/MCB.21.13.4129-4139.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.