

Abstract
The cyclin D1 oncogene is frequently amplified/overexpressed in oral squamous cell carcinomas. Mice with overexpression of cyclin D1 targeted to the stratified squamous epithelia of the tongue, esophagus and forestomach develop a phenotype of epithelial dysplasia at these sites. In this study, we examined the effect cyclin D1 overexpression on susceptibility of mice to carcinogen-induced tumorigenesis, using 4-nitroquinoline-1-oxide (4NQO), an established potent oral carcinogen in mice. Cyclin D1 overexpressing mice and non-transgenic littermates were administered 4NQO (20 ppm or 50 ppm in the drinking water) for 8 weeks and monitored for an additional 16 weeks. Histopathological analyses of the tongue revealed significantly higher severity of dysplasia in the cyclin D1 overexpression mice, compared with non-transgenic controls and with untreated controls. Moreover, only the cyclin D1 overexpression mice developed neoplastic lesions in the oro-esophageal epithelia. Examination of the dysplastic and neoplastic lesions reveled abnormal proliferation. Our findings suggest that cyclin D1 overexpression enhances susceptibility to carcinogen-induced oral tumorigenesis. These results underscore the importance of cyclin D1 in the process of oral neoplastic development. Further, they emphasize the value of this transgenic model to study the pathogenesis of oral precancer and cancer and establish it as a model system to test candidate agents for chemoprevention of upper aero-digestive cancer.
Keywords: Mouth neoplasms/genetics, Cyclin D1/genetics, Disease Models, Animal Mice, Transgenic
Introduction
Squamous cell carcinoma (SCC) of the oral cavity is the sixth most common cancer worldwide. Oral SCC is an aggressive tumor that results in significant mortality and morbidity. Despite advances in surgical, radiotherapeutic and medical management, the survival rate for this disease has improved only marginally over the last two decades [1]. The major risk factors are tobacco use and alcohol consumption. Frequently, a clinically detectable preneoplastic lesion, usually a red/white lesion that manifests histopathological changes of epithelial dysplasia, precedes the development of a frank SCC. However, the molecular and genetic events that participate in the progression of dysplasia to neoplasia are not fully understood.
It is well established that cyclin D1 is an important early driver of oral neoplasia. The cyclin D1 oncogene is a key regulator of progression through the G1 phase of the cell cycle. It binds to and activates its kinase partners CDK4 and CDK6, resulting in the phosphorylation of the retinoblastoma protein, thereby effecting transcription of genes that promote progression to the S-phase of the cell cycle [2]. In addition, there is growing evidence that cyclin D1 has cdk-independent functions [3,4]. There is strong evidence that dysregulation of cyclin D1 plays an important role in oral SCC development. Cyclin D1 is frequently amplified and/or overexpressed in oral SCC [5-9]. These aberrations are also noted in precancerous oral epithelial dysplastic lesions [10,11], suggesting that cyclin D1 deregulation is an early event in the neoplastic process. The importance of cyclin D1 in oral dysplasia and cancer has been further substantiated by studies of transgenic mice. In these mice (L2-D1), the ED2-L2 promoter of the Epstein Barr virus is used to target overexpression of cyclin D1 in the squamous epithelia of the tongue, esophagus and forestomach [12]. These mice manifest increased cell proliferation in the basal and suprabasal epithelial strata and develop epithelial dysplasia at these sites [12,13]. When crossbred with constitutive p53-deficient mice, the L2-D1 mice develop invasive oro-esophageal SCC [14]. However, the biological basis for cyclin D1's oncogenic effects remains to be fully elucidated.
Local chronic exposure to carcinogens from tobacco and alcohol constitute the major etiological factors in most oral SCCs’ development. However, little is known about the influence of genetic factors that modulate the oro-esophageal epithelium's response to these environmental carcinogens. The experimental carcinogen 4-nitroquinoline 1-oxide (4NQO) has been used extensively to simulate these environmental exposures in rodents [15-18]. 4NQO is a quinoline derivative and causes a wide range of DNA damage, including DNA adducts, single-strand breaks, abasic sites, pyrimidine dimers, and oxidized bases [19]. Depending upon the dose and duration of treatment, 4NQO induces a spectrum of dysplastic and neoplastic lesions in the oro-esophageal epithelium, with morphological and molecular alterations that mimic those occurring in human oral epithelial preneoplastic and neoplastic lesions [15-17]. In the present study, we examined the interaction between overexpression of cyclin D1 and 4NQO in inducing oro-esophageal carcinogenesis. Our results demonstrate that cyclin D1 overexpression significantly increases susceptibility to 4NQO-induced dysplastic and neoplastic lesions.
Materials and methods
Generation and analyses of transgenic mice
Transgenic mice were created as previously described [12]. Specifically, a plasmid clone pL2-HD1 containing an 800-bp fragment of the L2 promoter juxtaposed to 1.3kb of the human cyclin D1 cDNA (HD1) sequence was microinjected into pronuclei of fertilized eggs from C57/BL6J mice. Injected eggs were transplanted to pseudopregnant foster female mice and allowed to develop to term. Litters were weaned and tail DNA was analyzed by polymerase chain reaction using two sets of PCR primers. The first primer set amplified a 198 bp region of the cyclin D1 cDNA: 5’-TCC TCT CCA AAA TGC CAG AG -3’ (forward) and 5’-TGA GGC GGT AGT AGG ACA GG-3’ (reverse). The second primer set amplified a 289 bp fragment of the L2 promoter region: 5’-CCT GTC TCC CAC CCA GTA ACT -3’ (forward) and 5’- ATC TCG AGA GTG AGG CAC AGC TG -3’ (reverse). PCR reactions were done in a Mastercycler EPS (Eppendorf, Westbury, NY) using the following reaction conditions: initial denaturation for 10 min at 94°C, followed by 35 cycles of denaturation at 94°C for 30 sec, annealing at 55°C for 30 sec and extension at 72°C for 45 sec. PCR-amplified DNA was analyzed on a 2% agarose gel. The presence of the transgene was confirmed by Southern blot analysis using the human cyclin D1 cDNA as a probe. Briefly, 10 μg of genomic DNA was digested with PstI or with a combination of BamHI and EcoRI and separated by electrophoresis on a 1% agarose gel, transferred onto nitrocellulose membrane and hybridized to 32P-labelled human cyclin D1 cDNA probe. Hybridized membranes were washed twice with 2X SCC followed by a high stringency wash with 0.1X SSC, and exposed to autoradiography film.
Analysis of cyclin D1 transgene expression
Expression of the cyclin D1 transgene in the target tissues was confirmed by Northern analysis. Total RNA was extracted from the tongue and esophagus using Trizol reagent (Invitrogen, Carlsbad, CA) following the manufacturer's protocol. Approximately 10 μg of total RNA was fractionated on a 1% denaturing agarose gel containing 0.7M formaldehyde, transferred onto HyBond N+ membranes (GE Healthcare Bio-sciences, Corp., Piscataway, NJ) with 20X SSC and baked in a vacuum oven for 2 hours. The 1.3 kb fragment of the human cyclin D1 cDNA was used as a template to generate random-primed α-32P-labeled probes and hybridized to the membrane at 42°C in hybridization buffer containing 40% formamide, 4x SSC, 7 mM Tris (pH 7.9), 0.8x Denhardt's solution, 20% dextran sulfate, 20 μg/ml herring sperm DNA, and 1% SDS. Hybridized membranes were washed twice with 2X SCC followed by a high stringency wash with 0.1X SSC, and exposed to autoradiography film.
Carcinogen treatment
Adult L2-D1 transgenic mice and non-transgenic littermates (approximately 5 months of age) were treated with 4NQO (Sigma, St. Louis, MO) in their drinking water at a concentration of 20 parts per million (ppm) or 50 ppm for a period of 8 weeks. The carcinogenic solutions were prepared every week and delivered in amber bottles. Following the treatment period, the mice were given normal water for an additional 16 weeks. During this post-treatment period, mice were weighed biweekly. Every four weeks the mice were anesthetized with a combination of ketamine and xylazine (80 mg/kg and 10 mg/kg bodyweight, respectively, by intraperitoneal injection) and examined for the presence of gross morphological changes on the visible regions of the tongue and oral cavity. Mice were euthanized at the end of the 16-week period and the tongues and esophagi were dissected and examined for the presence of overt tumors. The institutional animal care committee reviewed and approved all animal experimental studies.
Histopathological examination
At necropsy, the entire length of the esophagus was split open longitudinally. The tongue and the esophagus were examined for the presence of macroscopic topographical alterations. The tongue was then split longitudinally and one half of the tongue was placed in a cryomold containing tissue-freezing medium (Triangle Biomedical Sciences, Durham, NC) and frozen on dry ice. The other half of the tongue and the entire esophagus were fixed overnight in 10% neutral buffered formalin, transferred to 70% ethanol and embedded in paraffin. Five-μm thick sections were obtained from the paraffin-embedded blocks and stained with hematoxylin and eosin. Sections were examined for the presence of epithelial atypia, dysplasia and neoplasia using established criteria [20]. The examiner was first calibrated to normal histopathological appearances by examining sections from 6 wild-type, non-carcinogen treated mice. Subsequent sections were analyzed blinded to the genotype and carcinogen treatment. Depending on the degree of atypical cytological and architectural changes observed, dysplastic lesions were scored as mild, moderate or severe dysplasia.
Immunohistochemistry
Paraffin-embedded tissues were sectioned onto glass slides. Sections were deparaffinized in xylene, cleared through a graded ethanol series and then hydrated in distilled water. For epitope retrieval, slides were heated for 15 minutes in citrate buffer, pH 6.0, in an electric pressure cooker. Endogenous peroxidase was quenched with 3% H2O2 and the sections were incubated for 30 minutes in 5% normal serum to reduce non-specific binding. Sections were incubated at room temperature with antibodies to the following proteins: Ki-67 (DakoCytomation, Carpinteria, CA, Clone Tec3, 1:100 for 30 min); Keratin 5 (Covance Research Products, Berkeley, CA, Clone AF 138, 1:2000 for 1 hour); Cox-2 (Lab Vision Corporation, Freemont, CA, clone SP-21, 1:1000 for 2 hours); E-cadherin (Cell Signaling Technology, Danvers, MA, clone 24E10, 1:50 for 2 hours). Binding of the primary antibodies was detected using the appropriate biotinylated IgG and the avidin-biotin complex (Vector Laboratories, Burlingame, CA) using 3,3-diaminobenzidine as a chromagen (Zymed, Invitrogen Corporation, Carslbad, CA). Sections were counterstained in hematoxylin. The labeling index for Ki67 was performed as previously described [21]. The number of positive cells was counted using the NIH Image J software (US National Institutes of Health, Bethesda, Maryland, USA; http://rsb.info.nih.gov/ij/) and expressed as positive cells per unit area.
For immunohistochemical detection of p16, a mouse monoclonal anti-p16 antibody (Santa Cruz Biotechnology, clone F-12) was used with the Animal Research Kit (DAKO cytomation, Carpinteria, CA) following the manufacturer's instructions. Briefly, antigen retrieval was done as described above. The primary antibody was biotinylated and subsequently incubated with the sections for 15 minutes. Sections were rinsed in PBS and incubated with Streptavidin-peroxidase and subsequently with DAB. Sections were counterstained, dehydrated and mounted as described above.
Statistical Analysis
To determine the difference in severity of 4NQO-induced dysplasia between L2-D1 and wild-type mice, we used the nonparametric Mann-Whitney test.
Results
Generation and characterization of transgenic mice
We used the Epstein-Barr virus ED2-L2 promoter to target expression of cyclin D1 to the oro-esophageal epithelia. This promoter is known to interact with eukaryotic transcription factors in oro-esophageal squamous epithelial cells [22]. Three independent founders were generated and confirmed by Southern Blot analyses (Figure 1A) and by PCR (data not shown). To confirm expression of the transgene, RNA from the tongue was analyzed by Northern Blot analysis using the human cyclin D1 cDNA as a probe (Figure 1B) and by RT-PCR (data not shown). Based upon the robust cyclin D1expression, one of the lines was expanded and used for the experiments described below.
Fig. 1.
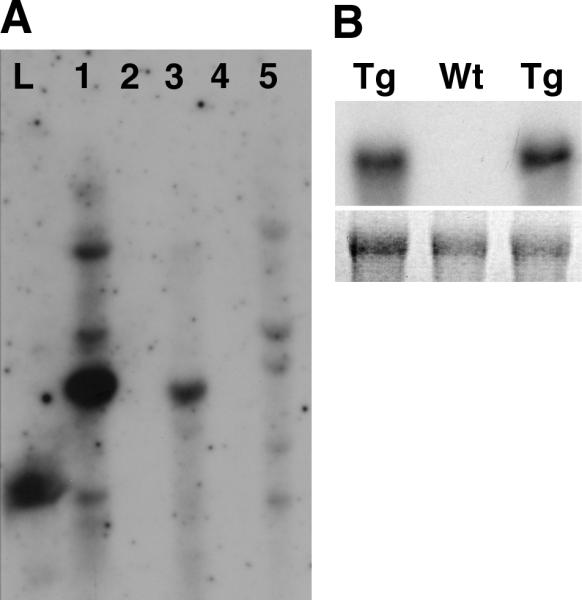
(A) Southern blot analysis of potential founder L2-D1 mice. Pst1-digested genomic DNA was transferred onto nitrocellulose membrane and hybridized to 32P-labelled human cyclin D1 probe. Founder mice are shown in lanes 1, 3 and 5. Lane L is DNA ladder. (B) Northern blot analysis of RNA from mouse tongues shows expression of cyclin D1 (top panel) in L2-D1 mice (Tg), but not in a non-transgenic littermate (Wt). The bottom panel shows the 18S band in the ethidium-bromide stained gel.
Cyclin D1 enhances susceptibility to 4NQO-induced oral dysplasia and neoplasia
We next examined whether cyclin D1 overexpression altered the susceptibility of the oral epithelium to carcinogenesis. We used the experimental carcinogen 4NQO to induce oro-esophageal preneoplastic and neoplastic lesions. Although it is not a natural tobacco-derived product, 4NQO induces several types of DNA damage, similar to those occurring as a result of tobacco exposure [23] and is a potent oral carcinogen in rodents [24]. In this study, we delivered the carcinogen via the drinking water. This route of delivery has been shown to reliably induce a wide spectrum of oro-esophageal dysplastic and neoplastic changes in mice [17]. Based upon previously established doses, we chose two different carcinogen doses—20 ppm and 50 ppm—delivered for a total of 8 weeks, following which the animals were monitored for an additional 16 weeks.
Overall, the carcinogenic regimens had no debilitating effects on the general health of the mice. At 8 weeks after the end of carcinogen treatment, 4NQO-treated L2-D1 mice started to developed gross morphological alterations on the tongue. The lesions ranged from discrete white keratotic patches to papillomatous/exophytic growths. At the end of the 16-week treatment, 66% of L2-D1 mice in the 20ppm group and 100% of L2-D1 mice in the 50ppm treated group had grossly visible lesions on the tongue and/or esophagus (Figure 2A). Notably, none of the 4NQO-treated wild-type controls or the non-carcinogen treated mice developed macroscopically visible lesions.
Fig. 2.

(A) Development of gross morphological lesions on the tongues of 4NQO-treated mice. Lesions include red/white patches and exophytic growths. (B) Degree of dysplasia induced by 4NQO in L2-D1 mice and non-transgenic wild-type littermates. Severity of 4NQO-induced dysplasia was significantly higher in L2-D1 mice, compared with wild-type littermates * p≤0.001, compared with wild-type mice treated with the same carcinogen dose. The numbers of mice are shown in Table I.
We next examined the tongues and esophagi for evidence of epithelial atypia, dysplasia and neoplasia. Treatment with 4NQO induced a range of dysplastic and neoplastic lesions in the tongues and esophagi of both the L2-D1 mice and their non-transgenic littermates (Figure 3). However, the degree of dysplastic change was significantly influenced by the cyclin D1 genotype and the carcinogenic dose (Figure 2B). Treatment with 20 ppm of 4NQO induced mild dysplasia in only 1 of the 6 wild-type mice (Table I). By contrast, at this dose, all the L2-D1 mice developed mild or moderate epithelial dysplasias in the tongue and esophagus. Treatment of L2-D1 mice with 50 ppm of 4NQO induced moderate to severe dysplasia. Notably, at this dose, 50% of the L2D1 mice developed SCC of the tongue and/or esophagus. By contrast, wild-type mice treated with 50 ppm of 4NQO showed lesser degrees of epithelial dysplastic change and none of the wild-type mice developed neoplastic changes (Table I). Consistent with the previously described phenotype of epithelial dysplasia induced by targeted cyclin D1 overexpression [12,14], examination of the untreated L2-D1 mice revealed atypical and in some cases mild dysplastic changes in the tongues and esophagi (Figure 3). Dysplastic/neoplastic changes were not observed in untreated non-transgenic littermate mice. In summary, at both doses examined, cyclin D1 overexpression in our transgenic model significantly increased the susceptibility to and severity of 4NQO-induced oro-esophageal dysplastic/neoplastic changes.
Fig. 3.

Hematoxylin and eosin stained sections of the tongue (A-E) and esophagus (F). (A) Wild-type mouse (B) Untreated L2D1 mouse (C) and (D) Severely dysplastic lesions in 4NQO-treated L2-D1 mice. (E) 4nQO-induced (papillary) SCC of the tongue. (F) SCC of the esophagus. (Scale bars: 200μm).
Table I.
Dysplastica and neoplastic lesions in the tongues and esophagi of L2-D1 and non-transgenic mice treated with 4NQO.
| Dose of 4NQO | Genotype | Number (n) | Numbers of mice with dysplasia and/or SCC (%) | |||
|---|---|---|---|---|---|---|
| Mild | Moderate | Severe | Neoplasia | |||
| 0 | Wild-type | 17 | 0 | 0 | 0 | 0 |
| L2-D1 | 17 | 10 (58%) | 0 | 0 | 0 | |
| 20 ppm | Wild-type | 6 | 1 (17%) | 0 | 0 | 0 |
| L2-D1 | 6 | 1 (17%) | 5 (83%) | 0 | 0 | |
| 50 ppm | Wild-type | 6 | 4 (67%) | 2 (33%) | 0 | 0 |
| L2-D1 | 6 | 0 | 2 (33%) | 4 (67%) | 3 (50%) | |
Dysplasia was graded as mild, moderate and severe as described in [20].
4NQO-induced dysplastic and neoplastic lesions are characterized by altered proliferation and differentiation
The stratified squamous epithelium of the oral cavity is composed predominantly of tightly packed keratinocytes with varying degrees of differentiation. The basal layer consists proliferating cells that differentiate as they move toward the surface. Precise temporal and spatial regulation of cell proliferation and differentiation is critical for the maintenance of epithelial homeostasis, which is deregulated in cancer [25]. Thus, we examined 4NQO-induced dysplastic lesions from L2-D1 mice for immunohistochemical evidence of deregulated epithelial cell proliferation and differentiation. To analyze changes in proliferation, we analyzed expression of Ki67, a well-defined marker of proliferation. Dysplastic lesions of the tongue showed increased expression of Ki67 (Figure 4). The labeling index, determined by the numbers of Ki67-positive cells per unit area was significantly higher in the dysplastic lesions compared with the non-dysplastic epithelium (75 ± 18 versus 20 ± 10, respectively; p≤0.001). We also examined the expression of keratin 5 (K5), which is normally expressed only in the basal cell stratum. In non-dysplastic tongue epithelium, expression of keratin 5 was limited to the basal cell layer. By contrast, 4NQO-induced dysplastic lesions showed strong expression of K5 through the basal and suprabasal layers (Figure 4). Thus, carcinogen-induced preneoplastic and neoplastic lesions were characterized by altered proliferation.
Fig 4.
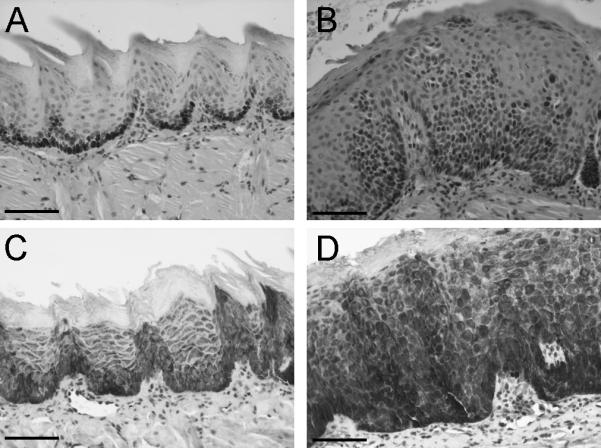
Immunohistochemical expression of Ki67 (A and B) and Keratin 5 (C and D) in the tongues of non-dysplastic (A and C) and dysplastic (B and D) regions of the tongue from L2-D1 mice. Note the increased numbers of Ki67-positive cells and the expanded zone of keratin 5-positive cells in dysplastic epithelium. (Scale bars: 100μm).
Molecular alterations in carcinogen-induced dysplastic and neoplastic lesions of L2D1 mice
We next examined the molecular alterations occurring in 4NQO-induced dysplastic and neoplasia lesions in the L2-D1 mice. The cyclin-dependent kinase inhibitor p16 is an important regulator of the G1 phase of the cell cycle—it binds to and inactivates the D-dependent cyclin dependent kinases CDK4 and CDK6. Inactivation of p16 by gene deletion, mutation or promoter hypermethylation is a frequent event in oral epithelial dysplasia and SCC [26,27]. Thus, we examined the expression of p16 in the 4NQO-induced dysplastic and neoplastic lesions in the L2-D1 mice. Immunohistochemical analysis showed that in wild-type mice, expression of p16 was restricted to the basal and parabasal layers of the tongue epithelium (Figure 5). Similarly, examination of the moderately to severely dysplastic lesions occurring in the L2-D1 mice revealed retention of p16 immunoreactivity—strong nuclear staining for p16 was observed in both the basal and parabasal layers. Interestingly, all three neoplastic lesions examined had retained expression of p16—strong nuclear p16-immunoreactivity was noted throughout the neoplastic lesion and in the adjacent dysplastic epithelium (Figure 5). Thus, loss of p16 expression did not seem to be necessary for carcinogen-induced of neoplastic transformation in our cyclin D1 animal model.
Fig 5.

Immunohistochemical expression of p16 in (A) normal, (B) dysplastic and (C) neoplastic regions of tongues from 4NQO-treated L2-D1 mice. Note that the expression of p16 is retained in the dysplastic and neoplastic lesions. Immunohistochemical expression of Cox2 in (D) normal, (E) dysplastic regions of the tongue and (F) SCC lesion from the esophagus. Immunohistochemical expression of E-cadherin (G—I) in a SCC lesion on the tongue. Panel H shows a magnified region of section indicated in (G) and demonstrates patchy loss of E-cadherin. Panel I shows that expression of E-cadherin in the islands of neoplastic epithelium.
Increased amounts of Cox2 are detected in both premalignant and malignant oral epithelial lesions. Although the role of Cox2 in this process is not understood fully, inhibition of Cox2 has gained considerable attention as a potential target for chemopreventive agents. Thus, we examined the expression pattern of Cox2 in the 4NQO-induced dysplastic and neoplastic lesions. In normal epithelium, expression of Cox 2 was sparse, but it was markedly increased in the dysplastic and neoplastic lesions, with moderate to strong cytoplasmic Cox 2 expression throughout the epithelium.
E-cadherin is a transmembranous glycoprotein that mediates cell-cell adhesion. It is a key molecule of the epithelial adherens junction. In vitro, E-cadherin suppresses the growth of carcinoma cells [28]. Further, loss of E-Cadherin mediated adhesion augments cell motility and is thought to mediate the transition from a preneoplastic to a neoplastic state [29]. Loss of E-cahderin expression is noted in several human malignancies, including oral SCC [30-32]. Thus, we examined the expression of E-cadherin in the 4NQO-induced SCCs and the adjacent dysplastic lesions. Adjacent normal tissue showed strong pericellular expression in the basal, parabasal and superficial layers (Figure 5). In 2 of the 3 SCCs examined, however, we detected heterogeneous expression of E-cadherin with areas of patchy loss or decrease in staining intensity in severely dysplastic areas immediately adjacent to the margins of the neoplastic lesion. However, the islands of neoplastic cells had strong pericellular expression of E-cadherin, clearly defining the intercellular margins (Figure 5), suggesting that the loss of E-cadherin expression was not permanent.
Discussion
Oral SCC is an aggressive neoplasm that causes significant morbidity and mortality. Environmental insults in the form of tobacco and alcohol use are the major risk factors for this disease. It is accepted generally that oral SCC develops by a multistep process with accumulation of mutations in key growth-regulating genes. Among these, the cyclin D1 oncogene is frequently amplified or overexpressed in oral and head and neck SCC [5,7,11,33]. Dysregulation of cyclin D1 occurs early in the preneoplastic process, underscoring the importance of this gene's contribution to the pathogenesis of oral cancer [10]. Even so, the contribution of cyclin D1 to modulating the risk of carcinogenesis is not well understood.
In this study, we show that cyclin D1 overexpression increases the susceptibility to chemical carcinogenesis in the oral and esophageal epithelia in mice. We used 4NQO, a well-established oral carcinogen in rodents [24,34]. When superimposed upon a background of increased cyclin D1 expression, the carcinogen induced marked preneoplastic and neoplastic changes. The severity of carcinogen-induced dysplasia was significantly higher in the cyclin D1 mice, compared with their wild-type littermates. The dysplastic lesions were characterized by increased proliferation as evidenced by increased expression of Ki67 and K5. Notably, our carcinogenic treatment induced malignant oro-esophageal neoplasms only in the cyclin D1 mice. Thus, our findings demonstrate that overexpression of cyclin D1 enhances the carcinogenic effects of 4NQO in the oro-esophageal epithelium.
Our studies are consistent with previous reports that demonstrate a role for cyclin D1 in modulating the susceptibility to carcinogens in other tissues. Previous studies using the L2-D1 transgenic model showed that cyclin D1 overexpression and N-nitrosomethylbenzylamine cooperate to accelerate dysplasia in the esophageal epithelium [35]. Likewise, targeted cyclin D1 overexpression in the skin enhanced the neoplastic effects of dimethylbenz[a]anthracene [36]. Taken together, our findings and previous studies strongly imply that cyclin D1 enhances carcinogenic effects. Our findings have important implications for human oral SCC: deregulation of cyclin D1 and other components in the cyclin D1 pathway is a frequent and early event in human oral SCC development [7,37]. Perhaps, precancerous lesions with cyclin D1 overexpression represent a subset that is at increased risk for malignant transformation.
The basis for cyclin D1's role in enhancing chemical carcinogenesis in our animal model is not known. One possibility is that the carcinogen induces additional genetic mutations, which in turn, co-operate with cyclin D1 to confer a selective growth advantage that results in clonal expansion of mutated preneoplastic or neoplastic cells. It is also possible that cyclin D1 may more directly modulate the cell's response to the DNA damaging effects of 4NQO—for example, cyclin D1 overexpression may permit the epithelial cells to override 4NQO damage-induced apoptotic signals and thus, favor accumulation of mutations necessary for development of dysplasia and neoplasia. Indeed, there is evidence for cyclin D1's involvement in modulating the cell's response to DNA damage. Cyclin D1 overexpression in a variety of cultured cells inhibits apoptosis [38-40] and confers resistance to cisplatin [41]. However, the pathways by which cyclin D1 modulates apoptosis in these various cell types is not understood fully. Notably, there is increasing evidence that a specific polymorphism of cyclin D1 (G/A870) influences the risk of oral SCC development [42,43]. This polymorphism results in an alternatively spliced protein product that lacks the threonine 286 phosphorylation site required for nuclear export and ubiqutin-mediated degradation [44,45]. It would be interesting to determine whether this polymorphic allele alters susceptibility to 4NQO-induced oral carcinogenesis. Indeed, transgenic mice that overexpress of a mutant form of cyclin D1 (D1T286A) that lacks nuclear export, develop mammary adenocarcinomas at an increased rate relative to mice that overexpress wild-type cyclin D1 [46]. Furthermore, inactivating mutations of Fbx4, a factor that directs cyclin D1 ubiquitination, are frequent in human cancers [47]. These Fbx4 result in cyclin D1 overexpression and underscore the importance cyclin D1 deregulation in carcinogenesis. There is mounting evidence that in addition to its role in CDK-mediated cell cycle regulation, cyclin D1 may have cell-cycle independent or CDK-independent functions [4,48]. Such activities include transcriptional regulation, cell differentiation and cellular migration [48] and it is certainly possible that these may contribute, in part, to cyclin D1's oncogenic effects.
Interestingly, immunohistochemical analyses of the carcinogen-induced lesions occurring in the cyclin D1 mice showed that p16 expression was retained in the dysplastic and neoplastic lesions. Given the increased expression of cyclin D1 in our animal model, perhaps loss of p16 expression may not offer any added growth advantage, and thus may not be required for dysplastic and neoplastic development in our animal model.
In summary, we have demonstrated the utility of a transgenic animal model to study the interactions between genetic alterations and susceptibility to chemical carcinogenesis. We have optimized the carcinogenic regimens to induce both preneoplastic and neoplastic lesions in a relatively short time frame. Molecular characterization of these lesions reveals similarities with alterations occurring in human oral SCC. Given the strong evidence for cyclin D1's role in oral neoplastic development, this model system provides a clinically relevant animal model to further study the process of oral carcinogenesis and in particular to dissect the biological role of the cyclin D1 oncogene in oral SCC. Furthermore, this model will also be of value to test potential chemopreventive agents or dietary factors that may modulate the oral neoplastic process.
Acknowledgements
The studies were funded by a grant from the National Institutes of Health DE0014773 (SM) and by institutional funds from the University of Connecticut School of Dental Medicine (SM). The work was supported also by the National Cancer Institute P01-CA098101 (HN, CM, AKR), National Institutes of Health R01-DK077005 (HN) and National Institutes of Health F32-DK07523 (CM).
Abbreviations
- 4NQO
4-nitroquinoline-1-oxide
References
- 1.Jemal A, Siegel R, Ward E, Murray T, Xu J, Thun MJ. Cancer Statistics, 2007. CA Cancer J Clin. 2007;57(1):43–66. doi: 10.3322/canjclin.57.1.43. [DOI] [PubMed] [Google Scholar]
- 2.Mallya SM, Arnold A. Cyclin D1 in parathyroid disease. Front Biosci. 2000;5:D367–371. doi: 10.2741/mallya. [DOI] [PubMed] [Google Scholar]
- 3.Baker GL, Landis MW, Hinds PW. Multiple functions of D-type cyclins can antagonize pRb-mediated suppression of proliferation. Cell cycle (Georgetown, Tex. 2005;4(2):330–338. [PubMed] [Google Scholar]
- 4.Ewen ME, Lamb J. The activities of cyclin D1 that drive tumorigenesis. Trends in molecular medicine. 2004;10(4):158–162. doi: 10.1016/j.molmed.2004.02.005. [DOI] [PubMed] [Google Scholar]
- 5.Akervall JA, Michalides RJ, Mineta H, et al. Amplification of cyclin D1 in squamous cell carcinoma of the head and neck and the prognostic value of chromosomal abnormalities and cyclin D1 overexpression. Cancer. 1997;79(2):380–389. [PubMed] [Google Scholar]
- 6.Bartkova J, Lukas J, Muller H, Strauss M, Gusterson B, Bartek J. Abnormal patterns of D-type cyclin expression and G1 regulation in human head and neck cancer. Cancer Res. 1995;55(4):949–956. [PubMed] [Google Scholar]
- 7.Izzo JG, Papadimitrakopoulou VA, Li XQ, et al. Dysregulated cyclin D1 expression early in head and neck tumorigenesis: in vivo evidence for an association with subsequent gene amplification. Oncogene. 1998;17(18):2313–2322. doi: 10.1038/sj.onc.1202153. [DOI] [PubMed] [Google Scholar]
- 8.Meredith SD, Levine PA, Burns JA, et al. Chromosome 11q13 amplification in head and neck squamous cell carcinoma. Association with poor prognosis. Arch Otolaryngol Head Neck Surg. 1995;121(7):790–794. doi: 10.1001/archotol.1995.01890070076016. [DOI] [PubMed] [Google Scholar]
- 9.Michalides R, van Veelen N, Hart A, Loftus B, Wientjens E, Balm A. Overexpression of cyclin D1 correlates with recurrence in a group of forty-seven operable squamous cell carcinomas of the head and neck. Cancer Res. 1995;55(5):975–978. [PubMed] [Google Scholar]
- 10.Rousseau A, Lim MS, Lin Z, Jordan RC. Frequent cyclin D1 gene amplification and protein overexpression in oral epithelial dysplasias. Oral Oncol. 2001;37(3):268–275. doi: 10.1016/s1368-8375(00)00114-7. [DOI] [PubMed] [Google Scholar]
- 11.Shintani S, Mihara M, Nakahara Y, et al. Expression of cell cycle control proteins in normal epithelium, premalignant and malignant lesions of oral cavity. Oral Oncol. 2002;38(3):235–243. doi: 10.1016/s1368-8375(01)00048-3. [DOI] [PubMed] [Google Scholar]
- 12.Nakagawa H, Wang TC, Zukerberg L, et al. The targeting of the cyclin D1 oncogene by an Epstein-Barr virus promoter in transgenic mice causes dysplasia in the tongue, esophagus and forestomach. Oncogene. 1997;14(10):1185–1190. doi: 10.1038/sj.onc.1200937. [DOI] [PubMed] [Google Scholar]
- 13.Mueller A, Odze R, Jenkins TD, et al. A transgenic mouse model with cyclin D1 overexpression results in cell cycle, epidermal growth factor receptor, and p53 abnormalities. Cancer Res. 1997;57(24):5542–5549. [PubMed] [Google Scholar]
- 14.Opitz OG, Harada H, Suliman Y, et al. A mouse model of human oral-esophageal cancer. J Clin Invest. 2002;110(6):761–769. doi: 10.1172/JCI15324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Hawkins BL, Heniford BW, Ackermann DM, Leonberger M, Martinez SA, Hendler FJ. 4NQO carcinogenesis: a mouse model of oral cavity squamous cell carcinoma. Head Neck. 1994;16(5):424–432. doi: 10.1002/hed.2880160506. [DOI] [PubMed] [Google Scholar]
- 16.Yuan B, Oechsli MN, Hendler FJ. A region within murine chromosome 7F4, syntenic to the human 11q13 amplicon, is frequently amplified in 4NQO-induced oral cavity tumors. Oncogene. 1997;15(10):1161–1170. doi: 10.1038/sj.onc.1201269. [DOI] [PubMed] [Google Scholar]
- 17.Tang XH, Knudsen B, Bemis D, Tickoo S, Gudas LJ. Oral cavity and esophageal carcinogenesis modeled in carcinogen-treated mice. Clin Cancer Res. 2004;10(1 Pt 1):301–313. doi: 10.1158/1078-0432.ccr-0999-3. [DOI] [PubMed] [Google Scholar]
- 18.Zhang Z, Wang Y, Yao R, Li J, Lubet RA, You M. p53 Transgenic mice are highly susceptible to 4-nitroquinoline-1-oxide-induced oral cancer. Mol Cancer Res. 2006;4(6):401–410. doi: 10.1158/1541-7786.MCR-06-0028. [DOI] [PubMed] [Google Scholar]
- 19.Winkle SA, Tinoco I., Jr. Interactions of 4-nitroquinoline 1-oxide with deoxyribodinucleotides. Biochemistry. 1979;18(18):3833–3839. doi: 10.1021/bi00585a001. [DOI] [PubMed] [Google Scholar]
- 20.Kramer IR, Lucas RB, Pindborg JJ, Sobin LH. Definition of leukoplakia and related lesions: an aid to studies on oral precancer. Oral Surg Oral Med Oral Pathol. 1978;46(4):518–539. [PubMed] [Google Scholar]
- 21.Dwivedi PP, Mallya S, Dongari-Bagtzoglou A. A novel immunocompetent murine model for Candida albicans-promoted oral epithelial dysplasia. Med Mycol. 2008:1–11. doi: 10.1080/13693780802165797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Nakagawa H, Inomoto T, Rustgi AK. A CACCC box-like cis-regulatory element of the Epstein-Barr virus ED-L2 promoter interacts with a novel transcriptional factor in tissue-specific squamous epithelia. J Biol Chem. 1997;272(26):16688–16699. doi: 10.1074/jbc.272.26.16688. [DOI] [PubMed] [Google Scholar]
- 23.Nagao M, Sugimura T. Molecular biology of the carcinogen, 4-nitroquinoline 1-oxide. Advances in cancer research. 1976;23:131–169. doi: 10.1016/s0065-230x(08)60545-x. [DOI] [PubMed] [Google Scholar]
- 24.Kanojia D, Vaidya MM. 4-nitroquinoline-1-oxide induced experimental oral carcinogenesis. Oral Oncol. 2006;42(7):655–667. doi: 10.1016/j.oraloncology.2005.10.013. [DOI] [PubMed] [Google Scholar]
- 25.Hanahan D, Weinberg RA. The hallmarks of cancer. Cell. 2000;100(1):57–70. doi: 10.1016/s0092-8674(00)81683-9. [DOI] [PubMed] [Google Scholar]
- 26.Papadimitrakopoulou V, Izzo J, Lippman SM, et al. Frequent inactivation of p16INK4a in oral premalignant lesions. Oncogene. 1997;14(15):1799–1803. doi: 10.1038/sj.onc.1201010. [DOI] [PubMed] [Google Scholar]
- 27.Reed AL, Califano J, Cairns P, et al. High frequency of p16 (CDKN2/MTS-1/INK4A) inactivation in head and neck squamous cell carcinoma. Cancer Res. 1996;56(16):3630–3633. [PubMed] [Google Scholar]
- 28.Frixen UH, Behrens J, Sachs M, et al. E-cadherin-mediated cell-cell adhesion prevents invasiveness of human carcinoma cells. The Journal of cell biology. 1991;113(1):173–185. doi: 10.1083/jcb.113.1.173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Alt-Holland A, Shamis Y, Riley KN, et al. E-Cadherin Suppression Directs Cytoskeletal Rearrangement and Intraepithelial Tumor Cell Migration in 3D Human Skin Equivalents. The Journal of investigative dermatology. 2008 doi: 10.1038/jid.2008.102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Downer CS, Speight PM. E-cadherin expression in normal, hyperplastic and malignant oral epithelium. Eur J Cancer B Oral Oncol. 1993;29B(4):303–305. doi: 10.1016/0964-1955(93)90053-h. [DOI] [PubMed] [Google Scholar]
- 31.Andrews NA, Jones AS, Helliwell TR, Kinsella AR. Expression of the E-cadherin-catenin cell adhesion complex in primary squamous cell carcinomas of the head and neck and their nodal metastases. Br J Cancer. 1997;75(10):1474–1480. doi: 10.1038/bjc.1997.252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Shinohara M, Hiraki A, Ikebe T, et al. Immunohistochemical study of desmosomes in oral squamous cell carcinoma: correlation with cytokeratin and E-cadherin staining, and with tumour behaviour. The Journal of pathology. 1998;184(4):369–381. doi: 10.1002/(SICI)1096-9896(199804)184:4<369::AID-PATH1236>3.0.CO;2-L. [DOI] [PubMed] [Google Scholar]
- 33.Todd R, Hinds PW, Munger K, et al. Cell cycle dysregulation in oral cancer. Crit Rev Oral Biol Med. 2002;13(1):51–61. doi: 10.1177/154411130201300106. [DOI] [PubMed] [Google Scholar]
- 34.Vered M, Yarom N, Dayan D. 4NQO oral carcinogenesis: animal models, molecular markers and future expectations. Oral Oncol. 2005;41(4):337–339. doi: 10.1016/j.oraloncology.2004.07.005. [DOI] [PubMed] [Google Scholar]
- 35.Jenkins TD, Mueller A, Odze R, et al. Cyclin D1 overexpression combined with N-nitrosomethylbenzylamine increases dysplasia and cellular proliferation in murine esophageal squamous epithelium. Oncogene. 1999;18(1):59–66. doi: 10.1038/sj.onc.1202296. [DOI] [PubMed] [Google Scholar]
- 36.Yamamoto H, Ochiya T, Takeshita F, et al. Enhanced skin carcinogenesis in cyclin D1-conditional transgenic mice: cyclin D1 alters keratinocyte response to calcium-induced terminal differentiation. Cancer Res. 2002;62(6):1641–1647. [PubMed] [Google Scholar]
- 37.Soni S, Kaur J, Kumar A, et al. Alterations of rb pathway components are frequent events in patients with oral epithelial dysplasia and predict clinical outcome in patients with squamous cell carcinoma. Oncology. 2005;68(4−6):314–325. doi: 10.1159/000086970. [DOI] [PubMed] [Google Scholar]
- 38.Albanese C, D'Amico M, Reutens AT, et al. Activation of the cyclin D1 gene by the E1A-associated protein p300 through AP-1 inhibits cellular apoptosis. J Biol Chem. 1999;274(48):34186–34195. doi: 10.1074/jbc.274.48.34186. [DOI] [PubMed] [Google Scholar]
- 39.Biliran H, Jr., Wang Y, Banerjee S, et al. Overexpression of cyclin D1 promotes tumor cell growth and confers resistance to cisplatin-mediated apoptosis in an elastase-myc transgene-expressing pancreatic tumor cell line. Clin Cancer Res. 2005;11(16):6075–6086. doi: 10.1158/1078-0432.CCR-04-2419. [DOI] [PubMed] [Google Scholar]
- 40.Biliran H, Jr., Banerjee S, Thakur A, et al. c-Myc-induced chemosensitization is mediated by suppression of cyclin D1 expression and nuclear factor-kappa B activity in pancreatic cancer cells. Clin Cancer Res. 2007;13(9):2811–2821. doi: 10.1158/1078-0432.CCR-06-1844. [DOI] [PubMed] [Google Scholar]
- 41.Nakashima T, Kuratomi Y, Yasumatsu R, et al. The effect of cyclin D1 overexpression in human head and neck cancer cells. Eur Arch Otorhinolaryngol. 2005;262(5):379–383. doi: 10.1007/s00405-004-0831-z. [DOI] [PubMed] [Google Scholar]
- 42.Buch S, Zhu B, Davis AG, et al. Association of polymorphisms in the cyclin D1 and XPD genes and susceptibility to cancers of the upper aero-digestive tract. Mol Carcinog. 2005;42(4):222–228. doi: 10.1002/mc.20086. [DOI] [PubMed] [Google Scholar]
- 43.Huang M, Spitz MR, Gu J, et al. Cyclin D1 gene polymorphism as a risk factor for oral premalignant lesions. Carcinogenesis. 2006;27(10):2034–2037. doi: 10.1093/carcin/bgl048. [DOI] [PubMed] [Google Scholar]
- 44.Lu F, Gladden AB, Diehl JA. An alternatively spliced cyclin D1 isoform, cyclin D1b, is a nuclear oncogene. Cancer Res. 2003;63(21):7056–7061. [PubMed] [Google Scholar]
- 45.Solomon DA, Wang Y, Fox SR, et al. Cyclin D1 splice variants. Differential effects on localization, RB phosphorylation, and cellular transformation. J Biol Chem. 2003;278(32):30339–30347. doi: 10.1074/jbc.M303969200. [DOI] [PubMed] [Google Scholar]
- 46.Lin DI, Lessie MD, Gladden AB, Bassing CH, Wagner KU, Diehl JA. Disruption of cyclin D1 nuclear export and proteolysis accelerates mammary carcinogenesis. Oncogene. 2008;27(9):1231–1242. doi: 10.1038/sj.onc.1210738. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Barbash O, Zamfirova P, Lin DI, et al. Mutations in Fbx4 inhibit dimerization of the SCF(Fbx4) ligase and contribute to cyclin D1 overexpression in human cancer. Cancer Cell. 2008;14(1):68–78. doi: 10.1016/j.ccr.2008.05.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Fu M, Wang C, Li Z, Sakamaki T, Pestell RG. Minireview: Cyclin D1: normal and abnormal functions. Endocrinology. 2004;145(12):5439–5447. doi: 10.1210/en.2004-0959. [DOI] [PubMed] [Google Scholar]