

Abstract
Human cytomegalovirus infection of monocytes stimulates a unique monocyte differentiation reprogramming resulting in polarization towards an M1 pro-inflammatory macrophage that simultaneously exhibits characteristics of an M2 anti-inflammatory macrophage. Our laboratory has previously shown that HCMV infection stimulates monocyte NF-κB and PI(3)K activities and now provides evidence that these cellular factors are essential for the HCMV-induced polarization of infected monocytes/macrophages. We find that the induction of NF-κB and PI(3)K activities following HCMV infection was required for the initiation of monocyte-to-macrophage differentiation. HCMV-infected monocytes treated with Bay11-7802 (an inhibitor of NF-κB activity) or LY294002 [an inhibitor of PI(3)K activity] prior to infection exhibited a small, round and monocyte-like undifferentiated morphology and the lack of CD68 upregulation (a macrophage differentiation marker). Detailed transcriptome analysis revealed 48%, 7% and 31% of HCMV-induced M1-associated genes were dependent on NF-κB, PI(3)K or both activities, respectively; while 100% of HCMV-induced M2-associated genes required both NF-κB and PI(3)K activities. Functionally, we demonstrated that NF-κB and PI(3)K activities were critical for the production of M1-and M2-associated cytokines/chemokines, in HCMV-induced differentiating monocytes. Supernatant from HCMV-infected monocytes pretreated with Bay11-7802 or LY294002 exhibited an 80% and 67% reduction in cell motility inducing activity. Overall, these data show that HCMV usurps monocyte NF-κB and PI(3)K signal transduction pathways to induce the unique polarization of HCMV-infected monocytes needed for the earliest steps in the viral dissemination and persistence strategy.
Keywords: Human Cytomegalovirus, Monocyte, Macrophage, Differentiation, NF-κB, Phosphatidylinositol 3-Kinase
Summary
Human cytomegalovirus is a ubiquitous β-herpesvirus that infects 50%–90% of the adult population (Mocarski et al., 2007). Infection of immunocompetent hosts by HCMV is generally asymptomatic or results in mild symptoms, although HCMV infection can cause mononucleosis (Mocarski et al., 2007) and is associated with chronic inflammatory diseases such as arthrosclerosis (Mueller et al., 2003; Westphal et al., 2006). In contrast, HCMV infection can have severe clinical consequences in immunosuppressed individuals including neonates, AIDS and organ transplant patients (Ho, 1977; Masur et al., 1996; Stagno et al., 1982) where, due to HCMV’s broad cellular tropism, multiple organ system disease can develop (Toorkey and Carrigan, 1989).
Systemic spread occurs during both symptomatic and asymptomatic infections (Toorkey and Carrigan, 1989) and is preceded by a cell-associated viremia suggesting the involvement of a cellular component in viral dissemination (Manez et al., 1996; Taylor-Wiedeman et al., 1994). Cells of the myeloid lineage are believed to be responsible for this systemic viral spread because monocytes from patients exhibiting viremia contain viral DNA. In addition, HCMV-infected macrophages (differentiated monocytes) are detected in the organs of these infected individuals (Sinclair and Sissons, 1996). We recently provided evidence that HCMV does not passively “hitchhike” on monocytes as they migrate into organ tissue, but rather plays an active role in the modulation of monocyte functions in order to stimulate the changes in the infected cell that favour hematogenous dissemination of the virus (Smith et al., 2004a). HCMV infection of monocytes induces a pro-inflammatory state characterized by the increased secretion of cytokines/chemokines, the acquisition of cellular motility, endothelial cell adhesion, and transendothelial migration, which provide the infected monocytes with the necessary biological tools to stimulate exit from the blood into organ tissue (Smith et al., 2004a). While functional changes in monocytes are observed following infection with other pathogenic agents, we found distinct functional changes in HCMV-infected monocytes (Chan et al., 2008a; Smith et al., 2004a). For example, in contrast to the pro-inflammatory phenotype acquired following LPS treatment (Smith et al., 2004a) or HIV infection (Tuttle et al., 1998), HCMV infection is, to date, the only pathogen that is able to rapidly stimulate monocyte-to-macrophage differentiation, a process essential for productive viral replication in these cells (Smith et al., 2004a). Transcriptionally, these HCMV-differentiated monocytes exhibited a distinct M1 (pro-inflammatory)/M2 (anti-inflammatory) reprogramming (Chan et al., 2008a). Functionally, infected monocytes produced both M1 and M2 cytokines/chemokines, which may promote recruitment of naïve monocytes to sites of infection to increase the pool of infected cells and/or to stimulate exit of the infected monocyte from the circulation into peripheral tissue (Chan et al., 2008a). This unusual M1/M2 reprogramming of infected monocytes is a direct consequence of HCMV’s ability to rapidly induce the mass activation of multiple cellular signalling pathways during viral entry (Chan et al., 2008b; Smith et al., 2007; Yurochko and Huang, 1999). However, the specific cellular signalling pathways utilized by HCMV to stimulate the distinct polarization of infected monocytes remain unresolved.
Because HCMV gene expression is not observed until 3–4 weeks post infection (Smith et al., 2004a), the involvement of de novo viral proteins in the initiation of monocyte polarization and differentiation is unlikely. Treatment of monocytes with UV-inactivated HCMV and purified viral glycoprotein gB induced similar rapid functional changes to that observed with live virus, suggesting that the regulation of cellular signalling pathways during viral entry triggers the polarization of HCMV-infected monocytes (Smith et al., 2004a; Yurochko and Huang, 1999). We have shown that the rapid activation of NF-κB and PI(3)K following HCMV infection led to increased monocyte cytokine production, motility, endothelial cell adhesion and transendothelial migration (Chan et al., 2008b; Smith et al., 2004b; Smith et al., 2007; Yurochko, 2008); however, the involvement of these factors in mediating monocyte-to-macrophage differentiation were not explored. To begin to address this issue, we examined the effects of inhibition of NF-κB and PI(3)K activities on the HCMV-induced differentiation of monocytes. Monocytes were isolated (Smith et al., 2004a) and plated onto fibronectin-coated coverslips for 1 hr at 37°C, followed by treatment with 5 μM of Bay11-7082 [a NF-κB inhibitor (Chan et al., 2008b)] or 25 μM LY294002 [a PI(3)K inhibitor (Chan et al., 2008b)] for an additional hour at 37°C prior to infection. No cytotoxic effects were observed at these concentrations of inhibitors for 48 hours post infection (hpi) (data not shown). Monocytes were then infected with gradient purified HCMV (the Towne/E strain; MOI 5) in the presence of inhibitors and examined for morphological changes associated with differentiation at 48 hpi. Consistent with previous studies (Smith et al., 2004a), we found that HCMV-infected monocytes exhibited accelerated rates of differentiation as demonstrated by the increase in granularity, cell spreading and size (Fig. 1A). We now show that NF-κB and PI(3)K activities are required for these early steps in HCMV-induced differentiation of monocytes, as pre-treatment of monocytes with either Bay11-7802 or LY294002 abrogated the morphological changes associated with differentiation. Quantitative analysis of cell size revealed a 4-fold increase in cell area following infection with HCMV at 48 hpi, which was inhibited in the absence of NF-κB or PI(3)K activity (Fig. 1B). In accord, Western blot analysis demonstrated the inhibition of HCMV-induced CD68 [a macrophage differentiation marker (Smith et al., 2004a)] protein expression in Bay11-7802 or LY294002 treated infected monocytes (Fig. 1C). Treatment of HCMV with neutralizing antibody prior to infection blocked viral-induced differentiation of infected monocytes, indicating that the pathogenic monocyte differentiation was not a bystander effect or due solely to the release of soluble mediators (data not shown). Consequently, the new data indicate the direct involvement of viral-induced NF-κB and PI(3)K activities in monocyte differentiation.
Figure 1.

HCMV-induces monocyte-to-macrophage differentiation in a NF-κB- and PI(3)K-dependent manner. Purified peripheral blood monocytes were treated with DMSO, Bay11-7082 (5 μM) or LY294002 (25 μM) for 1 hr. Cells were then mock infected or HCMV infected at a MOI of 5. (A) Photomicrographs were then taken at 24 hpi. (B) Utilizing these photomicrographs at 24 hpi, monocyte surface area was determined in square arbitrary units with Scion Image software. Results are plotted as means ± standard errors of the mean (SEM) for 75 cells isolated from 3 independent blood donors. (C) At 24 hpi, total cellular protein was harvested from monocytes and immunoblotting analysis performed with a CD68 specific antibody. Membranes were reprobed with an antibody against β-actin to verify equal loading. (A) Photomicrographs and (C) Western blot analysis are representative of 3 independent experiments from different donors.
Separate global transcriptome studies from our laboratory revealed that HCMV-infected monocytes polarize towards a M1 macrophage, while simultaneously exhibiting certain M2 macrophage characteristics (Chan et al., 2008a), and that NF-κB and PI(3)K are critical to the upregulation of several pro-inflammatory genes following infection with HCMV (Chan et al., 2008b). To address if NF-κB and PI(3)K biological activities are responsible for the HCMV-induced distinct M1/M2 reprogramming of monocytes, we combined our previous bioinformatic data (GEO accession number GSE11408 and GSE 9601) and re-examined the transcriptome of HCMV-infected monocytes for changes in the M1/M2 polarization that were dependent on NF-κB and/or PI(3)K activities. Microarray analysis was performed as previously described with the following modifications (Chan et al., 2008a; Chan et al., 2008b): gradient purified monocytes were pretreated with 5 μM of Bay11-7082, 50 μM of LY294002 or DMSO solvent control for 45 minutes before infection with HCMV. Monocytes were diluted in large volumes of medium to prevent homotypic aggregation of monocytes during the 4 hour infection period; thus, a MOI of 15 was used to ensure that all monocytes would be infected during this short incubation time. We have shown similar HCMV-induced phenotypic changes in monocytes using MOIs of 0.1–20 (Chan et al., 2008a; Chan et al., 2008b). Following the infection period, total RNA was harvested from mock-infected, HCMV-infected, Bay11-7082-pretreated HCMV-infected and LY294002-pretreated HCMV-infected monocytes with the RNA STAT-60 isolation kit (Tel-Test, Friendswood, TX). Affymetrix Human Genome U95Av2 arrays (Affymetrix, Santa Clara, CA), containing 12,626 characterized sequences (relating to 11,266 genes) were used, to examine the transcriptome modulation in primary human monocytes isolated from multiple human donors, and the obtained data analyzed using Affymetrix Microarray Suite version 5.0. We previously demonstrated the validity and reproducibility of hybridization signals from these 6 independent preparations of isolated monocyte RNAs by scatter plot analysis (Chan et al., 2008a; Chan et al., 2008b). Data Mining Tool version 3.0 was used to compile data from each of the replicates and one-way analysis of variance (ANOVA) tests performed to calculate p-values. Spotfire DecisionSite 8.1.1 (Somerville, MA) was used to group genes by ontology.
For compilation of the data from multiple replicates, the following criteria were applied. As determined by a predefined Affymetrix threshold algorithm to determine present, marginal or absent call, genes that had an absent call in more than two of six donors from HCMV-infected replicates were removed from the pool of genes. ANOVA tests were performed on the HCMV-infected versus mock-infected replicates, and a p-value of ≤0.05 was utilized for statistically significant genes among replicates (Chan et al., 2008a; Chan et al., 2008b). Moreover, genes with an average change of ≥1.5-fold in at least four of six HCMV-infected versus mock-infected samples were considered to be regulated by infection; thus, genes that may have been eliminated due to anomalous expression by a single donor were accepted. The HCMV-regulated cellular genes dependent upon NF-κB or PI(3)K activity were determined by identifying genes in HCMV-infected replicates that were downregulated ≥1.5-fold in Bay11-7082-pretreated or in LY294002-pretreated HCMV-infected samples when compared to untreated infected samples, respectively. A present call in the HCMV-infected replicates of at least 2 of 3 donors was required for a classification of gene expression dependence on NF-κB and/or PI(3)K activity. These criteria allowed us to identify those HCMV induced genes associated with M1 or M2 monocyte/macrophage polarization that require NF-κB and/or PI(3)K activity for their regulation.
In this current bioinformatic reanalysis, we show new data demonstrating precisely which HCMV-induced M1 and M2 genes specifically require NF-κB and/or PI(3)K activities for their induction following infection. We found that 48% of HCMV-induced M1 genes strictly required NF-κB activity, whereas only 7% strictly required PI(3)K activity (Table 1 and Fig. 2A,B). Moreover, both NF-κB and PI(3)K activity were involved in the upregulation of 31% of M1-associated genes following infection with HCMV. Thus, a total 79% of the HCMV-induced M1 genes were regulated in a NF-κB dependent manner versus 38% of the genes being regulated by PI(3)K activity, indicating that NF-κB activity plays a dominant role in biasing the HCMV-infected monocyte towards a M1 polarization state. It should be noted that other cellular signalling pathways must be involved during the early stages of infection as 14% of genes are regulated by neither NF-κB and PI(3)K, such as pentaxin-related gene (PTX3) and pre-B-cell colony enhancing factor 1 (PBEF1). In contrast, NF-κB and PI(3)K activities were required for the induction of all of HCMV-induced M2-associated genes, suggesting that the unique M2 characteristics acquired during the M1-polarization of HCMV-infected monocyte require the activation of both signalling pathways.
Table 1.
Expression of HCMV-induced M1- and M2-associated mRNAs that decreased 1.5-fold with Bay11-7082 or LY294002 treatmenta
| Category and Full Gene Name | Gene Title | Probe Set | HCMV-Induced Fold Change | Fold-Change Induced by Treatment with: |
|
|---|---|---|---|---|---|
| Bay11-7082b | LY294002c | ||||
| M1-Associated Genes | |||||
| Membrane Receptors | |||||
| Interleukin 15 receptor, alpha | IL15RA | 41677_at | 15.3 | −4.3 | n/c |
| Chemokine (C-C motif) receptor 7 | CCR7 | 1097_s_at | 3.0 | −3.5 | n/c |
| interleukin 2 receptor, gamma | IL2RG | 1506_at | 1.9 | −2.3 | −2.1 |
| Interleukin 7 receptor | IL7R | 1370_at | 1.7 | −2.3 | n/c |
| Cytokine and Chemokines | |||||
| Interleukin 6 (interferon, beta 2) | IL6 | 38299_at | 280.8 | −13.6 | −1.7 |
| Chemokine (C-X-C motif) ligand 10 | CXCL10 | 431_at | 69.3 | −162.0 | n/c |
| Interleukin 12B | IL12B | 563_at | 33.7 | −13.5 | −17.4 |
| Chemokine (C-C motif) ligand 20 | CCL20 | 40385_at | 16.5 | −11.6 | n/c |
| Chemokine (C-X-C motif) ligand 11 | CXCL11 | 35061_at | 15.2 | −15.1 | n/c |
| Tumor necrosis factor (TNF superfamily, member 2) | TNF | 1852_at | 14.6 | −4.1 | −2.2 |
| Tumor necrosis factor (ligand) superfamily, member 10 | TNFSF10 | 1715_at | 5.8 | −5.6 | n/c |
| Interleukin 15 | IL15 | 1036_at | 4.0 | −3.4 | n/c |
| Pre-B-cell colony enhancing factor 1 | PBEF1 | 33849_at | 1.9 | n/c | n/c |
| Chemokine (C-C motif) ligand 5 | CCL5 | 1403_s_at | 1.6 | −1.9 | n/c |
| Apoptosis-related Genes | |||||
| BCL2-related protein A1 | BCL2A1 | 2002_s_at | 4.9 | −1.6 | n/c |
| XIAP associated factor-1 | HSXIAPAF1 | 35583_at | 3.4 | −20.3 | −8.5 |
| Tumor necrosis factor receptor superfamily, member 6 | TNFRSF6 | 1440_s_at | 3.1 | −3.0 | n/c |
| Baculoviral IAP repeat-containing 3 | BIRC3 | 1717_s_at | 2.5 | −4.3 | n/c |
| Growth arrest and DNA-damage-inducible, alpha | GADD45A | 1911_s_at | 2.1 | n/c | n/c |
| Solute Carriers | |||||
| Solute carrier family 7, member 5 | SLC7A5 | 32186_at | 1.6 | n/c | n/c |
| Enzymes | |||||
| Adenylate kinase 3 | AK3 | 32331_at | 9.3 | −5.6 | −4.9 |
| Indoleamine-pyrrole 2,3 dioxygenase | INDO | 36804_at | 4.1 | −5.7 | −4.3 |
| 6-phosphofructo-2-kinase/fructose-2,6-biphosphatase 3 | PFKFB3 | 37111_g_at | 3.6 | −2.7 | n/c |
| 2′-5′-oligoadenylate synthetase 2, 69/71kDa | OAS2 | 39263_at | 3.4 | −3.8 | −3.6 |
| Proteasome (prosome, macropain) activator subunit 2 | PSME2 | 1184_at | 1.8 | n/c | −1.5 |
| Proteasome (prosome, macropain) subunit, beta type, 9 | PSMB9 | 38287_at | 1.7 | −2.1 | n/c |
| Extracellular Matrix | |||||
| Pentaxin-related gene, rapidly induced by IL-1 beta | PTX3 | 1491_at | 13.5 | n/c | n/c |
| Inhibin, beta A | INHBA | 40357_at | 9.9 | n/c | −6.8 |
| DNA-binding Factors | |||||
| Interferon regulatory factor 1 | IRF1 | 669_s_at | 3.3 | −1.9 | n/c |
| Interferon regulatory factor 7 | IRF7 | 36412_s_at | 2.1 | −1.9 | n/c |
| M2-Associated Genes | |||||
| Cytokine and Chemokines | |||||
| Chemokine (C-C motif) ligand 18 | CCL18 | 32128_at | 39.4 | −12.8 | −3.6 |
| Interleukin 1 receptor antagonist | IL1RN | 37603_at | 5.7 | −3.6 | −4.6 |
| Interleukin 10 | IL10 | 1548_s_at | 4.6 | −9.2 | −5.4 |
| Chemokine (C-C motif) ligand 23 | CCL23 | 36444_s_at | 3.5 | −8.4 | −3.2 |
Figure 2.
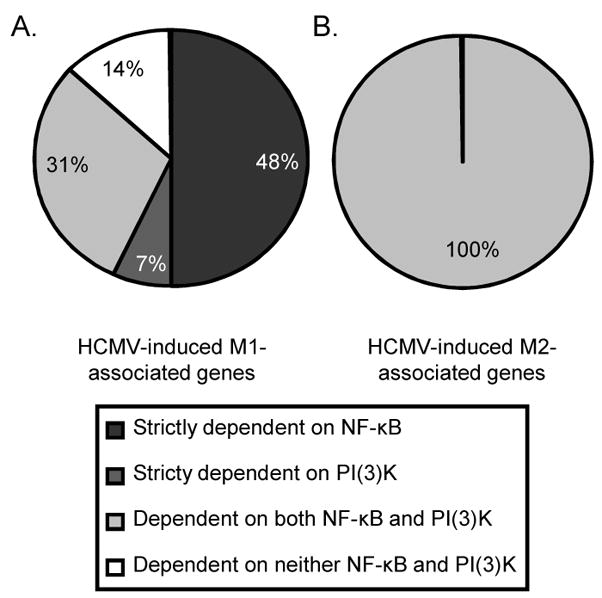
HCMV regulates M1/M2 reprogramming following infection in a NF-κB- and PI(3)K-dependent manner. Purified peripheral blood monocytes were treated with DMSO, Bay11-7082 (5 μM) or LY294002 (25 μM) for 1 hr. Pre-treated monocytes were then mock infected or HCMV infected (MOI 15) for 4 hrs and total RNA harvested for Affymetrix gene array analysis. The pie charts represent percentages of statistically significant (p≤0.05) (A) M1-associated or (B) M2-associated genes that were induced ≥1.5-fold following infection with HCMV and that were downregulated by NF-κB (≥1.5-fold), PI(3)K (≥1.5-fold), both or neither.
We have previously shown a unique HCMV-induced monocyte secretome, in which the release of both M1 and M2 cytokines/chemokines stimulated the motility of naïve monocytes (Chan et al., 2008a). Ontology analyses indicated that 100% of HCMV-induced M1-cytokines/chemokines were regulated by NF-κB, of which 33% were also regulated by PI(3)K activity. In contrast, both NF-κB and PI(3)K activities were required for the induction of 100% of the HCMV-induced M2-cytokines/chemokines (Table 1 and Fig. 3A). To determine if inhibition of NF-κB or PI(3)K activity blocked the secretion of these motility-inducing factors, phagokinetic track motility assays were performed using the following experimental design. Monocytes were pretreated with Bay11-7082 or LY294002 nonadherently for 45 minutes at 37°C and then either mock infected or HCMV infected. After the 45 min incubation with the virus, cells were washed thoroughly to remove unbound virus and drug, and the cells were incubated nonadherently for 6 hours at 37°C after which supernatants were harvested by centrifugation from each experimental variable. Monocytes isolated from the same donor, which were added to colloidal gold-coated coverslips, were then incubated with supernatants from mock-infected; LY294002-pretreated, mock-infected; Bay11-7082-pretreated, mock infected; HCMV-infected; LY294002-pretreated, HCMV-infected; and Bay11-7082-pretreated, HCMV-infected cells. Cells were incubated for 6 hours, and the tracks cleared by monocytes measured to determine the effects of secreted soluble factors on naïve monocyte motility. Treatment of uninfected monocytes with supernatants from HCMV-infected cells increased motility of naïve monocytes ~5.5-fold when compared to uninfected cells treated with mock-infected cell supernatants (Fig. 3B). Pretreatment of cells with Bay11-7082 or LY294002 prior to HCMV infection blocked 80% and 67% of the monocyte motility inducing activity from the virus-free supernatant, respectively. To exclude the possibility that the presence of contaminating Bay11-7082 or LY294002 in the supernatant was responsible for the reduction in motility, naïve monocytes treated with the supernatant from HCMV-infected monocytes incubated with either drug were also directly infected with HCMV and shown to exhibit increased motility (data not shown). Overall, the viral induction of monocyte NF-κB and PI(3)K activity is not only critical for distinct M1/M2 polarization during differentiation, but also for the control of the secretion of cell motility inducing factors. We argue that the secretion of these cytokines/chemokines by infected cells promotes recruitment of naïve monocytes to sites of infection to increase the total pool of potentially infected monocytes, thus enhancing the necessary early steps in the viral dissemination and persistence strategy.
Figure 3.

NF-κB and PI(3)K activities are responsible for the induction and secretion of M1- and M2-associated cytokines/chemokines. (A) Microarray analysis of M1-associated and M2-associated cytokines/chemokines that were regulated by NF-κB, PI(3)K or both. Cytokine/chemokine functional ontology was generated using Spotfire DecisionSite software based on the Gene Ontology Consortium database. Examined M1 and M2 genes are based on the transcriptional profiling work of Martinez et al (Martinez et al., 2006). The bar graph represents the percentages of genes regulated by the NF-κB pathway (dark grey), the PI(3)K pathway (white), and both pathways (light grey). (B) Monocytes were treated with DMSO, Bay11-7082 (5 μM) or LY294002 (50μM) for 45 min. Cells were then mock-infected or HCMV-infected (MOI 15) for 45 min and washed extensively to remove drugs and unbound virus. The infected cells were then incubated nonadherently for 6 hours and supernatants harvested from each of the samples following centrifugation. Supernatents were incubated with naïve monocytes for 6 hours and phagokenetic track motility assay performed (Chan et al., 2008a). The average area (arbitrary units2) of colloidal gold cleared by monocytes was determined for each experimental arm and plotted as a mean ± SEM. The results represent three independent experiments from separate human blood donors.
In summary, we show that the induction of NF-κB and PI(3)K activities in monocytes following infection with HCMV was required for early steps in the unique HCMV-induced monocyte-to-macrophage differentiation process. Specifically, NF-κB activity was critical to the biasing of the development of infected monocyte towards the M1 pro-inflammatory macrophage, while both NF-κB and PI(3)K activities dictated the acquisition of the M2 biological traits distinctly associated with the activation of monocytes following HCMV infection. This study, together with our previous results (Chan et al., 2008a; Chan et al., 2008b; Smith et al., 2004b; Smith et al., 2007; Yurochko and Huang, 1999), support our hypothesis that HCMV has evolved a strategy to navigate and manipulate specific pathways within the complex cellular signalling network of monocytes to stimulate precise functional changes in target cells that favour viral spread and persistence following primary infection. By deciphering how HCMV manipulates host signal transduction pathways, we gain insight into the molecular mechanism of HCMV pathogenesis (Ross, 1999; Watanabe et al., 1996) and provide potentially novel targets for therapeutic intervention.
Acknowledgments
This work was supported by a Malcolm Feist Cardiovascular Research Fellowship and the National Institutes of Health (AI56077 and 1-P20-RR018724).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Chan G, Bivins-Smith ER, Smith MS, Smith PM, Yurochko AD. Transcriptome Analysis Reveals Human Cytomegalovirus Reprograms Monocyte Differentiation toward an M1 Macrophage. J Immunol. 2008a;181(1):698–711. doi: 10.4049/jimmunol.181.1.698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chan G, Bivins-Smith ER, Smith MS, Yurochko AD. Transcriptome analysis of NF-κB- and phosphatidylinositol 3-kinase-regulated genes in human cytomegalovirus-infected monocytes. J Virol. 2008b;82(2):1040–6. doi: 10.1128/JVI.00864-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ho M. Virus infections after transplantation in man. Brief review Arch Virol. 1977;55(1–2):1–24. doi: 10.1007/BF01314475. [DOI] [PubMed] [Google Scholar]
- Manez R, Kusne S, Rinaldo C, Aguado JM, St George K, Grossi P, Frye B, Fung JJ, Ehrlich GD. Time to detection of cytomegalovirus (CMV) DNA in blood leukocytes is a predictor for the development of CMV disease in CMV-seronegative recipients of allografts from CMV-seropositive donors following liver transplantation. J Infect Dis. 1996;173(5):1072–6. doi: 10.1093/infdis/173.5.1072. [DOI] [PubMed] [Google Scholar]
- Martinez FO, Gordon S, Locati M, Mantovani A. Transcriptional profiling of the human monocyte-to-macrophage differentiation and polarization: new molecules and patterns of gene expression. J Immunol. 2006;177(10):7303–11. doi: 10.4049/jimmunol.177.10.7303. [DOI] [PubMed] [Google Scholar]
- Masur H, Whitcup SM, Cartwright C, Polis M, Nussenblatt R. Advances in the management of AIDS-related cytomegalovirus retinitis. Ann Intern Med. 1996;125(2):126–36. doi: 10.7326/0003-4819-125-2-199607150-00009. [DOI] [PubMed] [Google Scholar]
- Mocarski ES, Shenk T, Pass RF. Cytomegalovirus. In: Knipe DM, Howley PM, editors. Fields Virology. Vol. 2. Lippincott, Williams and Wilkins; Philadelphia: 2007. pp. 2701–2772. [Google Scholar]
- Mueller C, Hodgson JM, Bestehorn HP, Brutsche M, Perruchoud AP, Marsch S, Roskamm H, Buettner HJ. Previous cytomegalovirus infection and restenosis after aggressive angioplasty with provisional stenting. J Interv Cardiol. 2003;16(4):307–13. doi: 10.1034/j.1600-6143.2003.08060.x. [DOI] [PubMed] [Google Scholar]
- Ross R. Atherosclerosis--an inflammatory disease. N Engl J Med. 1999;340(2):115–26. doi: 10.1056/NEJM199901143400207. [DOI] [PubMed] [Google Scholar]
- Sinclair J, Sissons P. Latent and persistent infections of monocytes and macrophages. Intervirology. 1996;39(5–6):293–301. doi: 10.1159/000150501. [DOI] [PubMed] [Google Scholar]
- Smith MS, Bentz GL, Alexander JS, Yurochko AD. Human cytomegalovirus induces monocyte differentiation and migration as a strategy for dissemination and persistence. J Virol. 2004a;78(9):4444–53. doi: 10.1128/JVI.78.9.4444-4453.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith MS, Bentz GL, Smith PM, Bivins ER, Yurochko AD. HCMV activates PI(3)K in monocytes and promotes monocyte motility and transendothelial migration in a PI(3)K-dependent manner. J Leukoc Biol. 2004b;76(1):65–76. doi: 10.1189/jlb.1203621. [DOI] [PubMed] [Google Scholar]
- Smith MS, Bivins-Smith ER, Tilley AM, Bentz GL, Chan G, Minard J, Yurochko AD. Roles of phosphatidylinositol 3-kinase and NF-κB in human cytomegalovirus-mediated monocyte diapedesis and adhesion: strategy for viral persistence. J Virol. 2007;81(14):7683–94. doi: 10.1128/JVI.02839-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stagno S, Pass RF, Dworsky ME, Henderson RE, Moore EG, Walton PD, Alford CA. Congenital cytomegalovirus infection: The relative importance of primary and recurrent maternal infection. N Engl J Med. 1982;306(16):945–9. doi: 10.1056/NEJM198204223061601. [DOI] [PubMed] [Google Scholar]
- Taylor-Wiedeman J, Sissons P, Sinclair J. Induction of endogenous human cytomegalovirus gene expression after differentiation of monocytes from healthy carriers. J Virol. 1994;68(3):1597–604. doi: 10.1128/jvi.68.3.1597-1604.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Toorkey CB, Carrigan DR. Immunohistochemical detection of an immediate early antigen of human cytomegalovirus in normal tissues. J Infect Dis. 1989;160(5):741–51. doi: 10.1093/infdis/160.5.741. [DOI] [PubMed] [Google Scholar]
- Tuttle DL, Harrison JK, Anders C, Sleasman JW, Goodenow MM. Expression of CCR5 increases during monocyte differentiation and directly mediates macrophage susceptibility to infection by human immunodeficiency virus type 1. J Virol. 1998;72(6):4962–9. doi: 10.1128/jvi.72.6.4962-4969.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Watanabe T, Haraoka S, Shimokama T. Inflammatory and immunological nature of atherosclerosis. Int J Cardiol. 1996;54 Suppl:S51–60. doi: 10.1016/s0167-5273(96)88773-0. [DOI] [PubMed] [Google Scholar]
- Westphal M, Lautenschlager I, Backhaus C, Loginov R, Kundt G, Oberender H, Stamm C, Steinhoff G. Cytomegalovirus and proliferative signals in the vascular wall of CABG patients. Thorac Cardiovasc Surg. 2006;54(4):219–26. doi: 10.1055/s-2006-923891. [DOI] [PubMed] [Google Scholar]
- Yurochko AD. Human cytomegalovirus modulation of signal transduction. In: Stinski MF, Shenk T, editors. Human cytomegaloviruses. Vol. 325. Springer-verlag; Berlin Heidelberg: 2008. pp. 205–220. [DOI] [PubMed] [Google Scholar]
- Yurochko AD, Huang ES. Human cytomegalovirus binding to human monocytes induces immunoregulatory gene expression. J Immunol. 1999;162(8):4806–16. [PubMed] [Google Scholar]