

Abstract
3-acetamido-3,6-didexoy-α-d-glucose or Quip3NAc is an unusual dideoxy sugar found in the O-antigens of various Gram-negative bacteria and in the S-layer glycoprotein glycans of some Gram-positive bacteria. It is produced in these organisms as a dTDP-linked sugar, with five enzymes ultimately required for its biosynthesis. The focus of this investigation is on the enzyme QdtC, a CoA-dependent N-acetyltransferase that catalyzes the last step in the Quip3NAc biosynthetic pathway. For this analysis, three crystal structures were determined: the wild-type enzyme in the presence of acetyl-CoA, and two ternary complexes of the enzyme with CoA and either dTDP-d-Quip3N or dTDP-3-amino-3,6-didexoy-α-d-galactose (dTDP-d-Fucp3N). Each subunit of the trimeric enzyme is dominated by a left-handed β-helix motif with 11 turns. The three active sites are located at the subunit:subunit interfaces, and the two dTDP-sugar ligands employed in this study bind to the protein in nearly identical manners. Those residues responsible for anchoring the hexose moieties of the dTDP-sugars to the protein include Glu 141, Asn 159, Asp 160 from one subunit and His 134 from another subunit. To probe the roles of various amino acid residues in the catalytic mechanism of the enzyme, ten site-directed mutant proteins were constructed and their kinetic parameters measured. On the basis of these data, a catalytic mechanism is proposed for QdtC whereby the acetylation of the sugar amino group does not require a catalytic base provided by the protein. Rather, the sulfur of CoA functions as the ultimate proton acceptor.
Unusual deoxysugars are found throughout Nature, often in the lipopolysaccharides of Gram-negative bacteria (1) and on various antibiotics (1, 2), antifungals (3), anthelmintics (4), and antitumor drugs (5). One such sugar derivative, 3-acetamido-3,6-dideoxy-α-d-glucose or Quip3NAc, has been observed in the O-antigens of various Gram-negative bacteria including Escherichia coli O114 (6) and in the S-layer glycoprotein glycans of some Gram-positive bacteria (7). Nucleotide-activated sugar precursors such as dTDP-d-Quip3NAc serve as the building blocks for the formation of either the S-layer glycans or the O-antigens.
In Thermoanaerobacterium thermosaccharolyticum E207-71, a Gram positive, anaerobic, thermophilic organism, five enzymes are required for the biosynthesis of dTDP-d-Quip3NAc starting from glucose-1-phosphate (Scheme 1). Like most of the pathways for the synthesis of 3,6-dideoxyhexoses, the formation of this unusual sugar begins with the attachment of α-d-glucose-1-phosphate to a nucleotide via the action of glucose-1-phosphate thymidylyltransferase (RmlA). In the next step, the 6’-hydroxyl group is removed and the C-4’ hydroxyl group is oxidized to a keto-functionality yielding dTDP-4-keto-6-deoxyglucose. This reaction is catalyzed by dTDP-glucose 4,6-dehydratase (RmlB). Both the thymidylyltransferase and the dehydratase have been well characterized with respect to structure and function (8).
Three additional enzymes are ultimately required for the synthesis of dTDP-d-Quip3NAc: an isomerase, an aminotransferase, and an N-acetyltransferase (Scheme 1). The focus of this investigation is on the N-acetyltransferase, which is encoded by the qdtC gene and hereafter referred to as QdtC (9). The protein contains 263 amino acid residues, requires acetyl-CoA for activity, and, interestingly, can acetylate either dTDP-d-Quip3N or dTDP-3-amino-3,6-dideoxy-α-d-galactose (dTDP-d-Fucp3N). Here we present a combined structural and functional analysis of QdtC. For this investigation, three structures of QdtC were determined: one with bound acetyl-CoA and two as ternary complexes with CoA and either dTDP-d-Quip3N or dTDP-d-Fucp3N. On the basis of these structures, site-directed mutant proteins were subsequently constructed to probe the biochemical roles for five residues in the catalytic mechanism, namely His 123, His 134, Glu 141, Asn 159, and Asp 160. Taken together the results presented here reveal the overall trimeric structure of QdtC and allow for a novel catalytic mechanism to be proposed.
Materials and Methods
Cloning, Expression, and Purification
Genomic DNA from T. thermosaccharolyticum E207-71 was isolated by standard procedures. The qdtC gene was PCR-amplified from genomic DNA such that the forward primer 5’-AAAACATATG CCAAATAATATTTCTAAAAGTGCGATAATAAAAGAAGG and the reverse primer 5’-AAAACTCGAG GTTTTCTATTGAAATATTCTTTATCCATGTATCATAATCTGTTTC added NdeI site and XhoI cloning sites, respectively. The purified PCR product was A-tailed and ligated into a pGEM-T (Promega) vector for screening and sequencing. A QdtC-pGEM-T vector construct of the correct sequence was then appropriately digested and ligated into a pET31b(+) (Novagen) plasmid for protein production with a non-cleavable C-terminal His6-tag.
The QdtC-pET31 plasmid was used to transform Rosetta(DE3) E. coli cells (Novagen). The culture in TB media was grown at 19°C with shaking for 24 hours and subsequently induced with 0.05 mM IPTG. The cells were allowed to express protein at 19°C for 24 hours after induction. QdtC was purified by standard procedures using NiNTA resin. Following purification, the protein was dialyzed against 10 mM Tris-HCl and 200 mM NaCl at pH 8.0 and then concentrated to 17 mg/ml.
All point mutations of the QdtC-pET31 plasmid construct were created via the Stratagene QuikChange method and sequenced to verify that no other changes had been introduced into the gene. The ten mutant proteins, as listed, were expressed and purified in the same manner as that for the wild-type enzyme: H123N, H123A, H134N, H134A, E141Q, E141A, N159D, N159A, D160N, and D160A.
Production of dTDP-d-Quip3N and dTDP-d-Fucp3N
dTDP-d-Quip3N was synthesized from dTDP-glucose as the initiating ligand. A typical 10 mL reaction mixture contained the following: 20 mM HEPPS (pH 8.5), 5 mM MgCl2, 60 mg dTDP-glucose, 200 mg glutamate, 4 mg dTDP-glucose-4,6-dehydratase, 3 mg QdtA, and 15 mg QdtB (Scheme 1). The reaction was allowed to proceed at 37°C for 7 hours. All enzymes were removed via filtration with a 10-kDa cutoff centriprep concentrator, and the enzyme-free reaction products were diluted by 1:4 with water. Purification was achieved by chromatography using a 6 mL Resource-Q column and a 120 mL gradient from 0-250 mM ammonium bicarbonate at pH 8.5. The desired product peak was identified by mass spectrometry (M-H+ 546.3 amu). Fractions containing the amino sugar product were pooled and lyophilized until all traces of the buffer had been removed. dTDP-d-Fucp3N was produced in an analogous manner, with the exception that the enzymes FdtA and FdtB from Aneurinibacillus thermoaerophilus DSM10155 were used (10). Typical yields of these two compounds were ~50% based on the starting amount of dTDP-glucose.
Production of dTDP-d-Quip3Nac
After the QdtA/QdtB reaction outlined above had been allowed to proceed at 37°C for 7 hours, 5 mg of QdtC and 0.1 mmol of acetyl-CoA were added, and this reaction was allowed to proceed until all of the amino sugar was acetylated (~2 hrs). The enzymes were removed by filtration, the material diluted with water, and the dTDP-d-Quip3NAc was purified using a 6 mL Resource-Q column and a 120 mL gradient from 0-100 mM ammonium bicarbonate at pH 8.5. The peak containing the acetylated product was identified by mass spectrometry (M-H+ 588.4 amu). Fractions containing the acetylated sugar were pooled and lyophilized until all traces of the buffer had been removed. Reactions using purified dTDP-d-Quip3N as the initiating ligand gave 100% yield of the acetylated sugar when treated with acetyl-CoA and QdtC.
Structural Analysis of QdtC
Crystallization conditions were initially surveyed with either the apoprotein or protein incubated with 10 mM acetyl-CoA and 10 mM dTDP by the hanging drop method of vapor diffusion and using a sparse matrix screen developed in the laboratory. Diffraction quality crystals were subsequently grown via hanging drop by mixing in a 1:1 ratio the protein incubated with acetyl-CoA and dTDP and 22-26% monomethylether poly(ethylene glycol) 5000 and 2% ethylene glycol at pH 7.0. These crystals belonged to the space group P3 with unit cell dimensions of a = b = 67.3 Å, and c = 112.4 Å and contained two monomers per asymmetric unit.
X-ray data were measured at 100K using a Bruker AXS Platinum 135 CCD detector controlled with the Proteum software suite (Bruker AXS Inc.). The x-ray source wasCuKα radiation from a Rigaku RU200 x-ray generator equipped with Montel optics and operated at 50 kV and 90 mA. These x-ray data were processed with SAINT version V7.06A (Bruker AXS Inc.) and internally scaled with SADABS version 2005/1 (Bruker AXS Inc.). Prior to x-ray data collection, the crystals were transferred to a stabilization solution containing 30% monomethylether poly(ethylene glycol) 5000, 400 mM NaCl, 10 mM dTDP, 10 mM acetyl-CoA, and 15% ethylene glycol. Relevant x-ray data collection statistics are presented in Table 1. For the complexes with the dTDP-amino sugars, 40 mM of the respective nucleotide-linked sugar was substituted for dTDP in the stabilization solutions.
Table 1.
X-ray Data Collection Statistics.
| Enzyme Complexed with Acetyl-CoA | Gold Heavy Atom Derivative | Enzyme Complexed with CoA and dTDP-d-Quip3N | Enzyme Complexed with CoA and dTDP-d-Fucp3N | |
|---|---|---|---|---|
| resolution limits | 30.0-1.7 (1.8-1.7)b | 30-2.1 (2.2-2.1)b | 30.0-1.95 (2.05-1.95)b | 30.0-1.8 (1.9-1.8)b |
| number of independent reflections | 59238 (8384) | 32382 (3822) | 39913 (4815) | 50462 (6644) |
| completeness (%) | 93.9 (81.4) | 97.7 (89.7) | 95.1 (79.9) | 95.9 (82.6) |
| redundancy | 4.5 (1.3) | 4.0 (1.8) | 3.4 (1.4) | 3.8 (1.5) |
| avg I / avg σ(I) | 13.0 (2.6) | 13.1 (3.8) | 8.7 (2.3) | 10.7 (2.7) |
| Rsym(%)a | 6.4 (30.5) | 6.4 (16.7) | 8.4 (26.4) | 7.2 (27.3) |
Rsym = (• | Σ I - Ī | / • I) × 100.
Statistics for the highest resolution bin.
An initial structure was solved with one heavy atom derivative using 10 mM potassium gold(I) cyanide and a soak time of three days. Two gold binding sites were identified with the program SOLVE (11), giving an overall figure-of-merit of 0.41 to 2.1 Å resolution. Solvent flattening with RESOLVE (12, 13) generated an interpretable electron density map, which allowed for a preliminary model to be constructed using the software package COOT (14). There was no interpretable electron density corresponding to dTDP even though the crystals were grown in its presence. Consequently, the structure represents the QdtC/acetyl-CoA binary complex. Coordinates for the complex were refined by TNT (15) to an Rworking of 17.8% for all measured x-ray data from 30 to 1.7 Å resolution. A Ramachandran plot analysis shows that 85.6%, 13.6%, and 0.9% of the amino acid residues lie within the core, allowed, and generously allowed regions, respectively. Relevant refinement statistics are given in Table 2. This structure served as the search model for the subsequent structural analyses of the enzyme/acetyl-CoA/dTDP-linked sugar ternary complexes via molecular replacement with the software package PHASER (16).
Table 2.
Least-Squares Refinement Statistics.
| Enzyme Complexed with Acetyl-CoA | Enzyme Complexed with CoA and dTDP-d-Quip3N | Enzyme Complexed with CoA and dTDP-d-Fucp3N | |
|---|---|---|---|
| resolution limits (Å) | 30.0-1.7 | 30.0-1.95 | 30.0-1.8 |
| aR-factor (overall)%/no. reflections | 17.8/59147 | 18.5/39903 | 16.5/50377 |
| R-factor (working)%/no. reflections | 17.4/53219 | 18.3/35917 | 16.2/45405 |
| R-factor (free)%/no. reflections | 24.1/5928 | 24.7/3986 | 23.1/4972 |
| number of protein atoms | 4100b | 4116c | 4104 |
| number of heteroatoms | 618d | 395e | 585f |
| average B values | |||
| protein atoms (Å2) | 19.4 | 23.5 | 17.8 |
| ligands (Å2) | 33.6 | 37.7 | 20.5 |
| solvent (Å2) | 34.3 | 31.2 | 30.0 |
| weighted RMS deviations from ideality | |||
| bond lengths (Å) | 0.013 | 0.012 | 0.013 |
| bond angles (°) | 2.1 | 2.1 | 2.3 |
| trigonal planes (Å) | 0.007 | 0.006 | 0.008 |
| general planes (Å) | 0.015 | 0.011 | 0.013 |
| torsional anglesg (°) | 17.6 | 18.3 | 17.6 |
R-factor = (Σ | Fo - Fc | / Σ | Fo |) × 100 where Fo is the observed structure-factor amplitude and Fc is the calculated structure-factor amplitude.
These include multiple conformations for Asn 20 in subunit 2 of the asymmetric unit.
These include multiple conformations for His 90 in subunit 1 and Lys 108 in subunit 2.
Heteroatoms include two acetyl-CoA molecules and 498 waters.
Heteroatoms include two CoA molecules, 2 dTDP-D-Quip3N ligands, and 229 waters.
Heteroatoms include two CoA molecules, 2 dTDP-D-Fucp3N ligands, and 419 waters.
The torsional angles were not restrained during the refinement.
As discussed in the Results section, there were no interpretable electron densities for the CoA acetyl groups in either of the complexes. Hence the reported structures represent ternary complexes with the dTDP-linked sugars and CoA rather than acetyl-CoA. Least-squares refinement with TNT reduced the R-factor to 18.5% for all measured x-ray data from 30 to 1.95 Å resolution for the protein/CoA/dTDP-d-Quip3N complex and 16.5% for all measured x-ray data from 30 to 1.8 Å resolution for the protein/CoA/dTDP-d-Fucp3N complex. A Ramachandran plot analysis demonstrates that for the enzyme/CoA/dTDP-d-Quip3N complex, 85.0% 14.2%, and 0.9% of the amino acid residues lie within the core, allowed, and generously allowed regions, respectively. Likewise, the enzyme/CoA/dTDP-d-Fucp3N complex refined equally as well with 84.7%, 14.7%, and 0.7% of the amino acid residues located within the core, allowed, and generously allowed regions of the Ramachandran plot, respectively. Relevant refinement statistics are presented in Table 2.
Measurement of Enzymatic Activity
N-acetyltransferase activity was monitored spectrophotometrically by following the increase in absorbance at 412 nm due to the reaction of the sulfhydryl group of the CoASH product with 5,5’-dithiobis(2-nitrobenzoic acid) resulting in a disulfide interchange. This interchange leads to the formation of 5-thio-2-nitrobenzoic acid, which has a characteristic absorbance at 412 nm and an extinction coefficient of 14150 M-1 cm-1. The use of this compound for quantification of CoASH was first reported by Tomkins et al., (17), and our assay method was similar to that described in (18). Reactions were monitored continuously with a Beckman DU 640B spectrophotometer, and enzyme activities were calculated from the initial rates. Assay reactions were 100 μL in volume and contained, in addition to enzyme and substrates, 100 mM HEPES (pH 7.5) and 5 mM 5,5’-dithiobis(2-nitrobenzoic acid). The reactions were initiated by the addition of enzyme and monitored at 25°C. Kinetic data were fitted using Sigma Plot 8. Initial velocity patterns were evaluated by measuring the initial rates at five concentrations of each substrate, 0.01, 0.02, 0.05, 0.2 and 1.0 mM dTDP-Quip3N, and 0.007, 0.01, 0.02, 0.04, and 1.0 mM acetyl-CoA. Equation 1 was used to fit the data, and the double reciprocal plots clearly showed a series of intersecting lines, Figure 1, suggesting a sequential mechanism.
Figure 1.
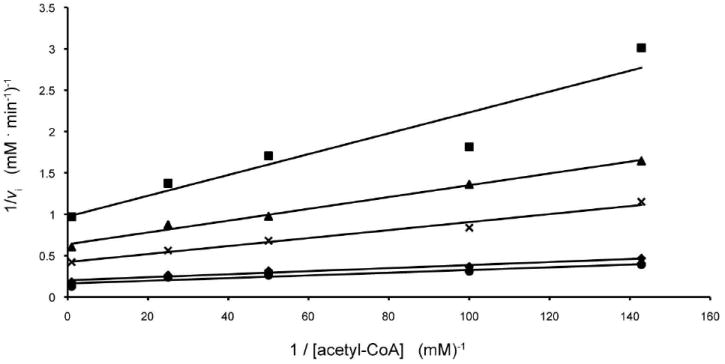
Double reciprocal plots of initial rates of formation of CoA. In this plot, the concentration of acetyl-CoA is varied at several fixed concentrations of dTDP-d-Quip3N equal to 0.01 mM (square), 0.02 mM (triangle), 0.05 mM (cross), 0.2 mM (diamond), and 1.0 mM (circle).
| (1) |
Calculated Km values were 0.018 ± 0.002 MM for acetyl-CoA and 0.11 ± 0.02 mM for dTDP-Quip3N.
Kinetic constants for the alternate substrate (dTDP-Fucp3N) and the mutant proteins were determined at saturating concentration of acetyl-CoA, 1.0 mM, while varying the dTDP-sugar concentrations between 0.01 - 54 mM. Individual substrate saturation kinetic data were fitted to equation 2.
| (2) |
The kinetics data are presented in Table 3.
Table 3.
Kinetic Parameters for the Forward Reaction with dTDP-d-QuiP3N and Acetyl-CoA.
| Protein |
Km (mM) dTDP-d-Quip3N |
kcat (s-1) | kcat/Km (M-1 s-1) |
|---|---|---|---|
| wild-type | 0.11 ± 0.02 | 8.7 ± 0.4 | 7.9 × 104 |
| wild-type using dTDP-d-Fucp3N | 0.36 ± 0.06 | 15.2 ± 0.7 | 4.2 × 104 |
| H134N | 0.60 ± 0.05 | 9.1 ± 0.3 | 1.5 × 103 |
| H134A | 4.50 ± 0.45 | 10.0 ± 1.0 | 2.2 × 103 |
| E141Q | 0.72 ± 0.09 | 5.8 ± 0.3 | 8.1 × 103 |
| E141A | 1.05 ± 0.08 | 44.1 ± 1.0 | 4.2 × 104 |
| H123N | 0.59 ± 0.08 | 12.3 ± 0.7 | 2.1 × 104 |
| H123A | 0.63 ± 0.04 | 12.3 ± 0.4 | 2.0 × 104 |
| D160N | 9.78 ± 0.67 | 12.2 ± 0.3 | 1.2 × 104 |
| D160A | 10.7 ± 0.8 | 0.25 ± 0.01 | 2.3 × 101 |
| N159D | 0.51 ± 0.08 | 20.3 ± 0.7 | 4.0 × 104 |
| N159A | 15.4 ± 1.5 | 18.9 ± 1.4 | 1.2 × 103 |
Results and Discussion
Structure of QdtC Complexed with Acetyl-CoA
The crystals employed in this investigation contained two subunits of QdtC, each of which packed along crystallographic three-fold rotational axes to generate the physiological trimers. The two subunits in the asymmetric unit are nearly identical and superimpose with a root-mean-square deviation of 0.15 Å. A ribbon representation of the QdtC trimer is displayed in Figure 2a. It is rather elongated with overall dimensions of ~73 Å × 76 Å × 64 Å. A total of ~4000 Å2 of surface area per subunit is buried upon trimerization. Each subunit participates in extensive interactions with the other two subunits forming the trimer. The individual acetyl-CoA binding sites, separated by ~24 Å as measured from the sulfurs, are shared between two subunits and are situated at one end of the trimer.
Figure 2.
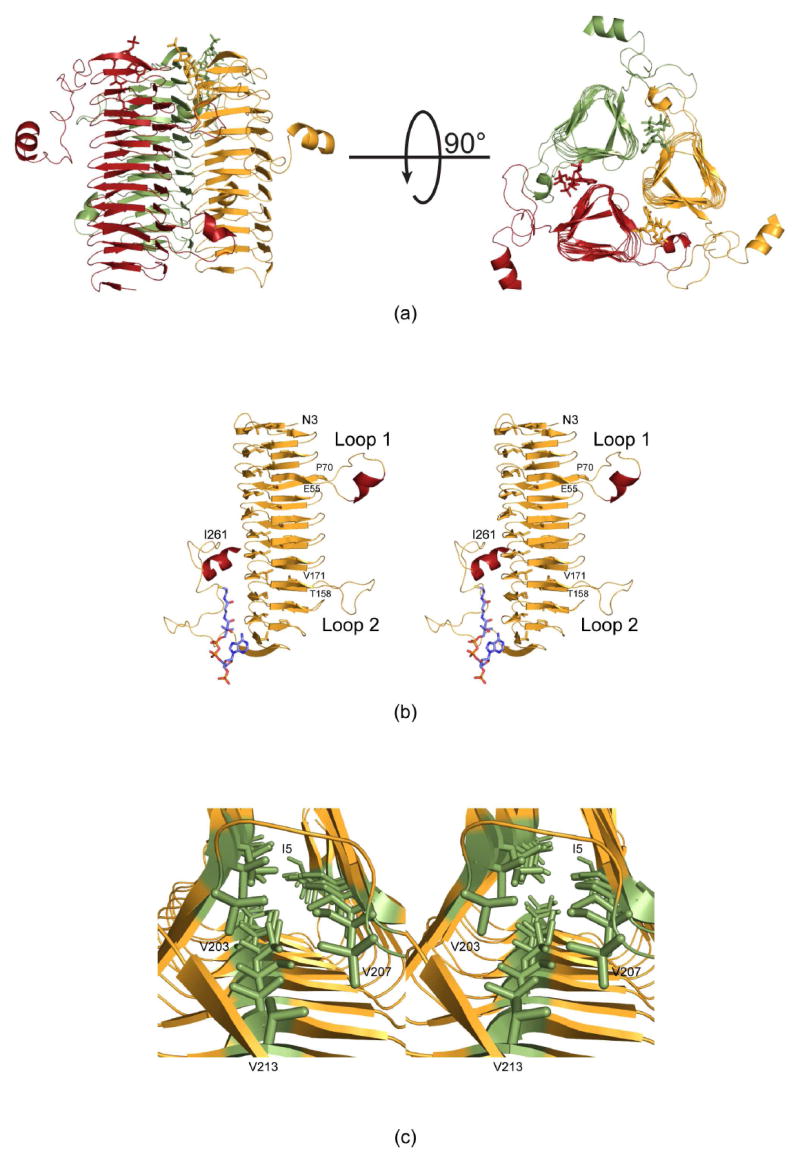
Molecular architecture of QdtC. (a) Two views of the trimeric enzyme are shown with the individual subunits color-coded in green, red, and yellow. The polypeptide chains are depicted in ribbon representations whereas the acetyl-CoA ligands are shown as sticks. As can be seen, the three active sites of the trimer are located between subunits. (b) A stereo view of one subunit is displayed. The β-strands and α-helices are displayed in orange and red, respectively. (c) A close-up view is shown that highlights the three hydrophobic walls. These result from the hexapeptide sequence that dominates the primary structure of QdtC. All figures were prepared with the software package PyMOL (23).
Shown in Figure 2b is a view of the individual subunit which is dominated by 32 β-strands that form a left-handed β-helix motif with 11 turns. The regularity of the β-helix is disrupted by two extended loops. The first, defined by Glu 55 to Pro 70, contains an α-helix defined by Val 58 to Asn 62. The second loop, delineated by Thr 158 to Val 171, projects towards the active site of a symmetry-related molecule and provides a platform for accommodating the acetyl moiety of acetyl-CoA. Following the last turn of the β-helix motif, the polypeptide chain from Ile 224 to Asp 253 adopts a random coil architecture, which is capped off by the second α-helix of the subunit formed by Tyr 254 to Ile 261. This α-helix extends away from the main body of the trimer (Figure 2a).
The type of left-handed β-helix motif observed in QdtC was first described in the elegant structural analysis of UDP-N-acetylglucosamine acyltransferase (LpxA) (19). Like LpxA, QdtC contains a hexapeptide repeat typically initiating with an isoleucine residue although some of the repeats begin with leucine, valine, or threonine. The first repeat begins with Ile 5 as indicated in Figure 2c. Due to this repeating motif, the interior of the β-helix is lined by three rows of mostly isoleucine residues. One row is defined by Ile 5, Ile 23, Ile 41, Ile 73, Ile 91, Ile 109, Ile 127, Ile 145, Ile 173, Ile 191, and Val 207. The second wall contains Ile 11, Ile 29, Ile 47, Ile 79, Thr 97, Ile 115, Ile 133, Leu 151, Ile 179, Val 197, and Val 213. Finally, the third row is formed by Ile 17, Ile 35, Leu 53, Ile 85, Ile 103, Ile 121, Val 139, Leu 157, Val 185, and Val 203. The regularity of the repeat breaks down at Val 203, which only contains four residues separating it from Val 207 (Figure 2c). The final hexapeptide repeat of the subunit is defined by Val 207 to Val 213.
Electron density corresponding to the bound acetyl-CoA is displayed in Figure 3a. Overall the electron density is well defined with the exception of that for the methyl group of the acetyl moiety. The average temperature factors for the cofactors in subunits 1 and 2 of the asymmetric unit are 38.2 Å2 and 25.7 Å2, respectively. As can be seen in Figure 3a, the acetyl-CoA adopts a curved conformation and nestles against the last four turns of the β-helix and the C-terminal loop. The ribose of the coenzyme assumes the C2’-endo pucker. The phosphoryl groups of the coenzyme project outwards towards the solvent, whereas the adenine ring, the pantothenate, and β-mercaptoethylamine units are quite buried within the QdtC trimer. Specifically a total of ~350 Å2 of the coenzyme’s surface area is buried upon binding to QdtC, representing ~35% of its total surface area.
Figure 3.
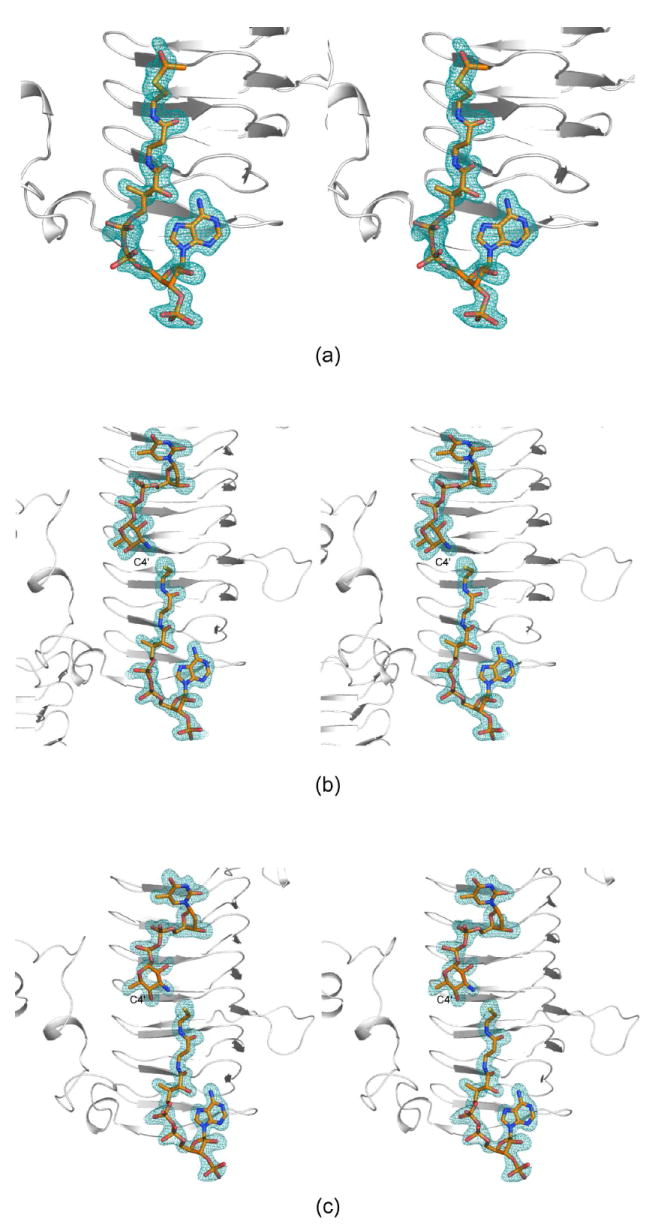
Representative electron density maps. (a) Electron density corresponding to acetyl-CoA in Subunit 1 of the asymmetric unit is depicted. The map was contoured at ~3σ and calculated with coefficients of the form (Fo-Fc), where Fo was the native structure factor amplitude and Fc was the calculated structure factor amplitude. (b) Electron density corresponding to acetyl-CoA and dTDP-d-Quip3N in subunit 2 is shown. The map was contoured at ~2.5σ As can be seen, CoA rather than acetyl-CoA was observed. (c) Electron density corresponding to acetyl-CoA and dTDP-d-Fucp3N in subunit 1 is displayed. The map was contoured at ~3.0σ Again, only CoA was observed.
Those amino acid residues located within approximately 3.5 Å of the coenzyme are shown in Figure 4a. The adenine group of CoA is anchored to the protein via two water molecules and the carbonyl oxygen of Ala 199 from one subunit of the trimer. The side chains of Thr 204 and Lys 205 from the second subunit served to position the 2-hydroxyl and the phosphoryl group of CoA, respectively, into the QdtC active site. Additional interactions occur between the carbonyl oxygens of the pantothenate and the backbone amide nitrogens of Ala 180 and Gly 198 from one subunit. The second subunit provides a hydrogen bond between the nitrogen of the β-mercaptoethylamine unit of CoA and the backbone carbonyl of Asp 160. A total of five water molecules lies within 3.2 Å of the coenzyme. In addition to all of these interactions, two intramolecular hydrogen bonds are formed between the hydroxyl of the pantothenate moiety and N7 and the amino group of the adenine ring of CoA.
Figure 4.
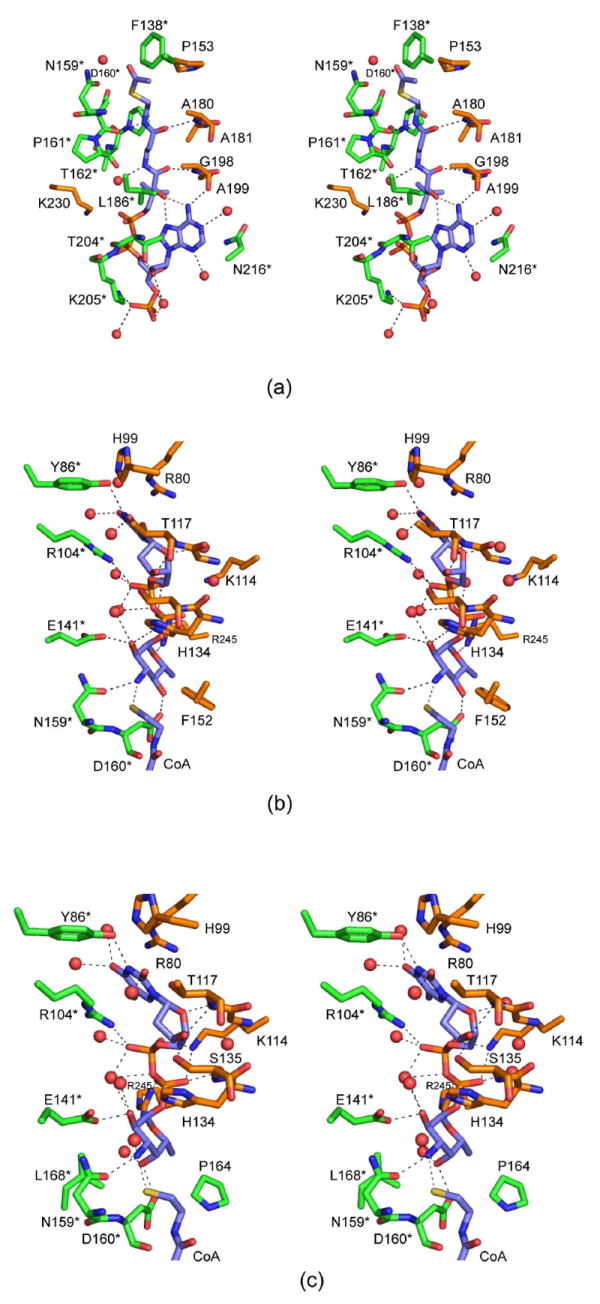
The QdtC active sites with bound ligands. (a) A close-up stereo view of the QdtC active site with bound acetyl-CoA is shown. The coenzyme is highlighted in blue bonds whereas the residues from two of the subunits in the trimer are displayed in gold and green. For the sake of clarity, only the side chains are displayed for those residues whose backbone atoms are not involved in hydrogen bonding to the ligand. Water molecules are depicted as red spheres. Possible hydrogen bonding interactions within 3.2 Å of CoA and the protein (or solvent) are indicated by the dashed lines. (b) A close-up view of the QdtC active site with bound CoA and dTDP-d-Quip3N is depicted. Given that the CoA binds in a nearly identical manner as that shown in (a), only the immediate region surrounding the nucleotide-linked sugar is shown. (c) A close-up view of the QdtC active site with bound CoA and dTDP-d-Fucp3N is displayed.
Structure of QdtC Complexed with dTDP-d-Quip3N and CoA
For the structural analysis of this ternary complex, the protein was first incubated with 10 mM acetyl-CoA and 40 mM of dTDP-d-Quip3N. The observed electron density corresponding to these ligands is displayed in Figure 3b. The nucleotide-linked sugar adopts an extended conformation and abuts four additional turns of the β-helix. It is more buried than the CoA with ~50% of its surface area covered upon protein binding.
Clearly the electron density presented in Figure 3b demonstrates that CoA, rather than acetyl-CoA is trapped in the active site, and that the nucleotide ligand is dTDP-d-Quip3N and not its acetylated version, dTDP-d-Quip3NAc. Most likely during the incubation period, the protein catalyzed the acetylation reaction thereby leading to CoA and dTDP-d-Quip3NAc. Given the widely differing concentrations of acetyl-CoA and dTDP-d-Quip3N used in the incubation (10 mM versus 40 mM), we speculate that all of the acetyl-CoA was hydrolyzed to CoA, which subsequently bound to the active site of QdtC in presence of excess of dTDP-d-Quip3N. As a consequence, an abortive complex was effectively trapped within the crystalline lattice.
The binding of the dTDP-linked sugar causes little overall change in the architecture of QdtC compared to that with only bound acetyl-CoA. Indeed the α-carbons between the two structures superimpose with a root-mean-square deviation of 0.12 Å. The manner in which CoA is accommodated in the active site of this ternary complex is similar to that observed in the acetyl-CoA/protein complex alone with some slight variations about the pyrophosphoryl groups. A close-up view of the active site surrounding the nucleotide-linked sugar is depicted in Figure 4b. As observed with acetyl-CoA, both subunits contribute to the binding of dTDP-d-Quip3N. Specifically, the backbone amide nitrogen of Thr 117 from one subunit hydrogen bonds to the ribose hydroxyl whereas Tyr 86 from the neighboring subunit interacts with N3 of the thymine ring. Arg 245 from one subunit and Arg 104 from the other participate in electrostatic interactions with the pyrophosphoryl groups of the substrate. The quinovose unit of the substrate, which adopts the 4C1 conformation, interacts primarily with one subunit, and in particular with the second loop that interrupts the regularity of the β-helix (Figure 2b). Its 2’-hydroxyl hydrogen bonds with the carboxylate of Glu 141 and the imidazole ring of His 134 (from the other subunit) whereas its C-4’ hydroxyl lies within hydrogen bonding distance to the carboxylate of Asp 160. The C-3’ amino group of the sugar is positioned within 2.9 Å of Oδ1 of Asn 159 and 2.9 Å of the CoA sulfhydryl group. Seven water molecules lie within 3.2 Å of the dTDP-d-Quip3N molecule.
Structure of QdtC Complexed with dTDP-d-Fucp3N and CoA
For this structure, the enzyme was first incubated in the presence of 10 mM acetyl-CoA and 40 mM of dTDP-d-Fucp3N prior to crystallization. Again, only CoA and dTDP-d-Fucp3N were observed in the electron density map (Figure 3c). The α-carbons for QdtC solved in the presence of either dTDP-d-Quip3N or dTDP-d-Fucp3N superimpose with a root-mean-square deviation of 0.14 Å, and the dTDP-sugar ligands interact with the protein in similar manners (Figures 3b and 3c). There is a slight movement of the hexose group in the active site such that in the case of dTDP-d-Fucp3N binding, the side chain of His 134 no longer lies within 3.2 Å of the hexose 2’-hydroxyl. On the whole, however, the active site residues move very little in response to the different configuration about the C-4’ hydroxyl of dTDP-d-Fucp3N. The carboxylate side chain of Asp 160 still interacts with the hexose C-4’ hydroxyl group, although the hydrogen-bonding geometry is not as good as that observed for bound dTDP-d-Quip3N (Figure 5). Previous activity assays have demonstrated that both dTDP-d-Quip3N and dTDP-d-Fucp3N are substrates for QdtC (7). As listed in Table 3, our kinetic data indicate a ~50% reduction in the catalytic efficiency of QdtC with dTDP-d-Fucp3N as a substrate relative to dTDP-d-Quip3N.
Figure 5.
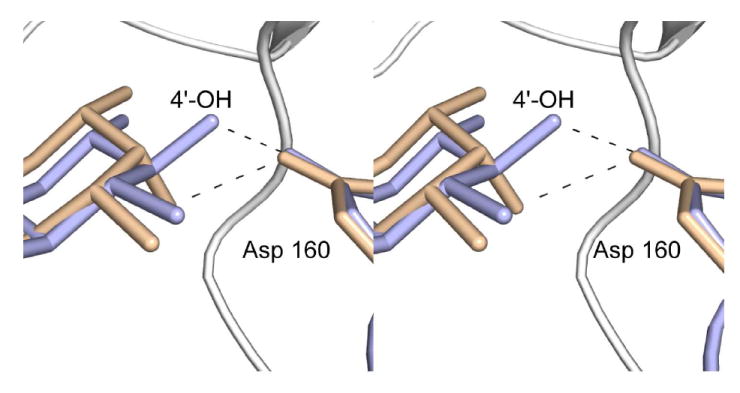
Differences in sugar binding to the QdtC active site. The dTDP-d-Quip3N and dTDP-d-Fucp3N ligands are displayed in light blue and wheat, respectively. Possible hydrogen bonds between the C-4’ hydroxyl groups of the sugars and the carboxylate of Asp 160 are indicated by the dashed lines.
Enzymatic Activities of Site-Directed Mutant Proteins and Implications for a Catalytic Mechanism
Recently two groups reported the crystal structure of PglD, an N-acetyltransferase that acetylates the C-4’ amino group of UDP-2-acetamido-4-amino-2,4,6-trideoxyglucose (20, 21). PglD is a smaller protein than QdtC with 203 versus 263 amino acid residues. Like QdtC, however, PglD is a trimer, and its three-dimensional architecture is dominated by a left-handed β-helix motif. The manners in which the nucleotide-linked sugars are accommodated within the active sites of PglD and QdtC are remarkably different, however, as shown in Figure 6. In PglD, His 125 has been implicated as the general base in the reaction mechanism (20, 21). This histidine residue is structurally conserved as His 123 in QdtC, but it is ~8 Å from the sugar amino group. Given the observed differences in dTDP-sugar binding in PglD versus QdtC, we were concerned that perhaps our ligand bound in an artifactual manner, and that His 123 might be the active site base. To test this hypothesis, two site-directed mutant proteins were constructed, H123N and H123A, and their Km and kcat values determined. In both cases the mutant proteins were active with catalytic efficiencies of 2.1 × 104 M-1 s-1 and 2.0 × 104 M-1 s-1, respectively (Table 3). This result highlights the dangers of making biochemical assumptions based simply on amino acid sequence alignments. His 125, which is the catalytic base in PglD, aligns with His 123 of QdtC on the basis of primary sequence analyses, but clearly the catalytic mechanisms for these two enzymes are different.
Figure 6.
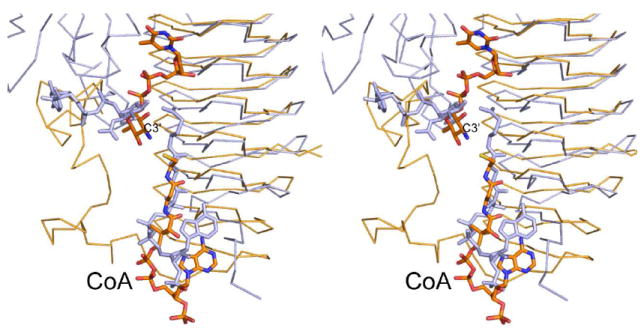
Superposition of QdtC onto PglD. The PglD structure with bound acetyl-CoA is highlighted in blue (PDB code 3BXY). The position of the nucleotide-linked sugar, also shown in blue, was obtained from the structure of the PglD solved in the presence of only the nucleotide-linked sugar (PDB code 3BSS). The ternary complex of QdtC with bound dTDP-d-Quip3N and CoA is depicted in gold bonds.
What residue then serves as the catalytic base in QdtC? There are three potential candidates, His 134, Glu 141, and Asp 160, that are located near the pyranosyl group, but they are not within hydrogen bonding distance of the sugar amino group. Again, we considered the possibility that an unusual conformation of the sugar might have been trapped in the active site and that in the Michaelis complex one of these residues resides near the sugar amino group. Thus, these residues were individually mutated to either an alanine or an asparagine in the case of His 134 and Asp 160 or a glutamine in the case of Glu 141. Again, all of these mutant proteins retained catalytic activity as indicated in Table 3. The most serious reduction in catalytic efficiency was observed for the D160A mutant protein (2.3 × 101 M-1 s-1). In this mutant protein, the Km increased to 10.7 ± 0.8 mM. As a comparison, the for Km wild-type enzyme was calculated to be 0.11 ± 0.02 mM. Assays in the absence of the nucleotide-linked sugar demonstrated no detectable acetyl-CoA hydrolysis for the D160A mutant protein. In addition, it should be noted that all of the mutant proteins were able to produce the acetylated sugar as demonstrated by an HPLC assay similar to that described in the Materials and Methods Section.
Finally, to complete the story, Asn 159, which lies within hydrogen bonding distance to the sugar C-3’ amino, was changed to either an aspartate or an alanine. Again the mutant proteins were catalytically active, albeit with reduced catalytic efficiencies (Table 3). For the N159A mutant form, the Km increased to 15.4 ± 1.5 mM. The reduction in enzymatic activities for all of these mutant proteins most likely results from perturbations of hexose positioning in the active site and not from the loss of an enzymatic base.
In light of these observations, we propose the following mechanism for QdtC, which does not invoke a general base provided by the protein. This mechanism is outlined in Scheme 2, and a stereo view of the potential attack geometry is presented in Figure 7. Unlike that suggested for PglD, whereby the nucleotide-linked sugar enters the active site in a protonated form (21), we suggest that QdtC binds the substrate in its unprotonated state. The carboxamide group of Asn 159 forms a hydrogen bond with the sugar C-3’ amino group, which helps to align the amino nitrogen for nucleophilic attack on the carbonyl carbon of acetyl-CoA. The other hydrogen on the amino group is directed at the sulfur of acetyl-CoA whereas the lone pair of electrons on the nitrogen is directed at the carbonyl carbon of the cofactor. As the amino nitrogen attacks, the bond between the carbonyl carbon and the sulfur of acetyl-CoA lengthens and eventually breaks. The sulfur of acetyl-CoA ultimately serves as the catalytic base by accepting a proton from the sugar amino group. The lack of a catalytic base in QdtC is reminiscent to the situation observed in homoserine kinase, a member of the GHMP superfamily (22). In this enzyme, it has been suggested that the γ-phosphoryl group of ATP, rather than an amino acid provided by the protein, ultimately serves as the base to remove the proton from the attacking hydroxyl group of homoserine. Strikingly, other members of the GHMP superfamily contain an aspartate residue in the appropriate position to function as a general base. It will be of interest to determine whether QdtC is simply an outlier like homoserine kinase, or rather its catalytic mechanism is a more common feature among sugar-modifying N-acetyltransferases. Structural and functional studies of additional N-acetyltransferases are presently in progress.
Figure 7.
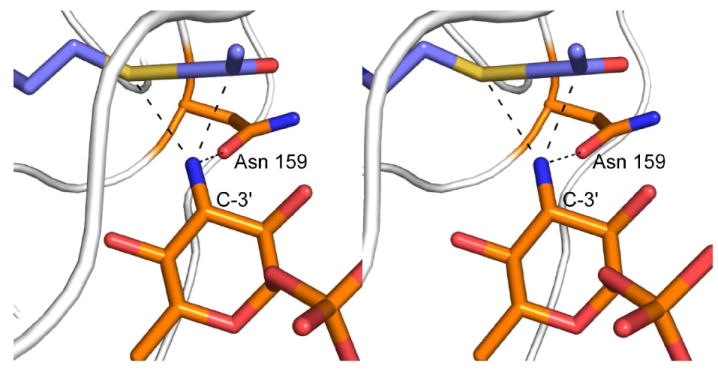
Hypothetical geometry of amino sugar attack at acetyl-CoA. According to the proposed mechanism, dTDP-d-Quip3N binds to the QdtC active site in an unprotonated form. One of the hydrogens on the sugar amino group is directed at the carboxamide group of Asn 159 whereas the other is directed at the sulfur of acetyl-CoA. The nitrogen of the amino group is sp3 hybridized, and the lone pair of electrons is directed towards the carbonyl group of acetyl-CoA. The model for acetyl-CoA was built on the basis of that observed in the wild-type structure.
Acknowledgments
We thank Dr. W. W. Cleland for helpful discussions and Dr. Grover L. Waldrop for critically reading the manuscript.
Abbreviations
- CoA
Coenzyme A
- Fucp3N
3-amino-3,6-dideoxy-α-d-galactose
- Fucp3NAc
3-acetamido-3,6-dideoxy-α-d-galactose
- HEPES
4-(2-hydroxyethyl)-1-piperazine-ethane sulfonic acid
- HEPPS
3-(4-(2-hydroxyethyl)-1-piperazine-propane sulfonic acid
- IPTG
isopropyl-β-d-thiogalactopyranoside
- NiNTA
nickel-nitrilotriacetic acid
- PCR
polymerase chain reaction
- Quip3N
3-amino-3,6-dideoxy-α-d-glucose
- Quip3NAc
3-acetamido-3,6-dideoxy-α-d-glucose
- TB
terrific broth
- dTDP
thymidine diphosphate
- dTMP
thymidine monophosphate
- Tris
tris-(hydroxymethyl)aminomethane
Footnotes
This research was supported in part by an NIH grant (DK47814 to H. M. H.)
X-ray coordinates have been deposited in the Research Collaboratory for Structural Bioinformatics, Rutgers University, New Brunswick, N. J. (accession nos. 3FSA, 3FSB, and 3FSC).
References
- 1.Trefzer A, Salas JA, Bechthold A. Genes and enzymes involved in deoxysugar biosynthesis in bacteria. Nat Prod Rep. 1999;16:283–299. doi: 10.1039/a804431g. [DOI] [PubMed] [Google Scholar]
- 2.Johnson DA, Liu H-w. Deoxysugars: Occurrence, genetics, and mechanisms of biosynthesis. Vol. 3. Elsevier; Amsterdam: 1999. [Google Scholar]
- 3.Chen SC, Sorrell TC. Antifungal agents. Med J Aust. 2007;187:404–409. doi: 10.5694/j.1326-5377.2007.tb01313.x. [DOI] [PubMed] [Google Scholar]
- 4.Yoon YJ, Kim ES, Hwang YS, Choi CY. Avermectin: biochemical and molecular basis of its biosynthesis and regulation. Appl Microbiol Biotechnol. 2004;63:626–634. doi: 10.1007/s00253-003-1491-4. [DOI] [PubMed] [Google Scholar]
- 5.Minotti G, Menna P, Salvatorelli E, Cairo G, Gianni L. Anthracyclines: molecular advances and pharmacologic developments in antitumor activity and cardiotoxicity. Pharmacol Rev. 2004;56:185–229. doi: 10.1124/pr.56.2.6. [DOI] [PubMed] [Google Scholar]
- 6.Feng L, Wang W, Tao J, Guo H, Krause G, Beutin L, Wang L. Identification of Escherichia coli O114 O-antigen gene cluster and development of an O114 serogroup-specific PCR assay. J Clin Microbiol. 2004;42:3799–3804. doi: 10.1128/JCM.42.8.3799-3804.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Pföstl A, Zayni S, Hofinger A, Kosma P, Schäffer C, Messner P. Biosynthesis of dTDP-3-acetamido-3,6-dideoxy-α-d-glucose. Biochem J. 2008;410:187–194. doi: 10.1042/BJ20071044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Dong C, Beis K, Giraud MF, Blankenfeldt W, Allard S, Major LL, Kerr ID, Whitfield C, Naismith JH. A structural perspective on the enzymes that convert dTDP-d-glucose into dTDP-l-rhamnose. Biochem Soc Trans. 2003;31:532–536. doi: 10.1042/bst0310532. [DOI] [PubMed] [Google Scholar]
- 9.Novotny R, Pfoestl A, Messner P, Schaffer C. Genetic organization of chromosomal S-layer glycan biosynthesis loci of Bacillaceae. Glycoconj J. 2004;20:435–447. doi: 10.1023/B:GLYC.0000038290.74944.65. [DOI] [PubMed] [Google Scholar]
- 10.Davis ML, Thoden JB, Holden HM. The x-ray structure of dTDP-4-keto-6-deoxy-d-glucose-3,4-ketoisomerase. J Biol Chem. 2007;282:19227–19236. doi: 10.1074/jbc.M702529200. [DOI] [PubMed] [Google Scholar]
- 11.Terwilliger TC, Berendzen J. Automated MAD and MIR structure solution. Acta Crystallogr D Biol Crystallogr. 1999;55(Pt 4):849–861. doi: 10.1107/S0907444999000839. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Terwilliger TC. Maximum-likelihood density modification. Acta Crystallogr D Biol Crystallogr. 2000;56(Pt 8):965–972. doi: 10.1107/S0907444900005072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Terwilliger TC. Automated main-chain model building by template matching and iterative fragment extension. Acta Crystallogr D Biol Crystallogr. 2003;59:38–44. doi: 10.1107/S0907444902018036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Emsley P, Cowtan K. Coot: model-building tools for molecular graphics. Acta Crystallogr D Biol Crystallogr. 2004;60:2126–2132. doi: 10.1107/S0907444904019158. [DOI] [PubMed] [Google Scholar]
- 15.Tronrud DE, Ten Eyck LF, Matthews BW. An efficient general-purpose least-squares refinement program for macromolecular structures. Acta Crystallogr Sect A. 1987;43:489–501. [Google Scholar]
- 16.McCoy AJ, Grosse-Kunstleve RW, Adams PD, Winn MD, Storoni LC, Read RJ. Phaser crystallographic software. J Appl Cryst. 2007;40:658–674. doi: 10.1107/S0021889807021206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Alpers DH, Appel SH, Tomkins GM. A spectrophotometric assay for thiogalactoside transacetylase. J Biol Chem. 1965;240:10–13. [PubMed] [Google Scholar]
- 18.Magalhaes ML, Blanchard JS. The kinetic mechanism of AAC3-IV aminoglycoside acetyltransferase from Escherichia coli. Biochemistry. 2005;44:16275–16283. doi: 10.1021/bi051777d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Raetz CR, Roderick SL. A left-handed parallel beta helix in the structure of UDP-N-acetylglucosamine acyltransferase. Science. 1995;270:997–1000. doi: 10.1126/science.270.5238.997. [DOI] [PubMed] [Google Scholar]
- 20.Rangarajan ES, Ruane KM, Sulea T, Watson DC, Proteau A, Leclerc S, Cygler M, Matte A, Young NM. Structure and active site residues of PglD, an N-acetyltransferase from the bacillosamine synthetic pathway required for N-glycan synthesis in Campylobacter jejuni. Biochemistry. 2008;47:1827–1836. doi: 10.1021/bi702032r. [DOI] [PubMed] [Google Scholar]
- 21.Olivier NB, Imperiali B. Crystal structure and catalytic mechanism of PglD from Campylobacter jejuni. J Biol Chem. 2008;283:27937–27946. doi: 10.1074/jbc.M801207200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Zhou T, Daugherty M, Grishin NV, Osterman AL, Zhang H. Structure and mechanism of homoserine kinase: prototype for the GHMP kinase superfamily. Structure Fold Des. 2000;8:1247–1257. doi: 10.1016/s0969-2126(00)00533-5. [DOI] [PubMed] [Google Scholar]
- 23.DeLano WL. The PyMOL Molecular Graphics System. DeLano Scientific; San Carlos, CA, USA: 2002. [Google Scholar]