

Abstract
Increased expression of autotaxin is linked to several malignancies including glioblastoma, breast, renal, ovarian, lung and thyroid cancers. Autotaxin promotes metastasis as well as cell growth, survival, and migration of cancer cells. These actions could depend on the non-catalytic effects of autotaxin on cell adhesion, or the catalytic activity of autotaxin, which converts lysophosphatidylcholine into lysophosphatidate in the extracellular environment. Both lysophosphatidylcholine and lysophosphatidate have been reported to stimulate migration through their respective G-protein coupled receptors. The present study determines the roles of autotaxin, lysophosphatidylcholine and lysophosphatidate in controlling the migration two cancer cell lines MDA-MB-231 breast cancer cells, which produce little autotaxin and MDA-MB-435 melanoma cells that secrete significant levels of autotaxin. Lysophosphatidylcholine alone was unable to stimulate the migration of either cell type unless autotaxin was present. Knocking down autotaxin secretion, or inhibiting its catalytic activity, blocked cell migration by preventing lysophosphatidate production and the subsequent activation of LPA1/3 receptors. We conclude that inhibiting autotaxin production or activity of could provide a beneficial adjuvant to chemotherapy for preventing metastasis in patients with high autotaxin expression in their tumors.
Keywords: lysophosphatidate, lysophosphatidylcholine, metastasis
Introduction
The ability of cancer cells to migrate and invade surrounding tissues is the main determinant of whether metastases will develop. Most aggressive cancers will metastasize and this results in a poorer prognosis for patients requiring treatment. The ability to prevent the formation of metastases after surgical intervention, or chemotherapy would provide a powerful tool in decreasing the morbidity and mortality associated with cancer.
Autotaxin (ATX) is a secreted glycoprotein whose level of expression in tumors is strongly correlated with their aggressiveness and invasiveness (1). ATX was first identified in human melanoma A2058 cells (2) and it has also been detected in several other tumor cell lines (3–6). ATX expression in breast and other cancers is strongly associated with tumor cell survival, growth, migration, invasion, and metastasis (1, 7–11). However, the mechanisms by which ATX modifies cell signaling to stimulate angiogenesis, cancer cell migration and metastasis are still not fully understood. A non-catalytic effect of ATX was shown to occur in oligodendrocytes where ATX acts as a matrixcellular protein through integrin-dependant focal adhesion assembly (12, 13). ATX may thus mediate cell to extracellular matrix interactions and that may be linked to cancer aggressiveness. Another mechanism of ATX action is through the conversion of extra-cellular lysophosphatidylcholine (LPC) to lysophosphatidate (LPA) (14), which could change the balance of cell activation by LPC versus LPA.
LPC is an abundant extracellular lipid that can be found in concentrations up to 200 μM (15). It is produced by the liver, which secretes mainly polyunsaturated LPC (15). Alternatively, mainly saturated LPC is produced by lecithin:cholesterol acyltransferase, which circulates in high-density lipoproteins (16). It has been proposed that LPC can stimulate cell signaling and cell migration through its action as an extracellular ligand for G2A (G2 accumulation) and GPR4 (G protein-coupled receptor-4) receptors (17–22). Alternatively, LPC can signal after its conversion to LPA (7, 14).
LPA is a potent signaling molecule that stimulates cell proliferation, neurite retraction, chemotaxis, and wound healing (23, 24). These effects of LPA are mediated by at least six G- protein coupled receptors on the cell surface: LPA1/EDG2, LPA2/EDG4, LPA3/EDG7, LPA4/GPR23/p2y9, LPA5/GRP92 and LPA6/p2y5 (25–27). The expression of these LPA receptors is cell-specific, and each can elicit different responses upon LPA binding. Initial evidence that LPA can be involved in carcinogenesis came from the identification of its role as an activating factor in ovarian cancer. LPA is present in high concentrations in the ascites fluid of ovarian tumors and it stimulates cell proliferation and metastasis (28, 29). These actions are mediated mainly through LPA1 and LPA3 receptors (11, 30–32). LPA stimulates angiogenesis by elevating levels of vascular endothelial growth factor (8). LPA also increases the synthesis of macrophage migration inhibitory factor, a tumor promoter, in a colon cancer line (33) and it decreases the expression of the tumor suppressor, p53, in lung cancer cells (34).
Two main pathways have been identified for the production of extracellular LPA (35). During inflammation, secretory PLA2 is produced, which generates LPA from phosphatidate on exposed membranes (36). The second, and more significant source of extracellular LPA, is through the action of ATX on extracellular LPC (14).
In the present work, we examined the effects of LPC and LPA on the migration MDA-MB-231 breast cancer cells, which produce very little ATX and MDA-MB-435 melanoma cells (37), which secrete abundant ATX. We found that MDA-MB-231 cells migrate in response to LPC only if ATX is present. Also, knocking down ATX expression in MDA-MB-435 cells, or inhibiting the catalytic effect of ATX, blocked cell migration in the presence of LPC, but not LPA. We propose that inhibiting the production, or activity of ATX could decrease new tumor growth and metastasis following surgical excision of a tumor given that LPC is abundant in the environment of malignant tumors and it can readily be converted to LPA by tumor-derived ATX.
Results
Differential Effects of Lysophosphatidylcholine and Lysophosphatidate on the Migration of MDA-MB-231 Breast Cancer Cells and MDA-MB-435 Melanoma Cells
We first compared the migration of MDA-MB-231 breast cancer cells and MDA-MB-435 cells, which are now known to be derived from M14 melanoma cells (37). MDA-MB-231 and MDA-MB-435 showed a similar migratory response to LPA over 6 h, with maximum migration occurring with about 0.5 μM LPA. This LPA concentration elicited a migratory response similar to that obtained with 5% charcoal treated-FBS (Fig. 1A). MDA-MB-231 cells showed no significant stimulation of migration over the 6 h incubation with any LPC concentration employed (Fig. 1B). By contrast, MDA-MB-435 cells showed a stimulation of migration in response to LPC concentrations of ≥5 μM in the bottom chamber.
FIGURE 1.

Effects of lysophosphatidate and lysophosphatidylcholine on the migration of MDA-MB-435 and MDA-MB-231 cells. Panel A, shows the migration of MDA-MB-435 (○), and MDA-MB-231 (■) cells after a 3 h incubation with different concentrations of LPA. Migration in response to 5% charcoal-stripped FBS was used as a positive control. Panel B, shows migration when the cells were incubated for 6 h with different concentrations of LPC. Results are expressed relative to the number of cells migrating with only basal media (RPMI 1640 with 0.1% BSA and 0.2% FBS-C), which was given a value of 1. Typically, basal migration consisted of approximately 75 cells per field for a 3 h incubation and 120 cells per field for a 6 h incubation. Results are means ± SEM from at least 3 independent experiments. Statistically significant differences between basal migration and agonist-stimulated migration are indicated by * (p < 0.01) and between cell lines are indicated by: † (p < 0.01).
ATX Activity Is Required For Lysophosphatidylcholine-Induced Cell Migration
The results in Fig. 1 could be explained in two ways. First, the MDA-MB-231 cells may not express putative receptors that respond to LPC, whereas MDA-MB-435 cells express these receptors. Second, and more likely, MDA-MB-435 could express ATX that produces LPA from LPC, which stimulates cell migration, whereas MDA-MB-231 cells do not express ATX. To test the latter hypothesis, we measured ATX expression in both cells lines. The MDA-MB-435 cells expressed significantly more (p<0.05) ATX mRNA than the MDA-MB-231 cells as shown by real-time RT-PCR analysis (Fig. 2A). Furthermore, concentrated medium from MDA-MB-231 cells showed very low ATX activity compared to MDA-MB-435 medium (Fig. 2B). Western Blot analysis of medium from MDA-MB-435 cells (Fig. 2C) showed marked expression of ATX, which migrated with the recombinant ATX standard at about 100 kDa as expected (7). MDA-MB-435 cells can, therefore, be used as a convenient source of ATX for future experiments described below. By contrast, ATX was barely detectable in equivalent amounts of medium from MDA-MB-231 cells.
FIGURE 2.

MDA-MB-435 cells secrete significant amounts of active ATX into the extracellular medium compared to MDA-MB-231 cells. Panel A shows the relative mRNA levels of ATX compared to GAPDH in MDA-MB-435 and MDA-MB-231 cells. Results are means ± SEM from at least 3 independent experiments. Panel B shows the results from the fluorescence assay for ATX activity of concentrated conditioned media collected from MDA-MB-435 (○), or MDA-MB-231 (■) cells. Results are means ± SEM from at least 3 independent experiments. Panel C shows the Western Blot analysis for ATX of concentrated media collected from 3 separate dishes of MDA-MB-435 or MDA-MB-231 cells. Recombinant ATX from Dr T Clair was used as a standard (StdATX).
We could not detect any significant ATX activity or protein expression in the lysates from MDA-MB-435 and MDA-MB-231 cells (results not shown), which implies that ATX is mainly a secreted protein as shown previously (14).
Concentrated Medium from MDA-MB-435 Cells Stimulates Cell Migration in the Presence of Lysophosphatidylcholine Because of its ATX Activity
To determine why MDA-MB-435 and MDA-MB-231 cells respond differently to LPC, we measured the effects of adding concentrated media from these cells to the bottom well of the migration chamber. MDA-MB-231 cells did not show any significant migration in the presence of LPC and their own concentrated medium. By contrast, a migratory response similar to that obtained with 5% charcoal-treated FBS, or 0.5 μM LPA was obtained when concentrated medium from MDA-MB-435 and LPC were added to the bottom chamber. This latter effect was completely abolished by the addition of 1 μM VPC8a202, an ATX inhibitor (38), to the wells (Fig. 3A).
FIGURE 3.
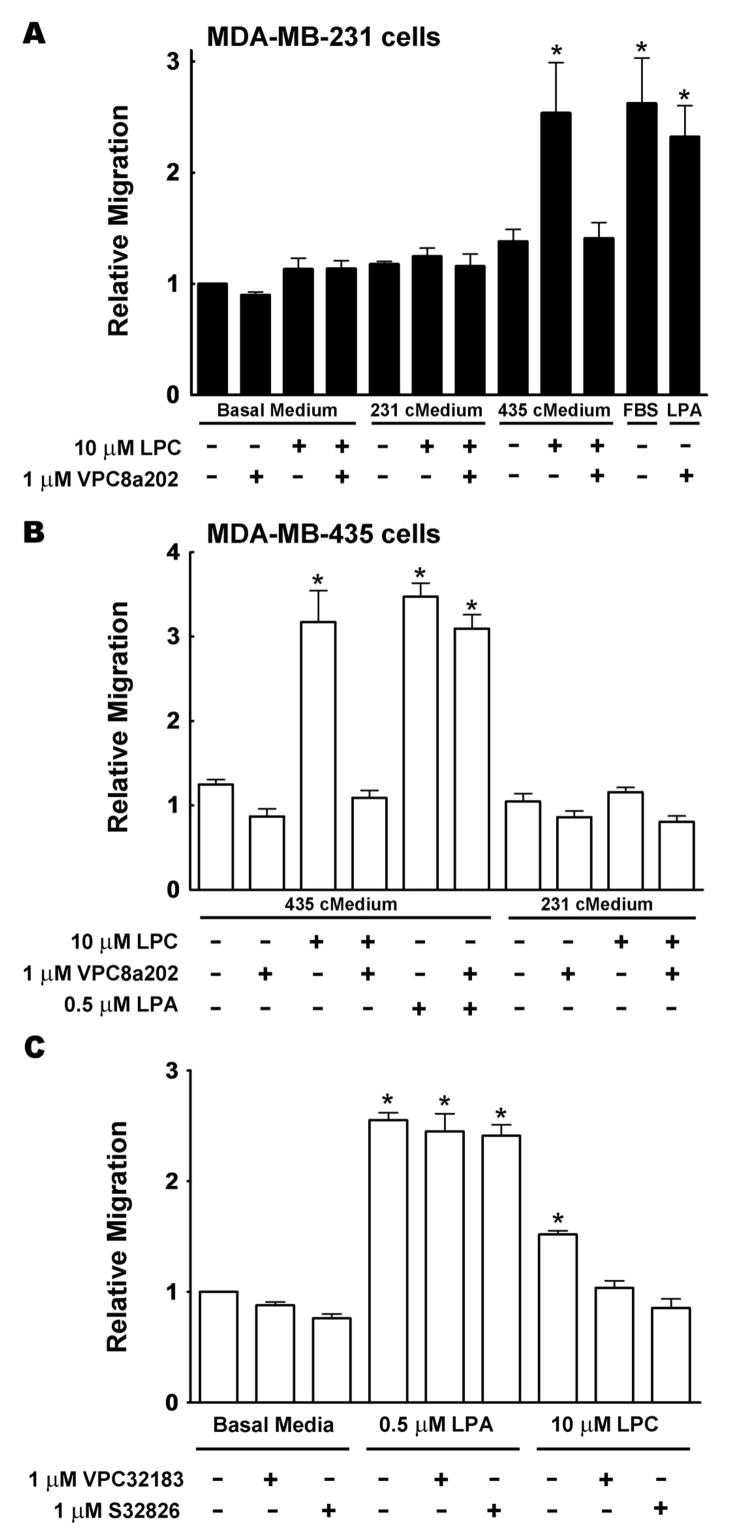
Stimulation of migration by lysophosphatidylcholine depends on autotaxin activity. Panel A shows the migration of MDA-MB-231 cells over 3 h in the presence of basal medium or concentrated medium prepared from either MDA-MB-231 cells (231 cMedium), or from MDA-MB-435 cells (435 cMedium). These experiments were performed in the presence or absence of 10 μM LPC, or 1 μM of the ATX inhibitor, VPC8a202. Migration of MDA-MB-231 cells with only basal media (RPMI 1640 with 0.1% BSA and 0.2% FBS) present was given a value of 1. Migration in the presence of 5% FBS and 0.5 μM LPA plus VPC8a202 were used as controls. Panel B, shows results from similar experiments using MDA-MB-435 cells over a 3 h period. Panel C shows the effects of the ATX inhibitors, VPC8a202 and S32826, on the migration of MDA-MB-435 cells over 3 h in the absence of concentrated medium. Results are means ± SEM from at least 3 independent experiments. Statistically significant differences are indicated by: * (p < 0.01).
A similar experiment was performed with MDA-MB-435 cells. Addition of concentrated medium from MDA-MB-231 cells had no significant effect on the migration of MDA-MB-435 cell in the presence, or absence of LPC (Fig. 3B). By contrast, a significant stimulation of migration was observed when concentrated medium from MDA-MB-435 cells was added to the bottom chamber with LPC. This response was abolished by the addition of 1 μM of the ATX inhibitor, VPC8a202. We also used 1 μM S32826 ([4-(tetradecanoylamino)benzyl]phosphonic acid), another ATX inhibitor (39), and found that it also blocked LPC-induced migration of MDA-MB-435 cells (Fig. 3C). As controls, we showed that VPC8a202 and S32826 did not significantly affect the LPA-induced stimulation of migration for either cell line (Figs 3A–C), thus demonstrating that the effects were specific for LPC-induced migration.
These results support the conclusion that the stimulatory effect of LPC on the migration of MDA-MB-435 and MDA-MB-231 cells depends on the catalytic activity of ATX. To provide further evidence to support this conclusion, we established that VPC8a202 and S32826 inhibited ATX activity under the conditions of the FS-3 fluorescence assay by about 93 and 90%, respectively. We also performed more detailed kinetic studies on the inhibition of ATX activity by VPC8a202 using the natural substrate, LPC. Our results show that VPC8a202 acts as a competitive inhibitor of ATX, as expected from a substrate analogue, with a Ki of about 390 nM (Fig. 4). This result indicates that concentrations of VPC8a202 in the μM range are effective at inhibiting ATX and thus supports our interpretation that the observed migratory effects of LPC reflect the conversion of LPC to LPA by ATX.
FIGURE 4.

Kinetics of inhibition of ATX activity by VPC8a202. The initial rate of recombinant ATX activity was measured colorimetrically at different concentrations of LPC in presence and absence of 0.5 μM VPC8a202. Activity was expressed as the release of choline during 18 h and it is represented by the absorbance of the product at 555 nm (A555). Each point is the average of three measurements (S.D. values for the reaction rates were less than 5% of every measurement and are not depicted). Consumed substrate was less than 10% in every case thus ensuring an initial rate of reaction. Results were fitted to a straight line by linear regression. The apparent Km for LPC was about 588 μM.
To further validate the results with the ATX inhibitors on migration, we also knocked down ATX expression using siRNA. Conditioned media from siATX treated cells showed an 85% decrease in ATX activity compared to the siControl (siCTRL) treated cells (Fig. 5A). Furthermore, expression of the ATX protein in conditioned media from siATX treated cells was markedly reduced to levels that were undetectable by Western blotting (Fig. 5B).
FIGURE 5.
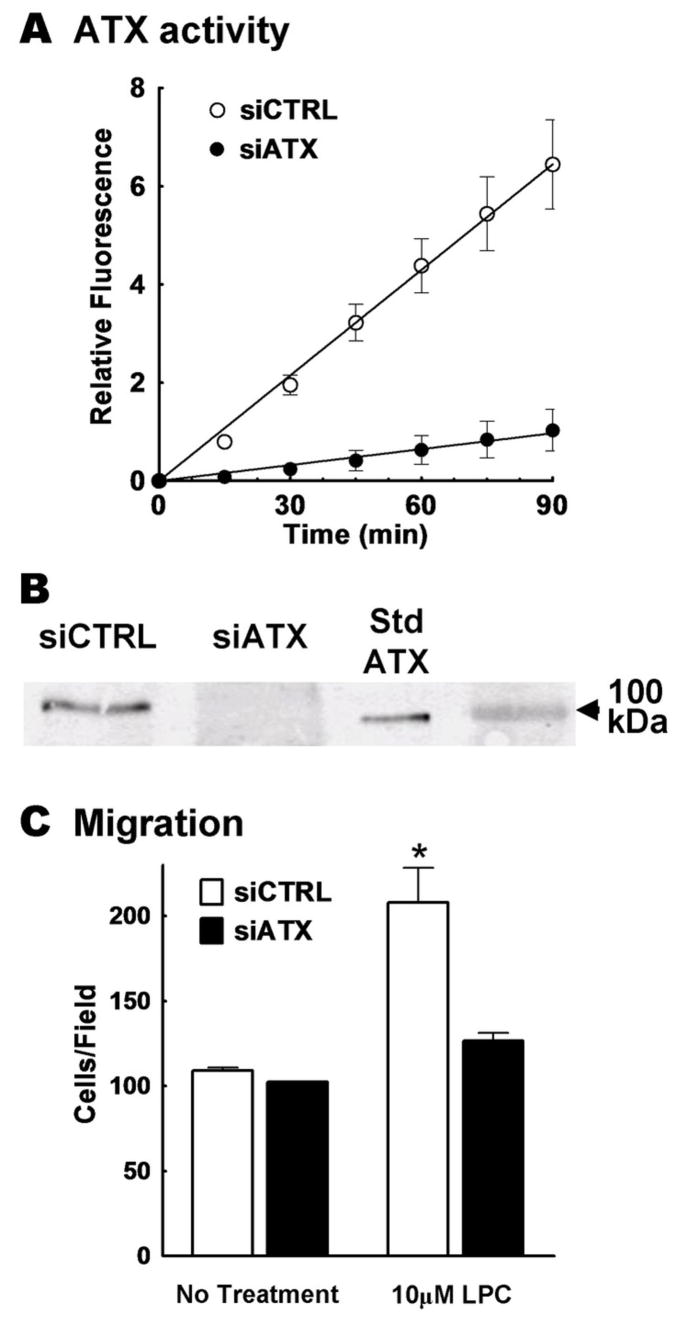
Knock-down of ATX with siRNA significantly decreases the migratory potential of concentrated medium from MDA-MB-435 cells. Concentrated medium was collected from 3 dishes of MDA-MB-435 cells treated with siRNA for ATX (siATX) and 3 dishes treated with control siRNA (siCTRL). Panel A shows a fluorescence assay representing ATX activity in siCTRL concentrated medium (○) and in siATX concentrated medium (●). Error bars indicate the SEM of 3 experimental values. Panel B, shows the Western Blot analysis of a representative sample of concentrated media that were probed for ATX. Recombinant ATX was used as a standard (StdATX). Panel C compares the migration of MDA-MB-435 cells over a 3 h incubation in the presence or absence of LPC and concentrated medium collected from MDA-MB-435 cells that were treated with siCTRL (white bars) or siATX (dark bars). Results are means ± SEM from 3 independent experiments. Statistically significant differences are indicated by * (p < 0.01).
There was no stimulation of the migration of MDA-MB-435 cells when we added only concentrated media from cells treated with siATX, or siCTRL. Also, addition of LPC with concentrated media from siATX-treated cells did not cause any significant stimulation of migration. By contrast, addition of LPC to concentrated medium from cells treated with the siCTRL elicited a migratory response (Fig. 5C).
We also tested the stimulation of migration by LPC, or LPA on the siRNA treated cells directly, but using a 6 h incubation for the migration assay. The siCTRL and the siATX treated cells migrated to the same extent in the absence of agonist. Addition of 10 μM LPC stimulated the migration of siCTRL treated cells, but not siATX-treated cells. As a control, we showed that the siATX treated cells still showed a normal migration with 0.5 μM LPA. The results emphasize that these cells are not migration deficient, but simply no longer respond to LPC (Fig. 6).
FIGURE 6.

Knockdown of ATX with siRNA in MDA-MB-435 cells abolishes stimulation of migration by lysophosphatidylcholine. A 6 h incubation was used to measure the migration of MDA-MB-435 cells that were treated with siATX or siCTRL. Migration in basal media (RPMI 1640 with 0.1% BSA and 0.2% FBS-C) was compared to migration in the presence of 10 μM LPC, or 0.5 μM LPA. Results are means ± ranges from 2 independent experiments.
These results establish that the LPC effect in stimulating cell migration is dependent on the catalytic activity of ATX and LPA production, which could then stimulate migration through LPA receptors. Support for this idea is presented in Fig. 7, which shows that there was a significant decrease in migration when 1 μM VPC32183, an LPA1/3 receptor antagonist (40), was added to the concentrated media alone. Also, the migration obtained with LPC in the presence of concentrated medium from MDA-MB-435 cells was abolished by VPC32183. Likewise, the migratory stimulus provided by LPA was greatly reduced by the addition of VPC32183.
FIGURE 7.

The LPA receptor antagonist VPC32183 inhibits migration of MDA-MB-435 cells. A 3 h incubation was used to measure the migration of MDA-MB-435 cells in the presence of their own concentrated medium with either LPC or LPA, and in the presence or absence of the LPA1/3 receptor antagonist, VPC32183. Migration in the presence of agonists/antagonists was compared to the number of cells migrating in basal media (RPMI 1640 with 0.1% BSA and 0.2% FBS-C), which is given a value of 1. Results are means ± SEM from at least 3 independent experiments. Statistically significant differences are indicated by: * (p < 0.01).
Discussion
Increased expression of ATX has been linked to several malignancies such as glioblastoma, lung, breast, renal, ovarian, and thyroid cancers (6–10, 23). In this paper, we employed MDA-MB-435 melanoma cells and MDA-MB-231 breast cancer cells, which are both able to migrate well in response to serum, or LPA. We found that MDA-MB-435 cells secrete abundant ATX, whereas ATX secretion from MDA-MB-231 was barely detectable. Significantly, MDA-MB-435 cells migrated in response to LPC, whereas MDA-MB-231 did not. We, therefore, hypothesized that LPC does not induce migration of these cells per se, but rather its effects depend on its conversion to LPA by ATX. Therefore, decreased expression or inhibition of ATX should diminish the LPC response.
We verified this hypothesis as follows: a) addition of medium from MDA-MB-435, but not from MDA-MB-231 cells, enabled MDA-MB-231 cells to migrate in response to LPC; b) this effect was abolished by the suppression of ATX expression by siRNA for ATX, c) inhibition of ATX activity with two different ATX inhibitors, VPC32183 and S32826, blocked the stimulatory effect of LPC on migration and d) LPC-induced migration was inhibited by an LPA1/3 receptor antagonist. These results also establish that the main action of ATX on migration in MDA-MB-435 and MDA-MB-231 cells is through the conversion of LPC to LPA, rather than a non-catalytically action of its C-terminal region, which can modify cell adhesion.
Although LPC has been widely described to stimulate cell activation and migration directly through its putative action on G2A and GPR4 receptors (17), much of this work is now in question. G2A and GPR4 are now considered to be proton-sensing receptors whose response is negatively regulated by LPC (41, 42). We found no evidence that LPC itself is able to stimulate migration, but rather its action depends on conversion to LPA by ATX. We also concluded from a separate study that ATX activity is required for LPC to protect MCF-7 breast cancer cells against Taxol-induced apoptosis (N Samadi, C Gaetano, IS Goping and DN Brindley, unpublished results).
LPC is found in blood and extracellular fluids at concentration up to 200 μM. Secretion of ATX into the interstitial fluid surrounding a tumor can convert the relatively abundant LPC, which does not stimulate migration, into the potent bioactive modulator, LPA. This lipid provides a survival signal for cancer cells and it causes them to migrate and metastasize. This is consistent with the fact that high levels of ATX are often found in aggressive cancers. Our work demonstrates that inhibition of ATX expression, or blocking its catalytic activity, could provide very powerful tools to improve the efficacy of surgery and chemotherapy in the treatment of metastatic disease in patients where ATX expression is high. It is, therefore, hoped that our work will lead to the development of ATX inhibitors that can be used therapeutically as an adjuvant in the treatment of cancer.
Materials and Methods
Materials
Mouse anti-GAPDH was from Sigma-Aldrich (Oakville, ON, Canada). Rabbit anti-ATX and recombinant ATX were gifts from Dr. T. Clair (National Cancer Institute, Bethesda, MA). Secondary antibodies were: AlexaFluor© 680 goat anti-mouse IgG, A-21057 was from Invitrogen (Carlsbad, CA); IRDye 800 goat anti-rabbit IgG (Rockland Immunochemicals, Philadelphia, PA). VPC32183 was purchased from Avanti Polar Lipids (Alabaster, AL). Oleoyl-L-α-lysophosphatidic acid, sodium salt (LPA), oleoyl-L-α-lysophosphatidylcholine (LPC), fatty acid-free bovine serum albumin, activated charcoal (Norit®), Hoechst33258, horseradish peroxidase, choline oxidase, 4-aminoantipyrine, N-ethyl-N-(2-hydroxy-3- sulfopropyl)-m-toluidine and fibronectin were purchased from Sigma-Aldrich (Oakville, ON, Canada and St. Louis, MO). Fetal bovine serum (FBS) was from Medicorp, Inc. (Montréal, PQ, Canada). The ATX inhibitors, VPC8a202 and S32826 were synthesized as described previously (38, 39). Transwells® (polycarbonate, 13mm D, 123m pore size) were obtained from Corning, Corning, NY. On the day before the migration assays they were coated with 120 μL of 0.15 mg/mL fibronectin and allowed to dry.
Cell Culture
MDA-MB-435 and MDA-MB-231 cells were obtained from the American Type Culture Collection. The cells were cultured in RPMI 1640 medium (GIBCO) supplemented with 10% FBS and an antibiotic/antimycotic cocktail (penicillin, streptomycin, amphotericin B) (Invitrogen) at 5% CO2, 95% humidity, and 37 °C.
Collection and Concentration of Conditioned Media
Equal numbers of cells were plated onto 10 cm dishes and they were grown until 90% confluence. Dishes were washed and then 10 mL of RPMI 1640 containing 0.1% BSA (≥96% fatty acid free) was added. After 24 h cells were washed and 10mL of RPMI 1640 was added. The dishes were incubated for a further 24 h, the conditioned media was collected and cells on each dish were counted. Collected conditioned media was then centrifuged at 1500 × g for 10 min to remove whole cells and debris and it was then stored at −20 °C. Conditioned media was concentrated approximately 20- to 30-fold using Centricon® YM-10 centrifugal filter devices (Millipore). Filters were pre-rinsed with 2 mL of water to remove glycerine and samples were centrifuged at 5000 × g for 120 min, or until the desired final volume was obtained. Filters were then inverted and centrifuged at 1500 × g for 2 min to collect concentrated media. The final volume was determined by weighing.
mRNA Expression
Total RNA was extracted from cell lysates using the RNAqueous kit (Ambion) according the manufacture’s instruction. Extracted RNA was treated with DNAase (DNA-free kit, Ambion). RNA reverse transcription reaction was performed with Superscript II reverse transcriptase (Invitrogen) in the presence of random primers according to the manufacturer’s instructions. A control reaction without reverse transcriptase was performed in parallel to detect genomic DNA contamination. cDNA was calculated assuming 100% conversion from RNA. Real time-RT-PCR was performed by mixing 25 μl of master mix containing 2 × SYBR Green buffer mix and forward and reverse primers (Invitrogen) to a 3.5 μl of sample of cDNA in 96 well plates. A 316 bp fragment of the constitutively expressed housekeeping human glyceraldehyde 3-phospate dehydrogenase (GAPDH) was used to normalize the expression of ATX mRNA. Primers for human ATX were: sense, 5′-ACAACGAGGAGAGCTGCAAT-3′, and anti-sense, 5′-AGAAGTCCAGGCTGGTGAGA-3′. Primers for human GAPDH were: sense, 5′-ACAGTCAGCCGCATCTTCTT-3′ and antisense, 5′-GACAAGCTTCCCGTTCTCAG-3′. Samples of cDNA were assayed in triplicates on the 7500 Real Time PCR System (Applied Biosystems). The transcript number of human GAPDH was quantified, and each sample was normalized on the basis of GAPDH mRNA content.
Boyden Chamber Assay for Migration
Two million cells were seeded in a 25 cm2 flask with growth medium for at least 48 h. Starvation medium (RPMI 1640 containing 0.1% BSA, ≥96% fatty acid free) was added for the last 18 h before the migration experiment. Cells were washed twice with HBS, trypsinized, and collected in starvation media and then 1 mL of 0.1% trypsin inhibitor solution in HBS was added. Cells were centrifuged down, media were removed by aspiration, and cells were resuspended in starvation media and counted. Cells (300,000) were seeded into each of the fibronectin coated Transwell® filters and incubated for 45 min for attachment. Filters were then transferred into bottom chambers that contained 1.5 mL of starvation medium with 0.2% charcoal-treated FBS (FBS-C) and various agonists. This charcoal treatment removed >95% of the LPA as assessed after spiking the FBS with 32P-labeled LPA. This means that the concentration of LPA added in the diluted serum should have been <1 nM. Chambers were incubated at 37 °C and cells were allowed to migrate through the pores in the filter for 3 or 6 h. The basal level of migration was measured with 0.2% FBS-C in the bottom chamber and 5% FBS was used as a positive control for stimulated migration. After migration, cells were then fixed in 5% formaldehyde for 1 h, or overnight. Filters were then rinsed in water and placed in 1 μg/mL Hoechst 33258 stain for 2 h. Then the upper surface of the filters was cleaned with a moist cotton swab to remove cells that had not migrated. Filters were washed again and placed in PBS. Four to six random fields were photographed under a Leica DM IRB fluorescence microscope at 40 X magnification without knowing the treatment and the average numbers of cells per field was calculated.
Western Blot Analysis
Concentrated conditioned media were analyzed by SDS-PAGE according to Laemmli (43). Proteins were transferred at 450 mA for 5 h onto nitrocellulose membranes (Trans-Blot® Transfer Medium, Bio-Rad) in transfer buffer (192 mM glycine, 24 mM Tris base, pH 8.5 and 20% (v/v) ethanol). Membranes were blocked in 50% PBS and 50% Odyssey™ blocking buffer (Li-Cor Biosciences, Lincoln, NE) for a minimum of 1.5 h. All antibodies were diluted in 50/50 PBS/Odyssey™ blocking buffer with 0.1% (v/v) Tween 20. Membranes were incubated with anti-ATX (rabbit, 1:10,000) for 1 h. Membranes were then washed 4 times with PBS containing 0.1% Tween-20 and incubated in IRDye 800 goat anti-rabbit IgG (Rockland Immunochemicals, Philadelphia, PA, 1:10,000) for 1 h and washed 3 times in wash buffer and once in PBS alone. Membranes were scanned using the Odyssey™ Imager (Li-Cor).
Autotaxin Activity Assay
The assay of ATX activity in cells was based on the method described by Ferguson et al. (44) using a fluorogenic phospholipid ATX substrate, FS-3 (Echelon Biosciences, Salt Lake City, UT). FS-3 was diluted to 3.1 μM in a solution containing: 140 mM NaCl, 5 mM KCl, 1mM CaCl2, 1mM MgCl2, 50 mM Tris-HCl, pH 8.0, and 1 mg/mL BSA. The solution was heated at 60 °C for 10 min to destroy any enzymatic activity in the BSA and then cooled to 37 °C before use. Forty μl of FS-3 solution was added to 10 μl of cell lysate, or concentrated conditioned media in a black-wall, clear-bottom 96 well Costar® half-area plate. Measurements were then taken at appropriate intervals using a Fluoroskan Ascent fluorometer (Thermo Lab Systems) at an excitation wavelength of 485 nm and an emission wavelength of 527 nm.
For the kinetic studies, human recombinant ATX was subcloned into the mammalian expression vector cDNA3.1/V5His-TOPO (Invitrogen) and expressed as a C-terminus V5- and 6xHis-tagged protein in HEK-293 cells using PolyFect® (Qiagen) as a transfection reagent. ATX was purified from the culture medium using a nickel-Sepharose resin (Qiagen) according to manufacturer’s instructions and the buffer was changed to PBS using 30 kDa cutoff Centricon tubes (Millipore). ATX DNA was generated from an EST I.M.A.G.E. clone 5174518 using the following forward and reverse primers 5‚-CGC GCT AGC ATG GCA AGG AGG AGC TCG TTC-3‚; 5‚-AAT CTC GCT CTC ATA TGT ATG CAG-3‚ to amplify the ATX ORF. ATX activity was measured essentially as described by Umezu-Goto et al. (7) by determining the release of choline after incubation at 37 °C for 18 h in 100 μl of a buffer consisting of 100 mM Tris-HCl, pH 9.0, 500 mM NaCl, 5 mM MgCl2, 30 μM CoCl2, 0.05% Triton X-100 0.5 μM VPC8a202 and various concentrations of oleoyl-LPC (Avanti Polar Lipids, Alabaster, AL). Choline was detected colorimetrically at 555 nm after adding 100 μl of 50 mM Tris-HCl, pH 8.0, 5 mM MgCl2, 50 U/ml horseradish peroxidase, 18 U/ml choline oxidase, 5 mM 4- aminoantipyrine, and 3 mM N-ethyl-N-(2-hydroxy-3-sulfopropyl)-m-toluidine.
Knockdown of Autotaxin Expression Using siRNA
Knockdown of ATX was achieved using SMARTpool® siRNAs (Dharmacon Inc., Lafayette CO). About 800,000 cells were plated on 10 cm dishes with 15 mL of antibiotic-free RPMI 1640 containing 10% FBS. Cells were grown for two days until about 50% confluency. Before transfection, the medium was replaced with 7 mL of fresh antibiotic-free media. Ten μl of stock siRNA (50μM) was diluted in 1.5 mL of Opti-MEM Reduced Serum Medium (Invitrogen Life Technologies, Carlsbad CA). In a separate tube, 30 μl of Lipofectamine 2000 (Invitrogen) was mixed with 1.5 mL Opti-MEM and incubated at room temperature for 15 min. The siRNA and the Lipofectamine solutions were then combined and incubated for another 15 min at room temperature. Each dish of cells received 3 mL of the siRNA-Lipofectamine 2000 complex that was added drop-wise while swirling the dish. The final concentrations of Lipofectamine 2000 and siRNA were 1.4 μg/mL and 50 nM respectively. Cells were then incubated for 24 h at 37°C and the medium was collected as described above. Cells on each dish were trypsinized and counted so that equivalent amounts of concentrated media could be used in the migration assays.
Statistics
Results were presented as means ± SEM from at least 3 independent experiments, unless otherwise indicated. Statistical differences were calculated using GraphPad 4 software (Prism) by ANOVA with a Newman-Keuls post-hoc test and paired T-tests.
Acknowledgments
We thank Mr. J Dewald for excellent technical assistance, Drs F. Bamforth and GS Cembrowski and for their support of this study. We also thank Dr T. Clair for the production of recombinant ATX and the ATX antibody, and Dr JA Boutin (Institut de Recherches Servier), for supplying S32826. DNB is a recipient of a Medical Scientist Award from the Alberta Heritage Foundation for Medical Research. NS is a recipient of scholarships from Iranian Ministry of Health and the Bell McLeod Educational Fund from Department of Laboratory Medicine and Pathology, University of Alberta.
Grant support: This work was supported by grants to DNB from the Canadian Institute of Health Research (MOP 81137) and from NIH (R01 GM052722) to KRL. The provision of recombinant ATX and ATX antibody through Dr T. Clair was made possible by the Intramural Research Program of the NIH, National Cancer Institute, Center for Cancer Research.
Abbreviations
- ATX
autotaxin
- LPA
lysophosphatidate
- LPC
lysophosphatidylcholine
References
- 1.Yang SY, Lee J, Park CG, Kim S, Hong S, Chung HC, Min SK, Han JW, Lee HW, Lee HY. Expression of autotaxin (NPP-2) is closely linked to invasiveness of breast cancer cells. Clin Exp Metastasis. 2002;19:603–608. doi: 10.1023/a:1020950420196. [DOI] [PubMed] [Google Scholar]
- 2.Stracke ML, Krutzsch HC, Unsworth EJ, Arestad A, Cioce V, Schiffmann E, Liotta LA. Identification, purification, and partial sequence analysis of autotaxin, a novel motility-stimulating protein. J Biol Chem. 1992;267:2524–2529. [PubMed] [Google Scholar]
- 3.Kishi Y, Okudaira S, Tanaka M, Hama K, Shida D, Kitayama J, Yamori T, Aoki J, Fujimaki T, Arai H. Autotaxin is overexpressed in glioblastoma multiforme and contributes to cell motility of glioblastoma by converting lysophosphatidylcholine to lysophosphatidic acid. J Biol Chem. 2006;281:17492–17500. doi: 10.1074/jbc.M601803200. [DOI] [PubMed] [Google Scholar]
- 4.Euer N, Schwirzke M, Evtimova V, Burtscher H, Jarsch M, Tarin D, Weidle UH. Identification of genes associated with metastasis of mammary carcinoma in metastatic versus non-metastatic cell lines. Anticancer Res. 2002;22:733–740. [PubMed] [Google Scholar]
- 5.Stassar MJ, Devitt G, Brosius M, Rinnab L, Prang J, Schradin T, Simon J, Petersen S, Kopp-Schneider A, Zoller M. Identification of human renal cell carcinoma associated genes by suppression subtractive hybridization. Br J Cancer. 2001;85:1372–1382. doi: 10.1054/bjoc.2001.2074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Kehlen A, Englert N, Seifert A, Klonisch T, Dralle H, Langner J, Hoang-Vu C. Expression, regulation and function of autotaxin in thyroid carcinomas. Int J Cancer. 2004;109:833–838. doi: 10.1002/ijc.20022. [DOI] [PubMed] [Google Scholar]
- 7.Umezu-Goto M, Kishi Y, Taira A, Hama K, Dohmae N, Takio K, Yamori T, Mills GB, Inoue K, Aoki J, Arai H. Autotaxin has lysophospholipase D activity leading to tumor cell growth and motility by lysophosphatidic acid production. J Cell Biol. 2002;158:227–233. doi: 10.1083/jcb.200204026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.So J, Wang FQ, Navari J, Schreher J, Fishman DA. LPA-induced epithelial ovarian cancer (EOC) in vitro invasion and migration are mediated by VEGF receptor-2 (VEGF-R2) Gynecol Oncol. 2005;97:870–878. doi: 10.1016/j.ygyno.2005.03.004. [DOI] [PubMed] [Google Scholar]
- 9.Nam SW, Clair T, Campo CK, Lee HY, Liotta LA, Stracke ML. Autotaxin (ATX), a potent tumor motogen, augments invasive and metastatic potential of ras-transformed cells. Oncogene. 2000;19:241–247. doi: 10.1038/sj.onc.1203263. [DOI] [PubMed] [Google Scholar]
- 10.Nam SW, Clair T, Kim YS, McMarlin A, Schiffmann E, Liotta LA, Stracke ML. Autotaxin (NPP-2), a metastasis-enhancing motogen, is an angiogenic factor. Cancer Res. 2001;61:6938–6944. [PubMed] [Google Scholar]
- 11.Hama K, Aoki J, Fukaya M, Kishi Y, Sakai T, Suzuki R, Ohta H, Yamori T, Watanabe M, Chun J, Arai H. Lysophosphatidic acid and autotaxin stimulate cell motility of neoplastic and non-neoplastic cells through LPA1. J Biol Chem. 2004;279:17634–17639. doi: 10.1074/jbc.M313927200. [DOI] [PubMed] [Google Scholar]
- 12.Fox MA, Alexander JK, Afshari FS, Colello RJ, Fuss B. Phosphodiesterase-I alpha/autotaxin controls cytoskeletal organization and FAK phosphorylation during myelination. Mol Cell Neurosci. 2004;27:140–150. doi: 10.1016/j.mcn.2004.06.002. [DOI] [PubMed] [Google Scholar]
- 13.Dennis J, Nogaroli L, Fuss B. Phosphodiesterase-Ialpha/autotaxin (PD-Ialpha/ATX): a multifunctional protein involved in central nervous system development and disease. J Neurosci Res. 2005;82:737–742. doi: 10.1002/jnr.20686. [DOI] [PubMed] [Google Scholar]
- 14.Tokumura A, Majima E, Kariya Y, Tominaga K, Kogure K, Yasuda K, Fukuzawa K. Identification of human plasma lysophospholipase D, a lysophosphatidic acid-producing enzyme, as autotaxin, a multifunctional phosphodiesterase. J Biol Chem. 2002;277:39436–39442. doi: 10.1074/jbc.M205623200. [DOI] [PubMed] [Google Scholar]
- 15.Brindley DN. Hepatic secretion of lysphosphatidylcholine: a novel transport system for polyunsaturated fatty acids and choline. J Nutr Biochem. 1993;4:442–449. [Google Scholar]
- 16.Aoki J, Taira A, Takanezawa Y, Kishi Y, Hama K, Kishimoto T, Mizuno K, Saku K, Taguchi R, Arai H. Serum lysophosphatidic acid is produced through diverse phospholipase pathways. J Biol Chem. 2002;277:48737–48744. doi: 10.1074/jbc.M206812200. [DOI] [PubMed] [Google Scholar]
- 17.Radu CG, Yang LV, Riedinger M, Au M, Witte ON. T cell chemotaxis to lysophosphatidylcholine through the G2A receptor. Proc Natl Acad Sci U S A. 2004;101:245–250. doi: 10.1073/pnas.2536801100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Kim KS, Ren J, Jiang Y, Ebrahem Q, Tipps R, Cristina K, Xiao YJ, Qiao J, Taylor KL, Lum H, Anand-Apte B, Xu Y. GPR4 plays a critical role in endothelial cell function and mediates the effects of sphingosylphosphorylcholine. Faseb J. 2005;19:819–821. doi: 10.1096/fj.04-2988fje. [DOI] [PubMed] [Google Scholar]
- 19.Zhu K, Baudhuin LM, Hong G, Williams FS, Cristina KL, Kabarowski JH, Witte ON, Xu Y. Sphingosylphosphorylcholine and lysophosphatidylcholine are ligands for the G protein-coupled receptor GPR4. J Biol Chem. 2001;276:41325–41335. doi: 10.1074/jbc.M008057200. [DOI] [PubMed] [Google Scholar]
- 20.Rikitake Y, Hirata K, Yamashita T, Iwai K, Kobayashi S, Itoh H, Ozaki M, Ejiri J, Shiomi M, Inoue N, Kawashima S, Yokoyama M. Expression of G2A, a receptor for lysophosphatidylcholine, by macrophages in murine, rabbit, and human atherosclerotic plaques. Arterioscler Thromb Vasc Biol. 2002;22:2049–2053. doi: 10.1161/01.atv.0000040598.18570.54. [DOI] [PubMed] [Google Scholar]
- 21.Kabarowski JH, Zhu K, Le LQ, Witte ON, Xu Y. Lysophosphatidylcholine as a ligand for the immunoregulatory receptor G2A. Science. 2001;293:702–705. doi: 10.1126/science.1061781. [DOI] [PubMed] [Google Scholar]
- 22.Lin P, Ye RD. The lysophospholipid receptor G2A activates a specific combination of G proteins and promotes apoptosis. J Biol Chem. 2003;278:14379–14386. doi: 10.1074/jbc.M209101200. [DOI] [PubMed] [Google Scholar]
- 23.Mills GB, Moolenaar WH. The emerging role of lysophosphatidic acid in cancer. Nat Rev Cancer. 2003;3:582–591. doi: 10.1038/nrc1143. [DOI] [PubMed] [Google Scholar]
- 24.Brindley DN. Lipid phosphate phosphatases and related proteins: signaling functions in development, cell division, and cancer. J Cell Biochem. 2004;92:900–912. doi: 10.1002/jcb.20126. [DOI] [PubMed] [Google Scholar]
- 25.Lynch KR. Lysophospholipid receptor nomenclature. Biochim Biophys Acta. 2002;1582:70–71. doi: 10.1016/s1388-1981(02)00138-5. [DOI] [PubMed] [Google Scholar]
- 26.Lee CW, Rivera R, Gardell S, Dubin AE, Chun J. GPR92 as a New G12/13-and Gq-coupled Lysophosphatidic Acid Receptor That Increases cAMP, LPA5. J Biol Chem. 2006;281:23589–23597. doi: 10.1074/jbc.M603670200. [DOI] [PubMed] [Google Scholar]
- 27.Pasternack SM, von Kugelgen I, Aboud KA, Lee YA, Ruschendorf F, Voss K, Hillmer AM, Molderings GJ, Franz T, Ramirez A, Nurnberg P, Nothen MM, Betz RC. G protein-coupled receptor P2Y5 and its ligand LPA are involved in maintenance of human hair growth. Nat Genet. 2008;40:329–334. doi: 10.1038/ng.84. [DOI] [PubMed] [Google Scholar]
- 28.Xu Y, Fang XJ, Casey G, Mills GB. Lysophospholipids activate ovarian and breast cancer cells. Biochem J. 1995;309( Pt 3):933–940. doi: 10.1042/bj3090933. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Xu Y, Gaudette DC, Boynton JD, Frankel A, Fang XJ, Sharma A, Hurteau J, Casey G, Goodbody A, Mellors A, et al. Clin Cancer Res. 1995;1:1223–1232. [PubMed] [Google Scholar]
- 30.Van Leeuwen FN, Olivo C, Grivell S, Giepmans BN, Collard JG, Moolenaar WH. Rac activation by lysophosphatidic acid LPA1 receptors through the guanine nucleotide exchange factor Tiam1. J Biol Chem. 2003;278:400–406. doi: 10.1074/jbc.M210151200. [DOI] [PubMed] [Google Scholar]
- 31.Shida D, Kitayama J, Yamaguchi H, Okaji Y, Tsuno NH, Watanabe T, Takuwa Y, Nagawa H. Lysophosphatidic acid (LPA) enhances the metastatic potential of human colon carcinoma DLD1 cells through LPA1. Cancer Res. 2003;63:1706–1711. [PubMed] [Google Scholar]
- 32.Yamada T, Sato K, Komachi M, Malchinkhuu E, Tobo M, Kimura T, Kuwabara A, Yanagita Y, Ikeya T, Tanahashi Y, Ogawa T, Ohwada S, Morishita Y, Ohta H, Im DS, Tamoto K, Tomura H, Okajima F. Lysophosphatidic acid (LPA) in malignant ascites stimulates motility of human pancreatic cancer cells through LPA1. J Biol Chem. 2004;279:6595–6605. doi: 10.1074/jbc.M308133200. [DOI] [PubMed] [Google Scholar]
- 33.Sun B, Nishihira J, Suzuki M, Fukushima N, Ishibashi T, Kondo M, Sato Y, Todo S. Induction of macrophage migration inhibitory factor by lysophosphatidic acid: relevance to tumor growth and angiogenesis. Int J Mol Med. 2003;12:633–641. [PubMed] [Google Scholar]
- 34.Murph MM, Hurst-Kennedy J, Newton V, Brindley DN, Radhakrishna H. Lysophosphatidic acid decreases the nuclear localization and cellular abundance of the p53 tumor suppressor in A549 lung carcinoma cells. Mol Cancer Res. 2007;5:1201–1211. doi: 10.1158/1541-7786.MCR-06-0338. [DOI] [PubMed] [Google Scholar]
- 35.Xie Y, Meier KE. Lysophospholipase D and its role in LPA production. Cell Signal. 2004;16:975–981. doi: 10.1016/j.cellsig.2004.03.005. [DOI] [PubMed] [Google Scholar]
- 36.Fourcade O, Simon MF, Viode C, Rugani N, Leballe F, Ragab A, Fournie B, Sarda L, Chap H. Secretory phospholipase A2 generates the novel lipid mediator lysophosphatidic acid in membrane microvesicles shed from activated cells. Cell. 1995;80:919–927. doi: 10.1016/0092-8674(95)90295-3. [DOI] [PubMed] [Google Scholar]
- 37.Rae JM, Creighton CJ, Meck JM, Haddad BR, Johnson MD. MDA-MB-435 cells are derived from M14 melanoma cells--a loss for breast cancer, but a boon for melanoma research. Breast Cancer Res Treat. 2007;104:13–19. doi: 10.1007/s10549-006-9392-8. [DOI] [PubMed] [Google Scholar]
- 38.Cui P, Tomsig JL, McCalmont WF, Lee S, Becker CJ, Lynch KR, Macdonald TL. Synthesis and biological evaluation of phosphonate derivatives as autotaxin (ATX) inhibitors. Bioorg Med Chem Lett. 2007;17:1634–1640. doi: 10.1016/j.bmcl.2006.12.114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Ferry G, Moulharat N, Pradère J-P, Desos P, Try A, Genton A, Giganti A, Beucher-Gaudin M, Lonchampt M, Bertrand M, Saulnier-Blache J-S, Tucker GC, Cordi A, Boutin JA. S32826: A nonomolar inhibitor of autotaxin. Discovery, synthesis and applications as a pharmacological tool. J Pharmacol Expt Therap. 2008 doi: 10.1124/jpet.108.141911. In press. [DOI] [PubMed] [Google Scholar]
- 40.Lee S, Lynch KR. Brown recluse spider (Loxosceles reclusa) venom phospholipase D (PLD) generates lysophosphatidic acid (LPA) Biochem J. 2005;391:317–323. doi: 10.1042/BJ20050043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Seuwen K, Ludwig MG, Wolf RM. Receptors for protons or lipid messengers or both? J Recept Signal Transduct Res. 2006;26:599–610. doi: 10.1080/10799890600932220. [DOI] [PubMed] [Google Scholar]
- 42.Murakami N, Yokomizo T, Okuno T, Shimizu T. G2A is a proton-sensing G-protein-coupled receptor antagonized by lysophosphatidylcholine. J Biol Chem. 2004;279:42484–42491. doi: 10.1074/jbc.M406561200. [DOI] [PubMed] [Google Scholar]
- 43.Laemmli UK. Cleavage of structural proteins during the assembly of the head of bacteriophage T4. Nature. 1970;227:680–685. doi: 10.1038/227680a0. [DOI] [PubMed] [Google Scholar]
- 44.Ferguson CG, Bigman CS, Richardson RD, van Meeteren LA, Moolenaar WH, Prestwich GD. Fluorogenic phospholipid substrate to detect lysophospholipase D/autotaxin activity. Org Lett. 2006;8:2023–2026. doi: 10.1021/ol060414i. [DOI] [PMC free article] [PubMed] [Google Scholar]