

Abstract
The preparation of diffraction quality crystals remains the major bottleneck in macromolecular x-ray crystallography. A crystallization chaperone is an auxiliary protein, such as fragments of monoclonal antibodies, that binds to and increases the crystallization probability of a target molecule of interest. Such chaperones reduce conformational heterogeneity, mask counterproductive surfaces while extending surfaces predisposed to forming crystal contacts, and provide phasing information. Crystallization chaperones generated using recombinant technologies have emerged as superior alternatives that increase the throughput and eliminate inherent limitations associated with antibody production by animal immunization and the hybridoma technology.
Introduction
The major impediment to X-ray crystallography approaches in structural biology remains the production of high quality diffracting crystals. This situation is not the result of any lack of effort to produce new tools or methods to assist in crystallization. Despite extensive efforts using multiple strategies, the process of crystallization remains difficult. The probability of success is affected by many factors including the level of inherent conformational heterogeneity of the protein and the chemical character of the molecule's surfaces that often limits the number of productive lattice contacts available for crystallization.
Because crystallization involves an unfavorable loss of conformational entropy in the molecule to be assembled in the crystal lattice, methods that reduce the conformational entropy of the target while still in solution should enhance the likelihood of crystallization by lowering the net entropic penalty of lattice formation. The “surface entropy reduction” approach has proved to be highly effective [1]. Likewise, binding partners such as ions, small molecule ligands and peptides can reduce the conformational heterogeneity by binding to and stabilizing a subset of conformational states of a protein. Although such binding partners are effective, not all proteins have a known binding partner, and even when a binding partner is known, its affinity, solubility and chemical stability may not be compatible with crystallization trials.
An approach that holds promise, especially for membrane proteins and large protein complexes, is the use of crystallization chaperones. These crystallization chaperones come in the form of antibody fragments or other proteins that have been engineered to bind specifically to a given macromolecular target. The basis for the strategy is to increase the probability of obtaining well ordered crystals by (i) minimizing the conformational heterogeneity in the target by binding to a specific conformation and (ii) supplementing the amount of protein surface that can facilitate primary contacts between molecules in the crystal lattice. An additional attribute inherent in the crystallization chaperone approach is that the chaperone can provide initial model-based phasing information.
The idea of using engineered binding proteins as crystallization chaperones is not new [2,3]. Over a decade ago, fragments of monoclonal antibodies were used as crystallization chaperones [3-5]. This approach has been particularly effective in the determination of high-impact structures of membrane proteins [4]. Recently, the human β2-adrenergic G-protein-coupled receptor was crystallized as a complex with the antigen-binding fragment (Fab) derived from a monoclonal antibody (Figure 2a) [6,7]. The structure revealed that the Fab chaperone binds to an intracellular loop and a transmembrane helix in a conformationally specific manner. In these cases, crystallization chaperones seem to be able to stabilize detergent-solubilized membrane proteins, to reduce their conformational heterogeneity and to extend hydrophilic surfaces that can form effective crystal contacts.
Figure 2.
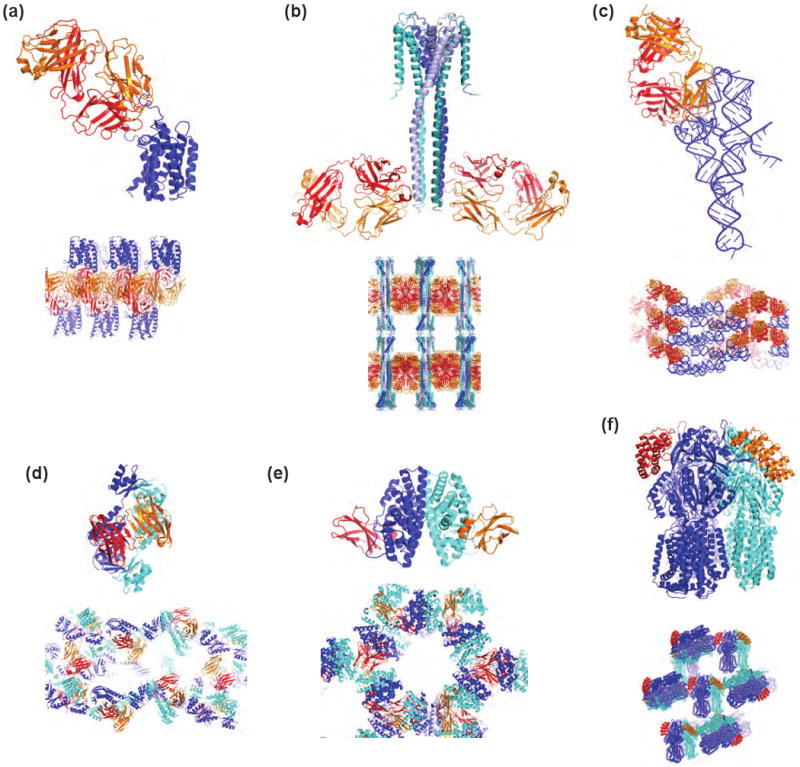
Recent structures determined using crystallization chaperones. (a) β2-adrenergic receptor with Fab (2R4R). (b) Full-length KcsA with a synthetic Fab (3EFF). (c) The P4P6 domain of group I intron with a synthetic Fab. (d) GspD with VHH (3EDJ). (e) Estrogen receptor ligand-binding domain with FN3 (2OCF). (f) AcrB with DARPIN (2J8S).
In addition to membrane proteins, Fab-assisted crystallography has been successfully applied to proteins that are too soluble to form crystals. Borrelia burgdorferi OspA is an extremely soluble protein, and it crystallized only in the form of Fab complexes [8]. Here, the Fab chaperone masks a large surface area rich in charged and highly flexible side chains (Arg, Glu and Lys) that have low propensities of forming crystal contacts [1].
These examples clearly demonstrate the effectiveness of chaperone-assisted crystallography using monoclonal antibody fragments. Unfortunately, this traditional approach is limited by its expense and throughput, which greatly reduce its potential as a broadly applicable method. Animal immunization is slow and the level of immunogenicity of individual targets is unknown. Only a small number of hybridoma cells can be screened. Further, the production of antibodies at the milligram scale is expensive and monoclonal antibodies need to be further fragmented by proteolysis. Consequently, still only a small number of structures have been determined using this otherwise powerful method.
Semi-synthetic crystallization chaperones
Here, I define “semi-synthetic” as a hybrid of animal immunization and recombinant techniques. Methods have now been well established to clone the cDNA for the Fv and Fab regions of monoclonal antibodies and produce them in E. coli. Indeed, the first application of chaperone-assisted crystallography to membrane proteins was performed using a recombinant Fv [5]. However, this approach has not been widely used, perhaps because it requires expertise and resources in separate areas including monoclonal antibody generation, cDNA cloning and bacterial expression of antibodies.
Another type of semi-synthetic approach has been used effectively for the generation of the antigen-binding domain of the camelid heavy chain antibodies (VHHs, also called “nanobodies”) [9,10]. Unlike conventional antibodies, these antibodies are devoid of the light chain, and the minimal antigen-binding unit is a single immunoglobulin domain of ∼125 residues (Figure 2D). Functionally, VHHs have a strong tendency to bind to preformed clefts, while conventional antibodies do not [11]. Thus Fab (or Fv) and VHH may be used as complementary chaperones that are targeted to distinct types of epitopes.
Typically, target-binding VHHs are identified from a phage-display cDNA library obtained from an immunized animal (llama, dromedary or camel). Phage display can search much larger antibody repertoire in a more expeditious manner than monoclonal antibody screening, significantly increasing the throughput [Sidhu, 2007 #1774]. Furthermore, recombinant expression of VHH is easier than the heterodimeric Fab or Fv. It should be noted that it would not be straightforward to employ an equivalent approach for cloning Fab/Fv fragments of the conventional antibodies because the heavy and light chains are encoded by separate genes and thus one must generate the right pairing of two genes to reconstitute a functional antibody.
Crystallographic studies of VHH-antigen complexes have demonstrated its potential as a crystallization chaperone. In one case, a ribonuclease A (RNaseA)-VHH complex collectively produced six crystal forms, exhibiting monoclinic, orthorhombic, triclinic and tetragonal symmetry and having either one or two complexes in the asymmetric unit [12]. Some of these crystal forms diffracted to an atomic resolution. Interestingly, an affinity-matured variant with sub-nM Kd was a less effective chaperone than another mutant with a ∼100 nM Kd [13], implying that it is not always better to seek the tightest binding chaperones. This study also demonstrated that a Met-enriched VHH chaperone generated by “shotgun Met scanning” with a total of five SeMet could provide abundant phasing power to solve the structure of the complex by the single-anomalous dispersion technique (SAD) without the need for introducing SeMet into the target protein.
Several protein structures have been determined de novo using VHH chaperones [14-17]. A bacterial “addiction antidoze” MazE protein was successfully crystallized as a VHH complex [14]. Remarkably, more than half of MazE is disordered in the crystal, shedding light on the extreme difficulty of crystallizing MazE in isolation. A VHH-EpsI:EpsJ peudopilin heterodimer (a component of the bacterial type 2 secretion systems) complex was crystallized in 15 days compared with 11 months and 17 variants required for crystallization without VHH [17]. In lattices of these two crystal structures, the VHH chaperone formed a layer supporting another layer of the target, thus suggesting VHH's critical role in forming crystal contacts. In the structure of the N-terminal domain of the secretin GspD, two VHHs forms an anti-parallel dimer and each VHH binds to a GspD monomer (Figure 2D) [16]. The resulting heterotetramer appears to reduce the conformational heterogeneity of elongated GspD, leading to successful crystallization. Taken together, these results demonstrate the effectiveness of VHH chaperones in crystallizing challenging targets. More broadly, they suggest that single-domain chaperones much smaller than Fab can be highly effective in this role.
Synthetic antibody chaperones
The crystallization chaperones described above were derived from natural antibodies generated by animal immunization, which exploits the versatility of the immune systems to generate antibodies against diverse antigens and to increase their affinity by somatic hypermutation. Although powerful, it has several shortcomings. One must maintain a group of animals that would eventually be sacrificed. The solvent conditions for an antigen cannot be controlled once it is injected into an animal. This is a major problem if one wishes to maintain the conformational integrity of a detergent-solubilized membrane protein or other type of labile molecule, to keep a weakly associated complex intact or to keep a protein in a specific conformational state (e.g. active versus inactive states).
Recent advances in recombinant technologies have made it possible to establish a fully recombinant method to generate antibody fragments without animal immunization. Such methods can circumvent many of the issues limiting antibodies derived from immunization and thus greatly expand the potential of antibody fragments as crystallization chaperones [18-21]. The technological engines of recombinant antibody generation that substitute for natural immune systems are the so-called molecular display techniques that establish phenotype-genotype linkage of protein variants and enable the generation of large protein libraries [22-24]. Libraries containing sequence diversity on the order of 1010 or even greater can be produced. Cycles of library selection with an immobilized antigen yield variants that bind to the antigen. Because of the phenotype-genotype linkage built in these display systems, the genes encoding selected variants are immediately available for determining sequences and for protein production.
Common formats for antibody display are single-chain Fv (scFv) in which the heavy chain V and light chain L domains are covalently connected with a flexible linker and Fab in which one of the two chains is displayed on the phage (or cell surface) and the other chain is produced as soluble protein and the heavy- and light-chain heterodimer spontaneously forms. While both are effective in identifying binding proteins, Fab has become the preferred platform for generating crystallization chaperones. The main reason for this preference seems to be the superior biophysical properties of Fab to scFv. The two additional immunoglobulin domains (the constant domains) present in the Fab format significantly enhance the stability of the heterodimer. scFv and Fv are more prone to aggregation and denaturation, which is particularly problematic in crystallization applications where proteins are highly concentrated.
Antibody display libraries can be produced using sequence diversity derived from natural sources or from “synthetic” diversity designed from the knowledge of the sequence, structure and evolution of antibodies. Sequence diversity of antibodies is concentrated in the complementarity determining regions (CDRs) that structurally correspond to six loops that collectively form contiguous surfaces for antigen recognition. In principle, with synthetic libraries of sufficient CDR diversity, antibody fragments can be engineered to recognize target molecules. However, in practice, antibody fragments obtained from synthetic libraries have typically had weaker affinity than their counterparts from hybridoma [25]. It should be noted that a typical antigen-binding site involves ∼30 positions of antibody residues and thus the total sequence space for such a binding site far exceeds the size of molecular display libraries. Consequently, it is unlikely that simple random search of the sequence space will produce highly functional antibodies.
A major breakthrough in balancing the requirements for sequence diversity with CDR residue coverage has been made by Sidhu et al. who have developed a series of synthetic antibody libraries based on “reduced genetic codes” and a single Fab framework [26,27]. Bioinformatic analyses of antibody CDR sequences and structures revealed clear biases that favor certain amino acid types, and these biases become even greater at positions that are directly involved in antigen binding [28,29]. Remarkably, Tyr is found to make ∼25% of the contacts to the antigen. Fellouse et al. showed that even a binary amino acid library in which four CDRs contained combinations of only Tyr and Ser produced high-affinity and high-specificity antibodies to several protein targets [30]. Subsequent improvements yielded an antibody library that includes additional diversity in one CDR (CDR-H3) but still otherwise the Tyr/Ser binary code, which produces highly functional synthetic Fabs that bind to a broad range of macromolecules without affinity maturation steps [20].
Synthetic Fabs have been used to determine the structure of the full-length form of the potassium channel KcsA (Figure 2b) [31]. The previously determined KcsA structure is for a truncated form lacking the C-terminal domain (residues 125-160) [32]. The C-terminal domain had been known to modulate KcsA function, but the full-length protein had been recalcitrant to crystallization despite intense effort. The synthetic Fab library of Fellouse et al. [20] was sorted using detergent-solubilized and biotinylated KcsA samples as the antigen. In vitro selection afforded by molecular display methods makes it straightforward to adjust solution conditions and temperature during library sorting to those that had been determined to maintain the integrity of detergent-solubilized membrane proteins. Fabs binding to the full-length KcsA were identified, and the KcsA channel was functional as a Fab complex, indicating that the Fab did not significantly perturb the structure.
The structure of full-length KcsA was determined at 3.8 Å [31]. Two molecules of synthetic Fab were bound to one molecule of the KcsA tetramer (Figure 2b). The structure reveal a well-defined four-helix bundle that projects ∼70 Å towards the cytoplasm, rationalizing the difficulty in crystallizing full-length KcsA without a crystallization chaperone. Extensive crystal contacts were formed between Fab chaperones, while KcsA molecules form only a small number of such contacts. Importantly, a helix that constricts the channel for ion passage (the inner bundle gate) in the full-length structure was tilted by 15° with respect to the counterpart in the truncated structure. As a result, the full-length channel has a tighter gate, and the residues corresponding to the narrowest point differ in the two structures by two turns of helix.
This synthetic Fab library was also effective in producing antibodies that bind to structured RNA [33]. Nucleic acids are usually not immunogenic in animal immunization primarily because they are quickly degraded in serum. In contrast, conditions for in vitro phage display selection have been developed that suppress such nuclease activities [33]. Synthetic Fabs that bind to the ΔC209 P4-P6 domain of Tetrahymena group I intron with mid-nM Kd were identified from the Fellouse library. The crystal structure of the RNA-Fab complex was determined at 1.95 Å resolution (Figure 2c), improving the resolution of the RNA structure and revealing, for the first time, the molecular interactions within an RNA-antibody interface. As in other cases, the Fab molecules formed layers in the lattice through extensive crystal contacts.
The ability to produce highly functional antibodies using synthetic diversity introduced in a single antibody scaffold is particularly useful for chaperone-assisted crystallography, because this capability allows one to optimize library design and scaffold improvement in a modular fashion. Libraries can be built on a stable and crystallization-friendly scaffold such as Herceptin Fab for which more than ten synthetic Fab-target complex structures have already been determined. Furthermore, mutations can be introduced to a scaffold that enhances the crystallizability, expression, stability and/or phasing power. Further, the amino acid diversity can be iteratively refined based on systematic analysis of CDR sequences of antibodies produced from the preceding generations of libraries [30]. This built-in evolution scheme of synthetic antibody design guarantees that chaperone libraries will continue to improve.
Non-antibody crystallization chaperones
Protein engineering technologies used for synthetic antibodies can be applied to non-antibody scaffolds to generate new types of crystallization chaperones. These “alternative” scaffolds have been developed to improve on properties that make antibody fragments less desirable [34]. These scaffolds are usually in the form of a single polypeptide free of disulfide bonds. They are highly stable and produced in much higher yield in bacteria, tens and sometimes hundreds of milligrams from a liter culture.
Designed ankyrin repeat proteins (DARPINs) are synthetic binding proteins built with ankyrin repeat modules [35]. Their use as crystallization chaperones was recently reviewed [36]. Each ankyrin repeat module consists of two helices and a loop, and typically three modules containing amino acid diversity are linked, which are terminated with two “end cap” modules. DARPINs with high affinity are selected using ribosome display [35] or phage display [37]. DARPINs are the only predominantly helical scaffold that has been used as a crystallization chaperone to date (Figure 2f).
Five protein structures using DARPIN chaperones have been reported [36]. A DARPIN chaperone successfully yielded a 2.3-Å structure of wild-type Polo-like kinase-1 (Plk-1), a highly recalcitrant target (a mutant had been crystallized) [38]. In this structure, the DARPIN generated against Plk-1 bound to an epitope that is enriched with Arg, Glu and Lys and masked these residues which are generally considered unfavorable for forming crystal contacts [1]. A DARPIN chaperone was also effective in crystallizing a detergent-solubilized membrane protein, the multidrug exporter AcrB, which resulted in higher resolution than AcrB crystallized without a chaperone and in the identification of new conformations of the subunits and substrate channels running through each subunit (Figure 2f). The crystal structures of DARPIN inhibitors of aminoglycoside phosphotransferase and caspase-2 both showed alternative conformations of the respective enzyme active sites, rationalizing the DARPINs' inhibitory activities [39,40]. These structures demonstrate the effectiveness of DARPINs as crystallization chaperones.
Fibronectin type III domain (FN3) is a widely used scaffold for generating binding proteins [41-43]. It has a monomeric β-sandwich fold similar to VHH but lacks disulfides. Loops at one end of the scaffold are diversified in combinatorial libraries from which binding proteins can be selected. The reduced genetic code libraries proved to be highly functional on the FN3 scaffold [41,44], demonstrating that the effectiveness of this approach in diversity design is not limited to the heterodimeric immunoglobulin scaffold. The crystal structures of FN3 chaperones bound to maltose-binding protein (MBP) [41,44], estrogen receptor ligand binding domain (PDB ID: 2OCF) and Abl1 SH2 domain (J. Wojcik, A. Koide and SK, in preparation) have been determined. Similar to the VHH chaperones, FN3 chaperones often form intermolecular β-sheets.
The disulfide-free nature of DARPIN, FN3 and similar molecular scaffolds makes it much easier to produce fusion proteins, because they can be expressed in the functional form regardless of the redox potential of the environment. In contrast, antibody fragments usually need to be produced under oxidizing environment such as the E. coli periplasm. One way to exploit the advantage of disulfide-free chaperones is to make a chaperone-target fusion protein. Such a fusion protein ensures the formation of 1:1 complex and dramatically enhances the effective affinity of the chaperone-target interaction by increasing the effective concentration of a chaperone. A similar fusion strategy has been utilized to crystallize peptide-protein complexes [45,46]. This strategy is conceptually similar to the use of fusion partners such as T4 lysozyme, which has been highly effective in crystallizing G-protein coupled receptors [47,48] and a SAM domain [49]. The FN3-MBP complexes were crystallized using this strategy [41,44]. Interestingly, although the complex formed a soluble oligomer in solution, the crystal structures contained a continuous helical rod assembly, indicating that domain swapping occurred during crystallization. Such chaperone fusion approaches may further decrease the entropic cost of crystallization and allow the exploration of broader range of crystal packing.
Exposed β-sheet edges of crystallization chaperones nucleate productive crystal contacts
Crystallization chaperones currently in use consist of β-sandwich folds except for DARPINs (Figure 2f). A strikingly common feature in the crystal structures determined with these β-rich chaperones is the formation of an intermolecular anti-parallel β-sheet between copies of crystallization chaperones. As a result, these chaperones create an additional level of symmetry such as dimerization of chaperone-target complexes (Figure 2d and 2d) and the formation of an extended layer of chaperones (Figure 2a-c). An exposed edge of a β-sheet is inherently predisposed for forming intermolecular interactions as seen in fibrils, and natural proteins employ negative design elements to eliminate uncontrolled self-assembly [50,51]. Thus, β-rich chaperones naturally possess capacity to self-assemble, which is clearly beneficial for crystallization.
The examination of the different crystal forms of the VHH-RNaseA complex shows that an edge strand of VHH is the primary element for the packing in most forms [12]. This β-strand acts as a “hot-spot” for the interactions that promote VHH dimerization through the formation of an intermolecular anti-parallel β-sheet (Figure 3). Two distinct modes of VHH dimerization were observed for the complex, which resulted in different space groups. This particular VHH was a mutant that contained a Leu-to-Met mutation in the edge β-strand, which probably introduces an additional level of flexibility into this region. Such local plasticity is an additional factor that appears to play a significant role in crystallization, and these results suggest a direction in chaperone scaffold engineering for better crystallizability.
Figure 3.
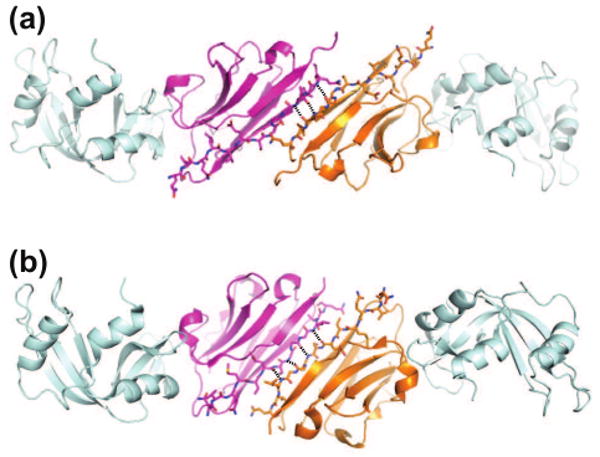
Crystal packing interactions through exposed edges of β-sheets often observed for β-rich crystallization chaperones. Two different modes of intermolecular β-sheet formation seen in RNaseA-VHH complex structures are shown. (a) 1BZQ (space group, P1) and (b) 2P48 (P3121). β-Strand A of the VHH chaperone is shown as stick models and intermolecular hydrogen bonds are shown as dashed lines. Also see Figure 2d for another example.
Do crystallization chaperones distort structure?
Concerns are often raised about the authenticity of structures determined through chaperone-assisted crystallography. One might think that chaperones can distort a protein into a nonnative structure [52]. Such a view however does not make thermodynamic sense. In order for a chaperone to bind to a particular conformation with reasonably high affinity, that conformation must exist at a detectable concentration. An antibody would pay a huge energetic penalty if it first binds to a low energy state and then distorts the structure into a state so high-energy that it does not appreciably exists in the absence of the bound antibody. Therefore, it is much more likely that immunization and library selection identify a high-affinity antibody that binds to and stabilizes one of the lower energy states.
In addition to full-length KcsA (Figure 2b), the structure of the isolated C-terminal domain in complex with another synthetic Fab, produced as a result of proteolysis during crystallization, was determined at 2.6 Å (Figure 4) [31]. The KcsA C-terminal domain structure was nearly identical between two crystal structures determined using different synthetic Fab chaperones with different binding stoichiometries and crystallized distinct solution conditions. In another example, the voltage sensor segment of the KvAP potassium channel was in a nonnative conformation in all three structures determined with a Fab or Fv chaperone and without a chaperone [52], strongly suggesting that other factors such as the detergent micelle are responsible for stabilizing the nonnative conformation. Furthermore, ubiquitin, a protein generally considered “rock solid”, shows significant conformational differences among the crystal structures of complexes with its natural binding partners, and recent analysis of solution NMR data in the free state revealed that the protein samples these conformations in the native state. [53]. Taken together, crystallization chaperones, like other types of ligands, can stabilize and freeze a particular conformation within the native ensemble. This capacity of crystallization chaperones to expand the structural annotation of the native state ensemble should be viewed as an advantage not as a shortcoming.
Figure 4.
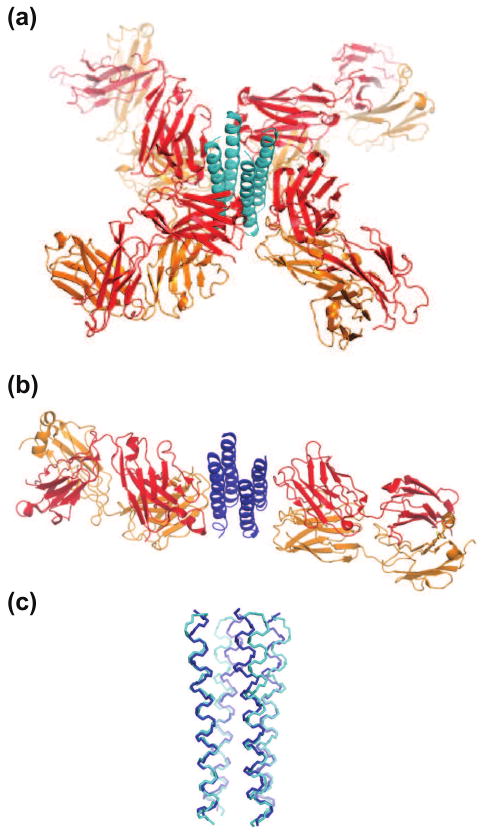
Comparisons of the KcsA cytoplasmic domain determined with two different Fab chaperones. (a) The full-length construct with “Fab2”. Only the C-terminal, cytoplasmic domain is shown for KcsA. (b) The truncated construct with “Fab4”. (c) Superposition of the two structures. The RMSD for the common Cα atoms is ∼1 Å.
Future perspectives
The examples described above convincingly show that chaperone-assisted crystallography helps reduce the crystallization bottleneck in structural biology. The successes of synthetic crystallization chaperones suggest that the technology will become faster and more economical. However, generating crystallization chaperones is a major undertaking, and thus it would not be the first line of attack in a crystallization project. Also chaperone-assisted crystallography will not substitute intimate knowledge of biophysical properties of target molecules. Because the challenging “high-hanging fruit” targets for which crystallization chaperones would be most desired are often flexible and/or fragile, each library selection experiment must be customized to ensure the target is kept in the intended conformation throughout selection steps. Nevertheless, with the flexibility in library selection schemes and exquisite control of experimental conditions, the range of structural biology problems that can be attacked with synthetic crystallization chaperones will continue to expand.
Figure 1.

The concept of crystallization chaperones. (a) A crystallization chaperone that binds to a specific conformation reduces conformational heterogeneity. (b) A crystallization chaperone can promote crystal lattice formation.
Acknowledgments
I would like to thank Serdar Uysal and Ryan Gilbreth for critical reading of the manuscript and assistance with figures. This work was supported by National Institutes of Health grants R01-GM72688, U54-GM74946, R21-CA132700 and R21-DA025725.
Footnotes
Reference Comments
6••, 7•• Day et al. [6] reports methods to produce conformation-specific monoclonal antibodies for GPCRs. Liposome-embedded antigens are used to stabilize the native conformation of GPCRs in animal immunization. Rasmussen et al. [7] reports the crystal structure of the first GPCR/Fab chaperone complex.
12• Tereshko et al. It reports a systematic study on the potential of VHH crystallization chaperones. It introduces shotgun Met scanning methodology to introduce additional Se-Met positions in crystallization chaperones.
16•• Korotkov et al. A VHH chaperone was used to crystallize two-domain protein with a flexible interdomain linker. The VHH chaperone produced an additional level of symmetry that appears to reduce interdomain motions.
17• Lam et al. A VHH chaperone was successfully used to accelerate structure determination of an otherwise recalcitrant target.
29•• Fellouse et al. This work establishes a potent synthetic Fab phage display library and a high-throughput selection methodology. They further determine the crystal structure of a Fab generated from this library in complex with a target, VEGF.
31•• Uysal et al. The first structure determination of an integral membrane protein using a synthetic Fab chaperone. It reveals the full-length KcsA structure, which has marked differences around the activation gate from the previously determined structure of a truncated form.
33•• Ye et al. This work demonstrates the capacity of synthetic Fab libraries in conjunction with molecular display to produce antibodies to antigens that are inaccessible by the conventional immunization methods and also it establishes synthetic Fabs chaperones for RNA crystallization.
36•• Sennhauser & Grutter. A comprehensive review of the use of DARPINs as crystallization chaperones.
41•, 45• These studies demonstrate the effectiveness of protein fusion approaches in crystallizing protein complexes.
47••, 48•• Two GPCR structures were determined by replacing most of the third cytoplasmic loop with T4 lysozyme and using lipidic cubic mesophase. The fused T4 lysozyme molecule reduced the conformational heterogeneity and provided many crystal contacts. This technique may represent a strategy generally applicable to crystallizing GPCR family members.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Derewenda ZS. Rational protein crystallization by mutational surface engineering. Structure (Camb) 2004;12:529–535. doi: 10.1016/j.str.2004.03.008. [DOI] [PubMed] [Google Scholar]
- 2.Kovari LC, Momany C, Rossmann MG. The use of antibody fragments for crystallization and structure determinations. Structure. 1995;3:1291–1293. doi: 10.1016/s0969-2126(01)00266-0. [DOI] [PubMed] [Google Scholar]
- 3.Iwata S, Ostermeier C, Ludwig B, Michel H. Structure at 2.8 A resolution of cytochrome c oxidase from Paracoccus denitrificans. Nature. 1995;376:660–669. doi: 10.1038/376660a0. [DOI] [PubMed] [Google Scholar]
- 4.Hunte C, Michel H. Crystallisation of membrane proteins mediated by antibody fragments. Curr Opin Struct Biol. 2002;12:503–508. doi: 10.1016/s0959-440x(02)00354-8. [DOI] [PubMed] [Google Scholar]
- 5.Ostermeier C, Iwata S, Ludwig B, Michel H. Fv fragment-mediated crystallization of the membrane protein bacterial cytochrome c oxidase. Nat Struct Biol. 1995;2:842–846. doi: 10.1038/nsb1095-842. [DOI] [PubMed] [Google Scholar]
- 6.Day PW, Rasmussen SG, Parnot C, Fung JJ, Masood A, Kobilka TS, Yao XJ, Choi HJ, Weis WI, Rohrer DK, et al. A monoclonal antibody for G protein-coupled receptor crystallography. Nat Methods. 2007;4:927–929. doi: 10.1038/nmeth1112. [DOI] [PubMed] [Google Scholar]
- 7.Rasmussen SG, Choi HJ, Rosenbaum DM, Kobilka TS, Thian FS, Edwards PC, Burghammer M, Ratnala VR, Sanishvili R, Fischetti RF, et al. Crystal structure of the human beta2 adrenergic G-protein-coupled receptor. Nature. 2007;450:383–387. doi: 10.1038/nature06325. [DOI] [PubMed] [Google Scholar]
- 8.Li H, Dunn JJ, Luft BJ, Lawson CL. Crystal structure of Lyme disease antigen outer surface protein A complexed with an Fab. Proc Natl Acad Sci USA. 1997;94:3584–3589. doi: 10.1073/pnas.94.8.3584. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Hamers-Casterman C, Atarhouch T, Muyldermans S, Robinson G, Hamers C, Songa EB, Bendahman N, Hamers R. Naturally occurring antibodies devoid of light chains. Nature. 1993;363:446–448. doi: 10.1038/363446a0. [DOI] [PubMed] [Google Scholar]
- 10.Muyldermans S, Cambillau C, Wyns L. Recognition of antigens by single-domain antibody fragments: the superfluous luxury of paired domains. Trends Biochem Sci. 2001;26:230–235. doi: 10.1016/s0968-0004(01)01790-x. [DOI] [PubMed] [Google Scholar]
- 11.De Genst E, Silence K, Decanniere K, Conrath K, Loris R, Kinne J, Muyldermans S, Wyns L. Molecular basis for the preferential cleft recognition by dromedary heavy-chain antibodies. Proc Natl Acad Sci U S A. 2006;103:4586–4591. doi: 10.1073/pnas.0505379103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Tereshko V, Uysal S, Koide A, Margalef K, Koide S, Kossiakoff AA. Toward chaperone-assisted crystallography: protein engineering enhancement of crystal packing and X-ray phasing capabilities of a camelid single-domain antibody (VHH) scaffold. Protein Sci. 2008;17:1175–1187. doi: 10.1110/ps.034892.108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Koide A, Tereshko V, Uysal S, Margalef K, Kossiakoff AA, Koide S. Exploring the capacity of minimalist protein interfaces: interface energetics and affinity maturation to picomolar KD of a single-domain antibody with a flat paratope. J Mol Biol. 2007;373:941–953. doi: 10.1016/j.jmb.2007.08.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Loris R, Marianovsky I, Lah J, Laeremans T, Engelberg-Kulka H, Glaser G, Muyldermans S, Wyns L. Crystal structure of the intrinsically flexible addiction antidote MazE. J Biol Chem. 2003;278:28252–28257. doi: 10.1074/jbc.M302336200. [DOI] [PubMed] [Google Scholar]
- 15.Spinelli S, Desmyter A, Verrips CT, de Haard HJ, Moineau S, Cambillau C. Lactococcal bacteriophage p2 receptor-binding protein structure suggests a common ancestor gene with bacterial and mammalian viruses. Nat Struct Mol Biol. 2006;13:85–89. doi: 10.1038/nsmb1029. [DOI] [PubMed] [Google Scholar]
- 16.Korotkov KV, Pardon E, Steyaert J, Hol WG. Crystal Structure of the N-Terminal Domain of the Secretin GspD from ETEC Determined with the Assistance of a Nanobody. Structure. 2009;17:255–265. doi: 10.1016/j.str.2008.11.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Lam AY, Pardon E, Korotkov KV, Hol WG, Steyaert J. Nanobody-aided structure determination of the EpsI:EpsJ pseudopilin heterodimer from Vibrio vulnificus. J Struct Biol. 2008 doi: 10.1016/j.jsb.2008.11.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Bradbury AR, Marks JD. Antibodies from phage antibody libraries. J Immunol Methods. 2004;290:29–49. doi: 10.1016/j.jim.2004.04.007. [DOI] [PubMed] [Google Scholar]
- 19.Sidhu SS, Fellouse FA. Synthetic therapeutic antibodies. Nat Chem Biol. 2006;2:682–688. doi: 10.1038/nchembio843. [DOI] [PubMed] [Google Scholar]
- 20.Fellouse FA, Esaki K, Birtalan S, Raptis D, Cancasci VJ, Koide A, Jhurani P, Vasser M, Wiesmann C, Kossiakoff AA, et al. High-throughput Generation of Synthetic Antibodies from Highly Functional Minimalist Phage-displayed Libraries. J Mol Biol. 2007;373:924–940. doi: 10.1016/j.jmb.2007.08.005. [DOI] [PubMed] [Google Scholar]
- 21.Hoogenboom HR. Selecting and screening recombinant antibody libraries. Nat Biotechnol. 2005;23:1105–1116. doi: 10.1038/nbt1126. [DOI] [PubMed] [Google Scholar]
- 22.Gai SA, Wittrup KD. Yeast surface display for protein engineering and characterization. Curr Opin Struct Biol. 2007;17:467–473. doi: 10.1016/j.sbi.2007.08.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Sidhu SS, Koide S. Phage display for engineering and analyzing protein interaction interfaces. Curr Opin Struct Biol. 2007;17:481–487. doi: 10.1016/j.sbi.2007.08.007. [DOI] [PubMed] [Google Scholar]
- 24.Lipovsek D, Pluckthun A. In-vitro protein evolution by ribosome display and mRNA display. J Immunol Methods. 2004;290:51–67. doi: 10.1016/j.jim.2004.04.008. [DOI] [PubMed] [Google Scholar]
- 25.Hawkins RE, Russell SJ, Winter G. Selection of phage antibodies by binding affinity. Mimicking affinity maturation. J Mol Biol. 1992;226:889–896. doi: 10.1016/0022-2836(92)90639-2. [DOI] [PubMed] [Google Scholar]
- 26.Sidhu SS, Li B, Chen Y, Fellouse FA, Eigenbrot C, Fuh G. Phage-displayed antibody libraries of synthetic heavy chain complementarity determining regions. J Mol Biol. 2004;338:299–310. doi: 10.1016/j.jmb.2004.02.050. [DOI] [PubMed] [Google Scholar]
- 27.Lee CV, Liang WC, Dennis MS, Eigenbrot C, Sidhu SS, Fuh G. High-affinity human antibodies from phage-displayed synthetic Fab libraries with a single framework scaffold. J Mol Biol. 2004;340:1073–1093. doi: 10.1016/j.jmb.2004.05.051. [DOI] [PubMed] [Google Scholar]
- 28.Mian IS, Bradwell AR, Olson AJ. Structure, function and properties of antibody binding sites. J Mol Biol. 1991;217:133–151. doi: 10.1016/0022-2836(91)90617-f. [DOI] [PubMed] [Google Scholar]
- 29.Zemlin M, Klinger M, Link J, Zemlin C, Bauer K, Engler JA, Schroeder HW, Jr, Kirkham PM. Expressed murine and human CDR-H3 intervals of equal length exhibit distinct repertoires that differ in their amino acid composition and predicted range of structures. J Mol Biol. 2003;334:733–749. doi: 10.1016/j.jmb.2003.10.007. [DOI] [PubMed] [Google Scholar]
- 30.Fellouse FA, Li B, Compaan DM, Peden AA, Hymowitz SG, Sidhu SS. Molecular recognition by a binary code. J Mol Biol. 2005;348:1153–1162. doi: 10.1016/j.jmb.2005.03.041. [DOI] [PubMed] [Google Scholar]
- 31.Uysal S, Vásquez V, Tereshko V, Esaki K, Fellouse FA, Sidhu SS, Koide S, Perozo E, Kossiakoff AA. The crystal structure of full-fength KcsA in its closed conformation. Proc Natl Acad Sci USA. 2009 doi: 10.1073/pnas.0810663106. in press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Zhou Y, Morais-Cabral JH, Kaufman A, MacKinnon R. Chemistry of ion coordination and hydration revealed by a K+ channel-Fab complex at 2.0 A resolution. Nature. 2001;414:43–48. doi: 10.1038/35102009. [DOI] [PubMed] [Google Scholar]
- 33.Ye JD, Tereshko V, Frederiksen JK, Koide A, Fellouse FA, Sidhu SS, Koide S, Kossiakoff AA, Piccirilli JA. Synthetic antibodies for specific recognition and crystallization of structured RNA. Proc Natl Acad Sci U S A. 2008;105:82–87. doi: 10.1073/pnas.0709082105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Binz HK, Amstutz P, Pluckthun A. Engineering novel binding proteins from nonimmunoglobulin domains. Nat Biotechnol. 2005;23:1257–1268. doi: 10.1038/nbt1127. [DOI] [PubMed] [Google Scholar]
- 35.Binz HK, Amstutz P, Kohl A, Stumpp MT, Briand C, Forrer P, Grutter MG, Pluckthun A. High-affinity binders selected from designed ankyrin repeat protein libraries. Nat Biotechnol. 2004;22:575–582. doi: 10.1038/nbt962. [DOI] [PubMed] [Google Scholar]
- 36.Sennhauser G, Grutter MG. Chaperone-assisted crystallography with DARPins. Structure. 2008;16:1443–1453. doi: 10.1016/j.str.2008.08.010. [DOI] [PubMed] [Google Scholar]
- 37.Steiner D, Forrer P, Pluckthun A. Efficient selection of DARPins with sub-nanomolar affinities using SRP phage display. J Mol Biol. 2008;382:1211–1227. doi: 10.1016/j.jmb.2008.07.085. [DOI] [PubMed] [Google Scholar]
- 38.Bandeiras TM, Hillig RC, Matias PM, Eberspaecher U, Fanghanel J, Thomaz M, Miranda S, Crusius K, Putter V, Amstutz P, et al. Structure of wild-type Plk-1 kinase domain in complex with a selective DARPin. Acta Crystallogr D Biol Crystallogr. 2008;64:339–353. doi: 10.1107/S0907444907068217. [DOI] [PubMed] [Google Scholar]
- 39.Schweizer A, Roschitzki-Voser H, Amstutz P, Briand C, Gulotti-Georgieva M, Prenosil E, Binz HK, Capitani G, Baici A, Pluckthun A, et al. Inhibition of Caspase-2 by a Designed Ankyrin Repeat Protein: Specificity, Structure, and Inhibition Mechanism. Structure. 2007;15:625–636. doi: 10.1016/j.str.2007.03.014. [DOI] [PubMed] [Google Scholar]
- 40.Kohl A, Amstutz P, Parizek P, Binz HK, Briand C, Capitani G, Forrer P, Pluckthun A, Grutter MG. Allosteric inhibition of aminoglycoside phosphotransferase by a designed ankyrin repeat protein. Structure. 2005;13:1131–1141. doi: 10.1016/j.str.2005.04.020. [DOI] [PubMed] [Google Scholar]
- 41.Koide A, Gilbreth RN, Esaki K, Tereshko V, Koide S. High-affinity single-domain binding proteins with a binary-code interface. Proc Natl Acad Sci U S A. 2007;104:6632–6637. doi: 10.1073/pnas.0700149104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Xu L, Aha P, Gu K, Kuimelis R, Kurz M, Lam T, Lim A, Liu H, Lohse P, Sun L, et al. Directed evolution of high-affinity antibody mimics using mRNA display. Chem Biol. 2002;9:933–942. doi: 10.1016/s1074-5521(02)00187-4. [DOI] [PubMed] [Google Scholar]
- 43.Lipovsek D, Lippow SM, Hackel BJ, Gregson MW, Cheng P, Kapila A, Wittrup KD. Evolution of an Interloop Disulfide Bond in High-Affinity Antibody Mimics Based on Fibronectin Type III Domain and Selected by Yeast Surface Display: Molecular Convergence with Single-Domain Camelid and Shark Antibodies. J Mol Biol. 2007;368:1024–1041. doi: 10.1016/j.jmb.2007.02.029. [DOI] [PubMed] [Google Scholar]
- 44.Gilbreth RN, Esaki K, Koide A, Sidhu SS, Koide S. A dominant conformational role for amino acid diversity in minimalist protein-protein interfaces. J Mol Biol. 2008;381:407–418. doi: 10.1016/j.jmb.2008.06.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Appleton BA, Zhang Y, Wu P, Yin JP, Hunziker W, Skelton NJ, Sidhu SS, Wiesmann C. Comparative structural analysis of the Erbin PDZ domain and the first PDZ domain of ZO-1. Insights into determinants of PDZ domain specificity. J Biol Chem. 2006;281:22312–22320. doi: 10.1074/jbc.M602901200. [DOI] [PubMed] [Google Scholar]
- 46.Elkins JM, Papagrigoriou E, Berridge G, Yang X, Phillips C, Gileadi C, Savitsky P, Doyle DA. Structure of PICK1 and other PDZ domains obtained with the help of self-binding C-terminal extensions. Protein Sci. 2007;16:683–694. doi: 10.1110/ps.062657507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Cherezov V, Rosenbaum DM, Hanson MA, Rasmussen SG, Thian FS, Kobilka TS, Choi HJ, Kuhn P, Weis WI, Kobilka BK, et al. High-resolution crystal structure of an engineered human beta2-adrenergic G protein-coupled receptor. Science. 2007;318:1258–1265. doi: 10.1126/science.1150577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Jaakola VP, Griffith MT, Hanson MA, Cherezov V, Chien EY, Lane JR, Ijzerman AP, Stevens RC. The 2.6 angstrom crystal structure of a human A2A adenosine receptor bound to an antagonist. Science. 2008;322:1211–1217. doi: 10.1126/science.1164772. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Nauli S, Farr S, Lee YJ, Kim HY, Faham S, Bowie JU. Polymer-driven crystallization. Protein Sci. 2007;16:2542–2551. doi: 10.1110/ps.073074207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Richardson JS, Richardson DC. Natural beta-sheet proteins use negative design to avoid edge-to-edge aggregation. Proc Natl Acad Sci U S A. 2002;99:2754–2759. doi: 10.1073/pnas.052706099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Makabe K, Koide S. The promiscuity of beta-strand pairing allows for rational design of beta-sheet face inversion. J Am Chem Soc. 2008;130:14370–14371. doi: 10.1021/ja805011h. [DOI] [PubMed] [Google Scholar]
- 52.Lee SY, Lee A, Chen J, MacKinnon R. Structure of the KvAP voltage-dependent K+ channel and its dependence on the lipid membrane. Proc Natl Acad Sci U S A. 2005;102:15441–15446. doi: 10.1073/pnas.0507651102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Lange OF, Lakomek NA, Fares C, Schroder GF, Walter KF, Becker S, Meiler J, Grubmuller H, Griesinger C, de Groot BL. Recognition dynamics up to microseconds revealed from an RDC-derived ubiquitin ensemble in solution. Science. 2008;320:1471–1475. doi: 10.1126/science.1157092. [DOI] [PubMed] [Google Scholar]