

Abstract
Heterotrimeric G proteins (Gαβγ) transmit signals from activated G protein coupled receptors (GPCRs) to downstream effectors through a guanine nucleotide signaling cycle. Numerous studies indicate that the carboxy-terminal α5 helix of Gα subunits participate in Gα-receptor binding, and previous EPR studies suggest this receptor-mediated interaction induces a rotation and translation of the α5 helix of the Gα subunit [Oldham et al., Nat. Struct. Mol. Biol., 13: 772-7 (2006)]. Based on this result, an engineered disulfide bond was designed to constrain the α5 helix of Gαi1 into its EPR-measured receptor-associated conformation through the introduction of cysteines at positions 56 in the α1 helix and 333 in the α5 helix (I56C/Q333C Gαi1). A functional mimetic of the EPR-measured α5 helix dipole movement upon receptor association was additionally created by introduction of a positive charge at the amino-terminus of this helix, D328R Gαi1. Both proteins exhibit dramatically elevated basal nucleotide exchange. The 2.9 Å resolution crystal structure of the I56C/Q333C Gαi1 in complex with GDP-AlF4 − reveals the shift of the α5 helix toward the guanine nucleotide-binding site that is anticipated by EPR measurements. The structure of the I56C/Q333C Gαi1 subunit further revealed altered positions for the switch regions and throughout the Gαi1 subunit, accompanied by significantly elevated crystallographic temperature factors. Combined with previous evidence in the literature, the structural analysis supports the critical role of electrostatics of the α5 helix dipole and overall conformational variability during nucleotide release.
Gα subunits of heterotrimeric G proteins (Gαβγ) are members of a family of over 100 different guanine nucleotide binding proteins identified in eukaryotic cells. These G proteins transmit signals through a guanine nucleotide signaling cycle, with the protein-bound guanine nucleotide regulating the activation state of the Gα subunit (1–4). Structural and biochemical studies of Gα and small G proteins indicate that these proteins encode the information about their activation state in the conformation of loops at the protein surface. Aptly known as the switch regions, these regions switch between conformations in response to the identity of the bound nucleotide (5). As a result, the binding affinities for both upstream and downstream signaling proteins are dramatically affected.
GTP-bound G proteins can readily activate downstream pathways through direct protein-protein interactions. Hydrolysis of GTP to GDP by the Gα subunit returns the protein back to its inactive, Gβγ-bound state. The uncatalyzed rate of nucleotide exchange is slow, but is dramatically enhanced by the interaction with an activated guanine-nucleotide exchange factor (GEF). For small G proteins, a soluble GEF catalyzes the release of GDP, while for heterotrimeric G proteins, a 7-transmembrane helix G protein coupled receptor (GPCR) acts as a GEF to catalyze nucleotide release.
Unlike many of the small G proteins which have a single domain and a guanine nucleotide binding site that is accessible to solvent, the guanine nucleotide in the Gα subunits of heterotrimeric G proteins is nestled tightly between a Ras-like GTPase domain and a helical domain (6–11), with no obvious exit tunnel for the nucleotide. As a result, the rate limiting step of heterotrimeric G protein signaling is release of GDP catalyzed by an activated GPCR (R*; (12)), which physiologically requires the formation of a transient Gαβγ-R* complex. The structure determination of R* in complex with its cognate Gαβγ heterotrimer in the nucleotide-free state would provide the most accurate information about the mechanism of nucleotide exchange for this system. Since current experimental limitations make this a daunting undertaking, the investigation of R*-catalyzed nucleotide exchange in G proteins has used alternative, low-resolution techniques. Many of the experiments have pointed to methods that can be used to stabilize Gα in its R*-bound conformation and provide new avenues to reveal mechanisms of nucleotide exchange in Gα.
The first hints of the mechanism of activation of Gα subunits by GPCRs came through mapping of the binding interface of the complex. The C-terminus of Gα subunits has long been known to interact with activated GPCRs. As early as the late 1980’s, screening of Gα-derived peptides revealed that peptides corresponding to the C-terminal α5 helix of Gα could bind to receptors with high affinity and mimic G protein stabilization of the active state of the receptor (13–15). Later studies employed photoactivatible crosslinkers to show that light-activated rhodopsin could crosslink to the C-terminus of Gαt (16). The use of chemical crosslinkers confirmed the binding of receptor to the C-terminal residues of the Gα subunit (17), and additionally identified residues within the amino terminus of Gαt that crosslinked to activated rhodopsin. Mapping of the receptor-G protein interaction using peptides corresponding to the cytoplasmic domain of the dopamine D2 receptor further verified the interaction between receptor and the C-terminus of the Gα subunit (18). As a complement to the peptide studies, C-terminal truncations in both Gαi and Gαo proteins have been shown to reduce affinity for GPCR, demonstrating the importance of this region in receptor affinity (19, 20). The C-terminus may also be important for receptor selectivity as chimeric Gα subunits that replace C-terminal residues in one Gα subtype for another affect receptor selectivity (21–23). Further, mutations within the carboxy terminal region of Gα subunits alter receptor-G protein coupling (24–26). A fluorescence-quenching approach revealed the binding site for a peptide derived from the carboxy terminus of Gα maps to a hydrophobic region on helix VI in activated rhodopsin (27, 28), demonstrating the utility of such approaches to provide detailed structural information, especially in the absence of a structure of an activated GPCR-G protein complex. Excitingly, a recent structure of opsin determined in complex with a peptide derived from the C-terminus of Gαt revealed the specific contacts between receptor and the last 10 amino acids of the Gα subunit (29).
However, the carboxy terminus is not the sole region of Gα that contacts the receptor; other studies have also implicated the α4-β6 loop (30, 31) and the amino terminus (13, 17, 30, 32–35). Together with the C-terminus, these regions can be mapped onto the structure of the Gαi1 subunit and form a contiguous, three-dimensional surface as would be anticipated for a protein binding interface (30, 36) (Figure 1a).
Figure 1.

Design of the Gαi1 mutants. (a) Location of GPCR binding site on Gαi1. The GTPase domain is shown in dark blue and the helical domain is shown in grey. The α5 helix is highlighted in yellow, the α1 helix is orange, and the GDP molecule is shown in a space-filling model colored red. The location of the GPCR binding site is indicated with a black donut. The GPCR is predicted to induce a conformational change that pushes the α5 helix toward the helical domain (down in this representation). (b) Design of Gαi1 mutations. A close-up view of helices α1 and α5 highlighting the locations of point mutations I56C/Q333C, and D328R. The I56C/Q333C double mutation is predicted to form a disulfide bond that constrains the α5 helix into its receptor-activated conformation, which is modeled as a blue cylinder. The D328R mutation is located at the N-terminus of the α5 helix and designed to mimic the movement of the α5 helix dipole. This charge reversal mutation was designed to maximize the effect of the electrostatics.
After the receptor binds its cognate G protein, the signal is transmitted from the receptor-binding site to the nucleotide-binding pocket of the Gα subunit. Electron paramagnetic resonance (EPR) spectroscopy measurements are consistent with the R*-Gα interaction causing a rotation and translation of the α5 helix of the Gα subunit toward the base of the guanine nucleotide (37) (Figure 1b). The insertion of a five amino acid flexible linker at the start of the C-terminal helix decouples receptor binding from the nucleotide release process (37), suggesting that the rigid-body roto-translation of the C-terminal helix changes the conformation of the β6-α5 linker containing the conserved TCAT sequence motif, which directly contacts the guanine ring of the nucleotide, to achieve receptor-mediated allosteric nucleotide release (38). In addition, the structure of opsin in complex with a Gα-mimicking peptide (29) cannot predict the magnitude of the rigid body movement of the C-terminal α5 helix toward the GDP-binding pocket during receptor association. However, this co-structure indicates an additional conformational change of the α5 helix of Gα is required to prevent steric clash of the Gα protein with the membrane upon receptor binding. This additional conformational change was proposed as a 40° tilt of the α5 helix of the Gα subunit relative to the remainder of the G protein. A less drastic change might involve the introduction of a small bend after the C-terminus of the α5 helix. This would prevent the exposure of hydrophobic side chains along the α5 helix to solvent and is consistent with both the crystal structure and the EPR spectroscopy measurements, which shows decreased mobility for residues along the α5 helix (residues 330, 331, 334, 340, 342, 344 and 349 in a Gαi protein show decreased mobility) upon receptor activation (37).
In light of the importance of receptor-catalyzed nucleotide exchange in the Gα subunit, the changes in inter-residue distances measured by EPR spectroscopy during receptor activation (37) were used to develop a conceptual model of Gαi1 conformation upon binding activated receptors (R*). This EPR data suggests that the roto-translation of the α5 helix plays a critical role in receptor-mediated G protein activation, which could be communicated to the nucleotide binding site via global structural effects, as well as communicated through specific electrostatic effects directed at the base of the α5 helix, which comes into closer proximity to the bound nucleotide upon roto-translation of the α5 helix. The roto-translation of the α5 helix indicated by EPR studies which accompany receptor activation (Figure 1b), when mapped onto existing crystal structures, results in a close proximity between side chains of Gln 333 (in the α5 helix) and Ile 56 suggesting that an engineered disulfide bond between these residues could stabilize the structural changes that are present when Gα is bound to receptor (37). The Gln at residue 333 is not conserved, and position 56 typically contains a hydrophobic Ile residue at this position (Leu in Gαs).
A rigid-body roto-translation of the α5 helix would also be expected to move the positive dipole of the α5 helix towards the guanine ring of the bound nucleotide upon receptor activation. To accentuate this effect, a positive charge was introduced into the base of the α5 helix of Gαi1 at residue 328, a residue conserved among Gαi family members (D328R Gαi1, Figure 1b). This residue is next to the TCAT motif involved in nucleotide binding (residues 324–327). The I56C/Q333C Gαi1 and the D328R Gαi1 proteins (as well as other mutant proteins for control experiments) were constructed; the proteins expressed at levels similar to wild-type Gαi1 and could be purified using similar protocols.
EXPERIMENTAL PROCEDURES
Construction, expression and purification of rat G αi1 mutant proteins
Gαi1 mutants were created using the Quikchange site-directed mutagenesis kit (Stratagene). The I56C/Q333C double mutant used primers 5′-AAGCAGATGAAAATTTGTCACGAGGCTGGC (I56C forward), 5′-GCCAGCCTCGTGACAAATTTTCATCTGCTT (I56C reverse), 5′-ACGAAGAATGTGTGTTTTGTGTTCGATGCT (Q333C forward) and 5′-AGCATCGAACACAAAACAC-ACATTCTTCGT (Q333C reverse). The D328R mutant used primers 5′-TTCACTTGCGCCACGCGCACGAAGAATGTGCAG (forward) and 5′-CTGCACATTCTTCGTGCGCGTGGCGCAAGTGAA (reverse); D328A, (forward) 5′-CACTTCACTTGCGCCACGGCTACGAAGAATGTGCAG- 3′, (reverse) 5′-CTGCACATTCTTCGTAGCCGTGGCGCAAGTGAAGTG- 3′; D328N (forward) 5′-CACTTCACTTGCGCCACGAATACGAAGAATGTGCAG- 3′, (reverse) 5′-CTGCACATTCTTCGTATTCGTGGCGCAAGTGAAGTG-3′, K330D (forward) 5′-CTTGCGCCACGGATACGGATAATGTGCAGTTTGTG-3′(reverse) 5′–CACAAACTGCACATTATCCGTATCCGTGGCGCAAG- 3′. All constructs contain an internal hexahistidine affinity tag inserted between residue 119 and residue 120 in the helical domain (39). Proteins were expressed and purified as described (39).
Nucleotide exchange of GDP as measured by intrinsic tryptophan fluorescence
The basal rate of GTPγS binding was determined by monitoring the relative increase in the intrinsic fluorescence (λex = 300 nm, λem = 345 nm) on a Varian Cary Eclipse fluorescence spectrophotometer of 500 nM wild-type or mutant Gαi1 subunits in 10 mM MOPS (pH 7.2), 130 mM NaCl and 2 mM MgCl2 for 40 min at 21°C after the addition of 10 μM GTPγS (37). Receptor-catalyzed exchange was performed on Gα subunits reconstituted with Gβ1γ1 (500 nM each) in the presence of 500 nM light activated rhodopsin from ureawashed Rod Outer Segments (ROS). Data were an average of three independent experiments and were normalized to the baseline (0%) and the fluorescence maximum (100%). The exchange rate was determined by fitting the data to an exponential association curve using Prism 4.0 (GraphPad Software).
Nucleotide exchange of GDP and GDP-AlF4− as measured by BODIPY-GTP γS fluorescence
Basal nucleotide exchange was measured as fold-increase in emission of 1μM BODIPY-GTPγS (λex = 490 nm, λem = 515 nm) in buffer containing 50mM Tris, 100mM NaCl, 1mM MgCl2, 10 μM GDP, pH 7.5, at 21°C, in the presence and absence of 75μM AlF4− before and after the addition of Gαi1 subunits (200 nM) to the cuvette containing BODIPY-GTPγS.
Rhodopsin binding assay and metarhodopsin II stabilization
The ability of labeled Gα subunits to bind rhodopsin in urea-washed ROS membranes was determined as described (35). Gαi1 (5 μM) with Gβγ (10 μM) and rhodopsin (50 μM) in a buffer containing 50 mM Tris (pH 8.0), 100 mM NaCl and 1 mM MgCl2 were incubated in the dark, after light activation, and after light activation with the addition of GTPγS (100 μM) for 30 minutes incubation at 4°C. Membranes were separated by centrifugation at 20,000 × g for 1hour, and the supernatants removed from pellets. For the dark fraction, the reaction was protected from light during centrifugation, and supernatant was removed in dim red light. The isolated fractions were boiled, visualized on Coomassie-stained SDS-PAGE, and quantified by densitometry using BioRad Multimager (37) by comparison of the amount of 37 kDa Gα in either pellet or supernatant to the total amount of Gα subunits in both fractions, and expressed as a percent of the total. Data are the average of 3 independent experiments. Stabilization of metarhodopsin II by increasing the concentration of Gα proteins (as detailed in Supplementary Figure 2) was performed essentially as described in (40) using an Olis DW2000 spectrophotometer before and after light activation of rhodopsin-G protein complexes.
Crystallization, data collection, structure determination, and refinement
Purified wild-type and I56C/Q333C Gαi1 were exchanged into crystallization buffer (20 mM HEPES pH 8.0, 1 mM EDTA, 10 mM MgCl2, 20 μM GDP, 16 mM NaF, and 40 μM AlCl3) and concentrated to 10 mg/mL prior to crystallization. Crystals of the Gαi1 I56C/Q333C double mutant were grown at 18°C by the hanging drop vapor diffusion method using 2 μL of 10 mg/mL protein and 2 μL of reservoir solution (16% PEG 3350, 240 mM Li(CH3COO), 0.1 M bis-Tris pH 6.5, and 10 mM SrCl2). Oxidizing agents were not added to the protein solution, and inclusion of DTT or β-mercaptoethanol inhibited crystal growth. For accurate comparison, crystals of the wild-type Gαi1 with a lipid modification were grown in the same space group using a modified reservoir solution containing 12% PEG 3350, 0.1 M Ca(CH3COO)2, and 0.1 M bis-Tris, pH 6.5. Crystals formed within three days and belong to the tetragonal space group P43212 with unit cell dimensions a=b=79.7 Å, c=114.5 Å, α=β=γ=90°. Crystals were cryo-cooled in reservoir solution containing 25% glycerol. Data for the I56C/Q333C Gαi1 subunit were collected at 93 K at wavelengths of 1.0 Å and 1.77 Å (Table 2) at the Advanced Photon Source IMCA-CAT beamline 17-ID on an ADSC CCD detector. Data for the wild-type protein were collected at the Advanced Photon Source LS-CAT ID-21-F at 93 K using a wavelength of 0.98 Å on a MAR CCD detector. Data were processed using the HKL (41) and CCP4 (42) suites of programs. Initial phases were calculated by molecular replacement using the program PHASER (43) using wild-type GDP-AlF4−-bound Gαi1 as the search model (accession code 1GFI; (6)). Sulfur-SAD phases resulting in a figure of merit of 0.32 were determined using SHARP (44). The molecular replacement and S-SAD phases were combined using SIGMAA (42). Model building was performed in O (45), and refinement performed in CNS (46), REFMAC (42, 47), and PHENIX (48).
Table 2.
Data collection, phasing and refinement statistics
GDP exchange rates in sec−1 for wild-type, I56C/Q333C, and D328R Gαi1 subunits. The exchange rate was determined by fitting the data to an exponential association equation Fλ = Fλmax(1 − e−kt). The value of k is given in sec−1 ± SEM.
| I56C/Q333C | Anomalous S I56C/Q333C | Wild-type | |
|---|---|---|---|
| Wavelength | 1.0 Å | 1.77 Å | 0.98 Å |
| Resolution | 2.9 Å | 3.5 Å | 3.0 Å (3.11–3.0 Å) |
| Completeness | 96.8 (80.6) | 100.0 (100.0) | 91.4 (92.6) |
| I/σ | 20.5 (1.9) | 62.9 (1.8) | 18.1 (3.6) |
| Rsyma | 0.05 (0.30) | 0.090 (0.30) | 0.08 (0.38) |
| Figure of Merit | – | 0.32 (17 sites) | – |
|
| |||
|
Model refinement statistics | |||
| Rcrystb | 0.249 | ||
| Rfreeb | 0.293 | ||
| RMSD bonds | 0.016 Å | ||
| RMSD angles | 1.7° | ||
Structural analyses
All structural comparisons of the GDP-AlF4−-bound I56C/Q333C Gαi1 subunit structure were performed against wild-type protein crystallized in a binary complex with GDP-AlF4− in both the same space group with the same crystallographic packing (P43212) and space group P3221 (PDBID 1GFI). Root mean squared deviations of Cα positions were calculated in O (45) and temperature factors were calculated in CNS (46). Figures and movies were made using PyMOL (DeLano Scientific LLC) and LSQMAN (49).
Surface plasmon resonance (SPR) biosensor measurements
SPR binding assays were performed at 25°C on a BIAcore 3000. N-terminally biotinylated versions of peptides KB-752 (SRVTWYDFLMEDTKSR; (50)) and KB-1753 (SSRGYYHGIWVGEEGRLSR; (51)), were diluted to 0.1 μg/ml in BIA running buffer [10 mM HEPES (pH 7.4), 150 mM NaCl, 10 mM MgCl2, and 0.005 % NP40], and coupled to separate flow cells of streptavidin biosensor chips (Biacore SA chips; GE Healthcare) using the MANUAL INJECT command to a surface density of approximately 750 resonance units. Prior to injection, the Gα subunits were diluted to a range of final concentrations used to establish KD in BIA running buffer containing either 100 μM GDP or 100 μM GDP, 30 μM AlCl3, and 10 mM NaF. 40 μL of 10–50 μM Gα subunits were then simultaneously injected over flow cells at 10 μl/min followed by a 200 second dissociation phase. Signal from binding to a non-Gα interacting, biotinylated peptide (C-terminal tail of mNOTCH1: PSQITHIPEAFK) was subtracted from all binding curves to correct for non-specific binding and buffer shifts created during injection. Surfaces were regenerated between each injection with two pulses of 10 μl of regeneration buffer (500 mM NaCl and 25 mM NaOH) at 20 μl/min. Binding curves and kinetic analyses were conducted using BIAevaluation software version 3.0 and plotted using GraphPad Prism version 4.0b. Binding affinities were calculated using simultaneous association (ka) and dissociation (kd) rates derived from generated sensorgram curves.
RESULTS
Identification of the I56C/Q333C and D328R G αi1 subunits as functional mimetics of R*-bound G αi1
As structural and functional mimetics of the receptor-activated form of Gα, I56C/Q333C and D328R Gαi1 proteins should exchange nucleotide at an accelerated rate, even in the absence of receptor, as compared to wild-type Gα subunits. To further characterize the mutant Gα subunits, the ability of each mutant to bind to activated receptors was compared to wild-type Gαi1.
(1) Rates of nucleotide exchange
The first experiment to evaluate if the I56C/Q333C and D328R Gαi1 subunits mimic R*-bound Gαi1 was the measurement of the rate of nucleotide exchange. Rhodopsin is the cognate receptor for Gt, which is a member of the Gi family of proteins. Rhodopsin can also efficiently catalyze nucleotide exchange in Gαil subunits (37, 52). Additionally, Gαi1 subunits can stabilize extra metarhodopsin II formation in a dose-dependent manner (Supplementary Figure 2). These studies are consistent with a functional interaction between these Gαil proteins and rhodopsin. The uncatalyzed rate of nucleotide exchange for the I56C/Q333C (0.013 sec−1) and D328R (0.018 sec−1) Gαi1 subunits are both increased relative to wild type under both uncatalyzed (0.002 sec−) and rhodopsin-catalyzed (0.008 sec−1) conditions (Figure 2 a–c, Table 1), consistent with the structural change in the I56C/Q333C Gαi1 subunit and the electrostatic change in the D328R Gαi1 subunit functionally mimicking receptor-bound Gα. Both mutant Gαi1 subunits can associate with receptor (see section 2, below) and the addition of activated rhodopsin to either mutant Gαi1 did not further enhance nucleotide exchange; this suggests that these mutant Gαi1 proteins are maximally activated in the absence of R*. To ensure rates of exchange in the D328R Gαil protein were not a non-specific effect of mutation we expressed control proteins D328N, D328A and K220D Gαil. The D328N and K330D mutations did not dramatically perturb nucleotide exchange as compared to wild type, and while the D328A mutant did exhibit increased basal exchange rates, it was not to the extent seen in either I56C/Q333C or D328R Gαil. In addition, the increases in exchange due to individual cysteine mutations in the background of a cysteine-depleted Gαil protein (37) at residues 56 and 333 (Supplementary Figure 3), when added together, accounts for only half of the fold-increase observed in the I56C/Q333C Gαil double mutant (Figure 2). Finally, unlike the I56C/Q333C or D328R Gαil proteins, nucleotide exchange in all of the control mutant Gαil subunits exhibited accelerated nucleotide exchange in the presence of activated receptor to accelerate nucleotide exchange, as does wild-type Gαil protein.
Figure 2.

Elevated rates of basal nucleotide exchange in Gαi1 mutants. (a) Basal Exchange. The basal rate of GTP-γS binding was determined by monitoring the relative increase in the intrinsic Trp211 fluorescence. Basal exchange for the wild-type is shown with a solid line (bottom trace), basal exchange for the I56C/Q333C double mutant is shown with a small-dashed line (------), and basal exchange for the D328R mutant is shown with a large-dashed line (——). (b) Receptor catalyzed nucleotide exchange. Nucleotide exchange for the receptor-catalyzed wild-type and mutant Gαi1 subunits was determined by monitoring the relative increase in the intrinsic Trp211 fluorescence performed in presence of 500 nM rhodopsin. The addition of rhodopsin does not significantly affect the rate of nucleotide exchange in the I56C/Q333C or D328R Gαi1 subunits but catalyzes a 4-fold rate enhancement for wild-type Gαi1 subunits (p=0.0005). (c) Quantitation of the initial rates of basal and rhodopsin-catalyzed nucleotide exchange monitored by the relative increase in the intrinsic Trp211 fluorescence for wild-type and mutant Gαi1 subunits. (d) Basal nucleotide exchange of GDP- or GDP-AlF4−-bound Gαi1 subunits as measured by the increase in emission from BODIPY-labeled GTPγS upon addition of GDP-bound Gαi1 or GDP-AlF4−-bound Gαi1 subunits. Relative increase was plotted as percentage of basal BODIPY-GTPγS fluorescence prior to addition of Gα subunits. Data are the average of three independent experiments (**p = 0.0027 and p=0.0001).
Table 1.
Summary of nucleotide exhange rates
Crystallographic data collection, phasing, and refinement statistics. Values in parenthesis indicate statistics for the highest resolution shell. aRsym = Σ |Ii − <I>|/ΣIi where I is intensity, “I” is the ith measurement, and <I> is the weighted mean of I. bRcryst, Rfree = Σ ||Fobs| − |Fcalc||/ΣFobs.
| Basal | Rhodopsin-catalyzed | |
|---|---|---|
| I56C/Q333C Gαi | 0.013 ± 0.001 sec−1 | 0.012 ± 0.0008 sec−1 |
| D328R Gαi | 0.018 ± 0.003 sec−1 | 0.019 ± 0.003 sec−1 |
| Wild type Gαi | 0.0021 ± 0.0001 sec−1 | 0.0081 ± 0.0009 sec−1 |
The structure of the complex between bound GDP and AlF4− in the nucleotide binding pocket of Gα has been correlated to the transition state associated with GTP hydrolysis in Gα proteins. The formation of a complex between bound GDP and AlF4− stabilizes the bound nucleotide through additional hydrogen-bonds from the nucleotide binding pocket to the GDP-AlF4− complex. To determine if the propensity for nucleotide release in the receptor-bound mimetics of Gαi1 subunits was sufficient to overcome these stabilizing forces and release GDP-AlF4−, fluorescently labeled GTPγS (BODIPY-GTPγS) was used to monitor nucleotide release by measuring the increase in BODIPY-GTPγS emission after the addition of Gα subunits. GDP-AlF4− -bound wild type and I56C/Q333C Gαi1 proteins both exhibit decreased binding of BODIPY-GTPγS when compared to their GDP-bound counterparts. This suggests additional contacts mediated by binding of AlF4−stabilizes bound GDP, reducing BODIPY-GTPγS binding in wild type and Gαi1 I56C/Q333C subunits (Figure 2d). Surprisingly, both GDP-AlF4− and GDP-bound D328R Gαi1 subunits bound BODIPY-GTPγS to a similar extent (Figure 2d) suggesting that AlF4− is not sufficient to stabilize nucleotide binding in the D328R Gαi1 protein. These results suggest that electrostatic perturbations at the base of the α5 helix exert distinct effects on both the affinity and duration of nucleotide binding.
(2) Binding to membrane-bound receptors
The ability of the GDP-Gαβγ subunits containing either wild-type, I56C/Q333C, or D328R Gαi1 to bind to rhodopsin-containing rod outer segment membranes in the dark, after light activation, and after addition of GTPγS to light activated complexes was investigated (Figure 3). Light activation significantly enhanced the membrane association of both wild-type and I56C/Q333C Gαi1 to a similar extent. Consistent with this being a more dramatic mimetic of receptor activated protein, D328R Gαi1 subunit had an even higher level of interaction with membrane bound receptors after light activation than did wild-type or I56C/Q333C Gαi1 proteins. As expected, subsequent addition of GTPγS dissociated the rhodopsin-Gαβγ complex for all three Gαi1 subunits, resulting in predominantly soluble GTPγS-bound Gαi1 subunits.
Figure 3.

Membrane localization and rhodopsin binding of the wild-type and mutant Gαi1 subunits. (a) Representative Coomassie stained SDS-PAGE analysis of wild-type (WT; top panel) I56C/Q333C (middle panel) and D328R (bottom panel) Gαi1 subunits reconstituted with excess Gβγ prior to binding to ROS in the dark, light, and after light activation followed by addition of GTPγS. DS, supernatant from dark sample; DP, pellet fraction from dark sample; LS, supernatant from light sample; LP, pellet from light sample; GS, supernatant from light and GTPγS activated sample; GP, pellet from light and GTPγS activated sample. (b) (c) and (d) and Quantitation of membrane binding. Each measurement is the average of three independent experiments. The D328R Gαi1 subunit showed a significant enhancement in binding to membrane fractions upon light activation as compared to either wild-type or I56C/Q333C Gαi1 subunits (**p = 0.0035 and p=0.0002, respectively).
Identification of global structural changes in the I56C/Q333C G αi1 by X-ray crystallography
To determine if the physical constraint of the α5 helix by the disulfide bond in the I56C/Q333C Gαi1 subunit propagated further structural changes, crystal structure of the I56C/Q333C Gαi1 was determined in complex with GDP-AlF4− to 2.9 Å resolution (Table 2). The structure of the I56C/Q333C Gαi1 is similar to wild-type Gαi1 (Figure 1a); however the superposition of the I56C/Q333C Gαi1 with wildtype protein results in root mean squared (RMS) deviation of 0.6 Å, which is higher than would be anticipated for the introduction of two cysteines. Excitingly, significant structural changes are located in regions of the GTPase domain previously identified for their importance in receptor binding and nucleotide binding, sensing, and exchange (Table 3).
Table 3.
Summary of structural features of I56C/Q333C Gαil
Summary of structural changes observed in I56C/Q333C Gαi1.
| Structural Element | Residues | Functional Role | Structural Change |
|---|---|---|---|
| P-loop | 41–45 | Phosphate-binding loop highly conserved in G proteins | 0.7 Å shift of G45 C α toward the phosphates |
| α1 helix | 44–58 | Walker A motif and connection to linker/β2- β3 loop | Translation toward α5 helix and nucleotide-binding pocket by 1.0 Å |
| α1-αA loop | 59–64 | Linker region between GTPase andhelical domains | Unfolding of αA helix at the α1-αA linker loop by 1.5 Å |
| β2-β3 hairpin | 188–197 | Connects α1/α5 with Switch II and β1 | Translation toward β1 strand by 1.3 Å; moves with α5 helix residues |
| β3–α4 loop | 202–207 | Part of Switch II; pulls away in GEF peptide and Gβγ-bound structures | Average 0.5 Å and maximum 1.25 Å movement away from AlF4− |
| α2 helix | 207–215 | Switch II, GEF peptide and Gβγ binding site | Unfolding and destabilization; pulls away from phosphate pocket by 0.8 Å |
| α4 helix | 294–299 | α4 helix upstream of α4-β6 loop involved in receptor interaction | Average 0.9 Å movement relative to wild type |
| α4–β6 loop | 312–316 | Known receptor contact region | Average 1.3 Å movement relative to wild type |
| β4-α3 loop | 234–238 | Switch III, known to move in activating peptide structure | Pulls away from Switch II by 0.8 Å |
| β6–α5 loop | 324–329 | TCAT motif directly interacts and stabilizes guanine ring | 0.5 Å movement towards the guanine ring of Cα and side chains |
| α5 helix | 329–345 | Known receptor contact and exchange mediator | Translation toward nucleotide-binding pocket by 0.6 Å |
The first statistically significant movement is at the site of introduction of the two cysteines. The initial σA-weighted 2|Fo|—|Fc| maps of the I56C/Q333C Gαi1 showed clear density between Cys 56 and Cys 333 indicating the presence of a disulfide bond (Figure 4a). This disulfide bond constrains the position of the α5 helix (residues 328–345) towards the guanine nucleotide-binding site along the helical axis (Figure 4b; average Cα displacement of 0.6 Å and a maximal Cα displacement of 0.8 Å over the 17 residue helix) and shifts the position of the preceding β6-α5 loop. The disulfide bond further constrains the four C-terminal residues of the α1 helix (residues 46–57) towards the α5 helix with an average displacement of 1.0 Å and a maximal displacement of 1.5 Å. This shifts positions of the Cα atoms of the α1-αA loop (residues 59–64) an average of 1.5 Å (Figure 4b). There is also a significant, albeit smaller, effect on the positions of the Cα atoms in the preceding P-loop (residues 41–45, Figure 4c). In the P-loop, a conserved glycine motif allows the backbone amide nitrogens to make hydrogen-bonding contacts with the nucleotide phosphates while the conserved Glu43 orients Arg 178 to further stabilize bound nucleotide. The Cα positions of the P-loop are shifted slightly toward the nucleotide with an average displacement of 0.65 Å and maximum displacement of 0.8 Å; however, the hydrogen-bonding interactions to phosphate and AlF4− are not significantly altered, and the side chains of Glu 43 and Arg 178 do not change conformation such that the latter remains within hydrogen-bonding contact of the phosphates. While AlF4− binding stabilizes the I56C/Q333C Gαi1 structure in a conformation similar to that observed for the wild type Gαi1 under the same conditions, the subtle differences between the two structures in the P-loop indicate a propensity towards conformational adaptability in the phosphate-binding region which may aid in nucleotide release. These subtle changes are detailed in figure 4f, the RMSD plot for this structure (as compared to wild-type protein). Figure 4f demonstrates that while subtle changes are present throughout the protein, there are particular regions which are significantly altered as compared to wild type, including residues in the α1-αA linker region (residues 56–64), the αB helix (residues 98–102), β2 and β3 strands (190–195), switch II region (residue 215–217) and the α4/β6 loop (311–314). These differences are not related to differences in space group between the two proteins, as these changes are still noted when comparing I56C/Q333C GαilGDP-AlF4− to wild type GαilGDP-AlF4− crystallized in the same space group.
Figure 4.


Structural changes in the I56C/Q333C Gαi1. All comparisons are made between the I56C/Q333C Gαi1 subunit and the wild-type Gαi1 subunit crystallized in complex with GDP-AlF4−. Coloring for panels (b) - (d) is the same as in Figure 1. (a) 2|Fo| - Fc| density (blue mesh) contoured at 1.2 σ shows a connection between the side chains at positions 56 and 333 and is consistent with the formation of a disulfide bond. (b) Movement of the α5 helix, the β2-β3 hairpin and the α1-αA linker that connects the GTPase (dark blue) and helical domains (cyan). (c) Movement of the P-loop. (d) Movement of the β2-β3 hairpin away from the α1 helix toward the β1 strand. The hairpin directly connects the second linker (residues 177–182) and switch II (residues 202–215; light green). (e) |Fo| - |Fc| simulated annealing omit density calculated using CNS and contoured at 3.0 σ after the removal of all atoms between residues 202–217 (α2 helix and switch II) highlights the shift between the I56C/Q333C (blue/green) and wild type (grey) Gαi1. The GDP molecule is in red and the AlF4− is in blue. (f) RMSD of each alpha carbon atom in I56C/Q333C Gαil structure as compared to wild-type Gαil (PDB 1GFI, selected regions overlaid in 4a-e).
While shifts in the positions of the α5 and α1 helices (containing residues 333 and 56, respectively) and their associated connecting loops were anticipated, several additional regions of the I56C/Q333C Gαi1 have unexpectedly altered positions when compared to wild-type Gαi1 (Table 3, Figure 4b–e). These regions include the β2–β3 hairpin (residues 188–197, Figure 4b, d), the β3-α2 loop (Figure 4d), the α2 helix (Switch II, residues 202–218, Figure 4d, e), and the β4–α3 loop (Switch III, residues 234–240). Each of these regions has previously been shown to have important functions connected with nucleotide sensing and exchange; the switch regions in particular exhibit distinct conformations in response to the identity of the bound guanine nucleotide, thereby altering the affinity of Gα for Gβγ and other binding partners.
In addition to visible changes in atomic positions, parameters such as crystallographic temperature factors provide a measure of relative mobility in crystallographic models, and these are generally used to identify regions of increased mobility in a given structure. A comparison of temperature factors between the wild-type and I56C/Q333C Gαi1 proteins crystallized in the same space group and at similar resolutions (3.0 Å for wild-type and 2.9 Å for the I56C/Q333C Gαi1) shows an average increase of 31 Å2 for the I56C/Q333C Gαi1 as compared to wild type (Supplementary Figure 1). This is consistent with the I56C/Q333C Gαi1 subunit having increased mobility and flexibility compared with wild-type Gαi1. The increase in temperature factors is even greater in several key regions of the protein, reaching 50 Å2 above wild type in the α1 and α5 helices and the switch regions.
Verification of structural changes of the switch regions of the I56C/Q333C and D328R G αi1 mutants by binding to nucleotide-state selective peptides
The ability of wild-type, I56C/Q333C and D328R Gαil subunits to bind to peptides selective for the activation state of Gαi proteins were analyzed by surface plasmon resonance (Figure 5). The peptides KB-1753 and KB-752 discriminate between active, GDPAlF4−-bound and inactive, GDP-bound Gαi1 subunits (50, 51). We found these peptides bind to wild-type and mutant Gαi1 subunits with the same selectivity (Figure 5) but with different binding kinetics and affinities (Table 4). The peptide KB-752, selective for GDP-bound Gα subunits, has been shown to bind to Gαil at the switch II region (50). KB-752 has an 8-fold higher affinity for GDP-bound wild-type Gαil subunits than it does for GDP-bound I56C/Q333C Gαi1 subunits, suggesting that the switch II region in the GDP-bound I56C/Q333C Gαi1 likely adopts a different conformation than is observed for GDP-bound wild-type Gαi1 subunits. This suggests that the altered position of switch II observed in the crystal structure is also present in solution. This peptide binds to GDP-bound D328R on the order of that seen in wild-type Gαi1 subunits, consistent with these proteins sharing a similar switch II geometry where the peptide binds. Conversely, GDP-AlF4−-bound I56C/Q333C and D328R Gαi1 subunits bind peptide KB-1753 (selective for activated Gαi1 subunits (51)) with a 3–4 fold higher affinity than wild-type Gαi1-GDP-AlF4−. This suggests that GDP-AlF4− bound I56C/Q333C Gαi1 and D328R Gαi1 subunits readily accommodates binding of the peptide selective for activated subunits.
Figure 5.

Surface plasmon resonance analysis of the binding of Gαi1 subunits to nucleotide-state selective peptides. Surface plasmon resonance demonstrates peptides KB-752 and KB-1753 discriminate between activation state of Gαi1 proteins. In each graph, the dashed line represents binding of the indicated Gα subunit to immobilized peptide KB-752, selective for inactive (GDP-bound) Gα subunits, while the solid line represents binding to immobilized peptide KB-1753, selective for activated (GTP-bound) Gα subunit. (a) Wild-type Gαi1in the presence of GDP; (b) Wild-type Gαi1 in the presence of GDP-AlF4−; (c) I56C/Q333C Gαi1 in the presence of GDP; (d) I56C/Q333C Gαi1 in the presence of GDP-AlF4−. (e) D328R Gαi1 in the presence of GDP; (f) D328R Gαi1 in the presence of GDP-AlF4−.
TABLE 4.
Comparison of kon, koff, and Kd values for wild-type, I56C/Q333C and D328R Gαi1. Values are from an average of 4 trials (wild type) or 2 trials (I56C/Q333C and D328R).
| KB-752 (selective for Gαi-GDP) | kon (1/M • s) | koff (s−1) | Kd (μM) |
|---|---|---|---|
| WT Gαi GDP | 21,100 ± 5,800 | 0.06 ± 0.01 | 2.84 ± 0.81 |
| I56C/Q333C Gαi GDP | 7,750 ± 250 | 0.22 ± 0.03 | 28.2 ± 2.9 |
| D328R Gαi GDP | 8,560 ± 650 | 0.01 ± 0.001 | 1.28 ± 0.02 |
| KB-1753 (selective for Gαi-GDP•AlF4) | |||
| WT Gαi GDP •AlF4− | 22,900 ± 2,300 | 0.025 ± 0.001 | 1.1 ± 0.1 |
| I56C/Q333C Gαi GDP•AlF4− | 16,800 ± 1,700 | 0.006 ± 0.0008 | 0.36 ± 0.01 |
| D328R Gαi GDP•AlF4− | 8,810 ± 1,390 | 0.003 ± 0.0004 | 0.26 ± 0.04 |
DISCUSSION
In the rate-determining step of G protein signaling, activated GPCRs catalyze the release of GDP in the Gα subunit of heterotrimeric G proteins (12). Gα lacking bound nucleotide is unstable in solution; in vivo Gα remains bound to R* until GTP binding, which result in predominantly soluble Gα-GTP subunits, capable of activating downstream effectors. Previous studies have mapped the binding site for the receptor onto a contiguous surface of the Gα subunit (30, 36) (Figure 1a), identified the three-dimensional constraints underlying the interaction between the C-terminus of the Gα subunit and receptor (29), and have characterized a rigid body movement of the α5 helix of Gα that accompanies receptor-catalyzed allosteric GDP release (37, 53). The latter study set the stage for the current work, where the known conformational changes in the α5 helix were used as a starting point to mimic the R*-bound conformation of Gα through a disulfide bond, or by replicating the movement of the α5 helix dipole.
The I56C/Q333C G αi1 subunit is a structural and functional mimic of the R*-bound G α subunit
An anticipated hallmark of an R*-bound Gα subunit is a dramatically decreased ability to bind to GDP, and a conformation predisposed to binding GTP. Enhanced nucleotide exchange in both mutant Gαi1 subunits occurs in the absence of receptor and is consistent with both of these mutant subunits acting as functional mimics of the R*-bound Gα subunit. In the I56C/Q333C Gαi1 variant, this is a result of structural changes that are anticipated for an R*-bound Gα subunit based on previous EPR studies. Even in the presence of GDP-AlF4−, the crystal structure of the I56C/Q333C Gαi1 subunit reveals altered positions for the switch regions, which shows switch II and switch III each have a conformation that is intermediate between those observed for the active (GTP-bound) and inactive (GDP-bound) forms of the Gα subunit.
This structure of the I56C/Q33C Gαi1 subunit also exhibits increased temperature factors relative to wild-type structures. Crystallographic temperature factors give a relative assessment of the average displacement of any given atom in a structure, which is usually interpreted as mobility. Elevated temperature factors throughout the molecule may also reflect an greater number of substructures in the ensemble, resulting in increased frequency of conformations facilitating nucleotide release. Temperature factors are generally limited to a range of 0 Å2 to 100 Å2 in soluble proteins, with a higher number correlating with increased motions. A 31 Å2 average increase in crystallographic temperature factors is observed in the I56C/Q333C Gαi1 as compared to wild-type crystallized in the same space group and at similar resolution (2.9 Å for the I56C/Q333C Gαi1 subunit and 3.0Å for the wild-type). These temperature factors are elevated to an even greater extent in the α1 and α5 helices and the switch I and switch II regions of the I56C/Q333C Gαi1, where they are an average of 50 Å2 above wild-type. This suggests that the I56C/Q333C variant is more conformationally variable than wild-type, especially in these four regions. An increase in protein mobility in Gαi1 proteins during nucleotide exchange has previously been noted in EPR experiments (53), while NMR spectra of nucleotide-free chimeric Gαt show significant line-broadening, which has been interpreted as an increase in protein dynamics (57). Increased dynamics may facilitate exit tunnel formation by moving the protein conformation into a higher energy landscape that can more easily convert the structure of the Gα subunit into the open state.
Increased flexibility in switch II and altered the P-loop conformations may also play a role in nucleotide exchange dynamics. The increased intrinsic nucleotide exchange rates of GDP-bound Gαi subunits compared to Gαt subunits is a distinctive feature of Gαi proteins. Another distinctive feature of Gαi subunits is the disorder in switch II regions, as compared to the more ordered switch II regions of Gαt-GDP proteins. These data are consistent with increased flexibility of this region contributing to increased nucleotide exchange in Gαi proteins.
There are a number of statistically significant structural changes clustered in the GTPase domain of the I56C/Q333C Gαi1 subunit (Figure 4f and Table 3). Many of these regions are implicated in nucleotide sensing, binding, exchange, or receptor interaction. The region linking the helical domain and GTPase domain, which in this structure shows a marked shift in position relative to wild-type, is predicted to lie at some distance from the regions of Gα already known to interact with receptors, such as the C-terminus and α4-β6 loop. Consistent with an allosteric mechanism of receptor-mediated G-protein activation, this region has been linked to specificity of receptor coupling, as mutation of the residue of Gαq homologous to residue 61 in the linker region of Gαil can alter the specificity of receptor coupling (55). The high RMSD deviation in this region, in addition to other regions known to be involved in receptor contacts, is consistent with an overall picture of specific structural changes and overall protein flexibility, which contribute to receptor-mediated G protein activation. While this data points to factors which may play a role in nucleotide release in Gαi proteins, further studies are currently underway to determine which of these conformational changes contribute to GDP release and which are compensatory shifts reflecting the fact that the protein is stabilized in its GDP-AlF4− transition state.
Forces contributing to allosteric GDP release in G α subunits: the D328R G αi1 subunit is a functional mimic of the R*-bound G α subunit
Structural and functional characterization of the D328R Gαi1 subunits suggest electrostatics from movement of the α5 helix dipole may contribute to increased nucleotide release. Shoemaker et al (54) points out the stabilizing role of a negatively charged residue (such as the native Asp at position 328) at the N-terminus of an α-helix with a positively helix dipole (Figure 1b). The negative charge can neutralize and stabilize the positively charged helix dipole. Mutation of Asp328 to positively charged Arg would be predicted to perturb this helix cap and de-shield the amide nitrogen. Consistent with this idea, the D328R mutant exhibits markedly increased nucleotide exchange while D328N has activity similar to wild type, suggesting that the Asn can participate in stabilization of the helix dipole. Substitution of D328 with Ala would not be predicted to have the same effect, since the amide nitrogen would be de-shielded. Indeed, the D328A mutant exhibits somewhat elevated nucleotide exchange as compared to wild type.
The use of electrostatics to initiate nucleotide release is a novel proposal for the mechanism of nucleotide exchange in G proteins. These results, combined with previous EPR studies, strongly suggest that the movement of the helix dipole during receptor binding contributes to decreased affinity of Gα for GDP by altering the local electrostatic distribution around the nucleotide. It is possible that even subtle changes in the electric field may act to reorient the GDP as a prerequisite to nucleotide release. In the absence of stabilizing contacts between a third phosphate or AlF4−, even complexation with AlF4− cannot overcome the electrostatic perturbation of the D to R mutation, allowing GDP-AlF4− to be exchanged for BODIPY-GTPγS. A decreased affinity for GDP in D328R Gαi1 subunits may result in a Gαi subunit exquisitely poised to bind GTP. Similarly, a nucleotide-free Gαi1 subunit is anticipated to bind to activated receptors with high affinity, and indeed D328R Gαi1 subunits display enhanced association with membranes containing activated receptors (Figure 3). This is consistent with ability of activated receptors to stabilize Gα subunits in the nucleotide-free state. Although Gαil subunits are known to bind to BODIPY-labeled GTPγS analog at a slower rate (and with relatively lower affinity) than unlabeled GTPγS (56), this reagent provides a useful tool to distinguish differences in nucleotide exchange between subunits which demonstrate elevated intrinsic nucleotide exchange.
Peptide binding studies are another tool which can be used to tease out differences between wildtype, D328R and I56C/Q333C Gαil subunits. The KB752 peptide which has been shown to bind to switch II region of inactive Gαil subunits binds to wild-type and D328R Gαil subunits with a similar affinity, not especially surprising given the mutation at residue 328 is in the α5 helix, removed from the switch II region. However, the I56C/Q333C mutation in Gαil subunits does substantially reduce the KB752 binding, indicating changes in the relative positions of these two helices can be allosterically communicated to the switch II region. On the other hand, both D328R and I56C/Q333C Gαil GDP-AlF4− subunits demonstrate a 3–4 fold enhancement in binding affinities to the KB1752 peptide (selective for activated subunits) relative to wild-type Gαi. This would be consistent with a conformation for both mutant proteins which favors GDP release and readily adopts the activated conformation, as would be expected for receptor-bound mimetics of activated Gα proteins. However, the differing binding affinities for the mutant subunits to peptides selective for the inactive state, as well as differences in ability of these proteins to exchange GDP-AlF4− for BODIPY-GTPγS, together suggest these mutations exert their effects on nucleotide release through distinct mechanisms.
Mechanisms of GEF-catalyzed GDP release from G proteins
For small G proteins with soluble GEFs, the GEFs bind across the switch I and switch II regions, and the conformational change induced by this interaction with switch II weakens GDP binding in two ways (5, 58). In the case of an Arf-GEF, the Sec7 domain utilizes the carboxylate from a conserved acidic residue the FG loop of the GEF to form a new hydrogen bond with a lysine in the phosphate-binding P-loop of the G protein, thereby introducing an electrostatic repulsion between the β-phosphate and negatively charged carboxylate group, and decreasing nucleotide affinity. Second, the binding of the Mg2+ ion in the nucleotide binding pocket is abrogated by steric interference with the new position of switch II. While GPCRs act as GEFs on heterotrimeric G proteins, they do not bind to the switch II region, which is in contact with Gβγ. Therefore, a different mechanism appears to have evolved to facilitate nucleotide exchange in Gα subunits. Here, nucleotide exchange is initiated with a roto-translation of the α5 helix, which exerts a coulombic effect that contributes to a reduction in its GDP-binding affinity. While the structure presented here is in complex with GDP-AlF4−, there are hints that this coulombic effect pulls the phosphates of the guanine nucleotide away from the P-loop. Other previously identified structural elements distributed throughout the GTPase domain respond to this roto-translation and nucleotide shift with subtle structural changes (Figure 6, Supplementary Movie 1).
Figure 6.
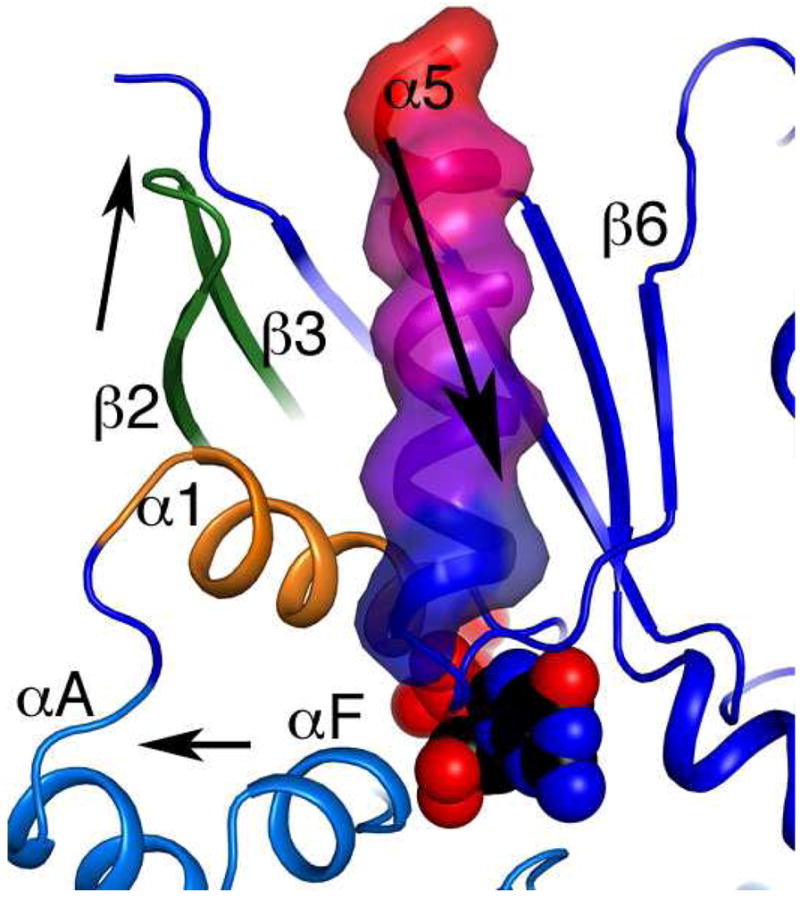
Proposed mechanism of receptor-catalyzed GDP release by Gαi1 mediated by movement of the α5 helix. Crystallographic and biochemical data implicate the receptor-induced movement of the α5 helix toward the bound GDP molecule in destabilization of guanine ring through coulombic attraction to the positive helix dipole. The movement of the α5 is coupled with a translation in the α1 helix (orange), the β2-β3 hairpin (dark green), and the αF helix (cyan). These concerted motions may be the first step of opening a tunnel to the guanine nucleotide binding pocket and allowing exchange.
The direction of nucleotide egress from the binding site has been debated using terms such as ‘front door,’ referring to egress toward the guanine ring, and ‘back door,’ referring to egress toward the phosphate corridor (59). After the initial roto-translation of the α5 helix, the negatively charged guanine ring may be attracted by a positively charged helix dipole, pulling the base of the guanine nucleotide toward the front door. Such an attraction would likely reorient the nucleotide and weaken the protein-phosphate contacts by pulling the phosphates away from the P-loop. Exit via the front door would require a conformational change in the TCAT motif, the side chain of Arg 176 (on the αF-β2 loop) and the side chain of Asp 272 (on the α4 helix).
Alternatively, the reorganization of the P-loop and decreased interactions with phosphates may allow nucleotide release through a back door route (Supplementary Movie 3), with the positively charged dipole shielded from bound nucleotide through helix capping via negatively charged D328. Exit via the back door requires a change in the position of the main chain of switch II and P-loop regions. Hints as to the nature of conformational changes near the back door exit route come from the I56C/Q333C Gαi1 GDP-AlF4− structure. Intriguingly, the position of the switch II region the I56C/Q333C Gαi1 structure can be modeled as a continuous motion into the dramatic conformational change observed in the analysis of the complex between the KB-752 and D2N peptides and the Gαi1 subunit (31) (supplementary movie 2). While the KB-752 peptide does not exhibit sequence similarity to Gα binding regions on any known GPCRs, conformational changes of the switch II region upon the binding of KB- 752 appear to correlate with increased nucleotide exchange activity in Gα subunits (50). In addition to the likely movement of the switch II region during back door nucleotide exchange, the structure of the I56C/Q333C Gαi1 suggests that a subtle shift in phosphate binding residues allows the γ-phosphate binding pocket to adopt a more open position (Figure 4c, Supplementary Movie 3). A larger conformational change may occur in the P-loop region during physiological GDP release than that seen in the GDP-AlF4−-bound I56C/Q333C Gαi1 subunit.
The results presented here do not unambiguously point to exit through either the front or back door, and egress through either route would require a unique arrangement of structural elements of the Gα subunit that has not yet been observed in any published Gα subunit crystal structure. Physiological nucleotide release from a front door exit route would likely involve the TCAT motif, αA-β2 loop, and α4 helix, while a back door egress would likely involve switch II and the P-loop. These data reported here do not discriminate the conformational movements required for GDP egress from those movements that are important for decreasing nucleotide affinity, which is a prerequisite for nucleotide release. If GTP, GTPγS, or GDP-AlF4− is bound in the active site, the additional stabilization through protein interactions to the third phosphate (or AlF4−) likely prevents the reorientation of the nucleotide, thus blocking the conformational changes that would be required for nucleotide egress. In the absence of the stabilizing influence imparted by a third phosphate or AlF4−, GDP-bound subunits may exhibit greater overall conformational flexibility, especially in the switch II and P-loop regions, which are involved in binding AlF4− or the γ phosphate of GTP. The lack of these additional contacts may contribute to allosteric changes at both the front door and the back door of the nucleotide binding site.
The Gα subunit differs from small G proteins in that receptor activation results in association of heterotrimeric Gαβγ with receptors. The Gβγ subunits have been implicated in receptor-mediated nucleotide exchange, and a number of proposals have suggested how these subunits affect GDP release (31, 60). While this study cannot address whether Gβγ-mediated structural changes follow the initial recognition event between the GPCR and the Gα subunit, the contribution of the roto-translation of the α5 helix to nucleotide exchange does not preclude the involvement of Gβγ in GDP release.
Conclusion
Physiological nucleotide exchange in Gα is likely initiated through a receptor-catalyzed roto-translation of the α5 helix. The data presented here suggest that following receptor binding the change in position of the α5 helix may be transmitted into subtle positional, electrostatic, and dynamic changes throughout Gα that may contribute to the efficiency of G protein activation.
Supplementary Material
Acknowledgments
Use of the Advanced Photon Source was supported by the U. S. Department of Energy, Office of Science, Office of Basic Energy Sciences, under Contract No. DE-AC02-06CH11357. Use of the IMCA-CAT beamline 17-ID at the Advanced Photon Source was supported by the companies of the Industrial Macromolecular Crystallography Association through a contract with the Center for Advanced Radiation Sources at the University of Chicago. Use of the LS-CAT Sector 21 was supported by the Michigan Economic Development Corporation and the Michigan Technology Tri-Corridor for the support of this research program (Grant 085P1000817). We thank G. Liao for expert technical assistance and Eric Dawson for assistance with computer modeling.
Footnotes
This work was supported by Pilot Project funds from the Vanderbilt Institute for Chemical Biology and a Young Investigator award from NARSAD to T.M.I., NIH grant EY06062 to H.E.H., NIH fellowship F30 MH074266 to AJK, and NIH grant GM082892 to D.P.S.
The atomic coordinates and structure factors for Gαi1 I56C/Q333C (code 3D7M) have been deposited in the Protein Data Bank, Research Collaboratory for Structural Bioinformatics, Rutgers University, New Brunswick, NJ (http://www.rcsb.org/).
SUPPORTING INFORMATION AVAILABLE three movies and three figures are available free of charge at http://pubs.acs.org.
References
- 1.Oldham WM, Hamm HE. Structural basis of function in heterotrimeric G proteins. Q Rev Biophys. 2006;39:117–166. doi: 10.1017/S0033583506004306. [DOI] [PubMed] [Google Scholar]
- 2.Bourne HR. How receptors talk to trimeric G proteins. Current Opinion in Cell Biology. 1997;9:134–142. doi: 10.1016/s0955-0674(97)80054-3. [DOI] [PubMed] [Google Scholar]
- 3.Oldham WM, Hamm HE. Heterotrimeric G protein activation by G-protein-coupled receptors. Nat Rev Mol Cell Biol. 2008;9:60–71. doi: 10.1038/nrm2299. [DOI] [PubMed] [Google Scholar]
- 4.Johnston CA, Siderovski DP. Receptor-mediated activation of heterotrimeric Gproteins: current structural insights. Mol Pharmacol. 2007;72:219–230. doi: 10.1124/mol.107.034348. [DOI] [PubMed] [Google Scholar]
- 5.Vetter IR, Wittinghofer A. The guanine nucleotide-binding switch in three dimensions. Science. 2001;294:1299–1304. doi: 10.1126/science.1062023. [DOI] [PubMed] [Google Scholar]
- 6.Coleman DE, Berghuis AM, Lee E, Linder ME, Gilman AG, Sprang SR. Structures of Active Conformations of Giα1 and the Mechanism of GTP Hydrolysis. Science. 1994;265:1405–1412. doi: 10.1126/science.8073283. [DOI] [PubMed] [Google Scholar]
- 7.Coleman DE, Sprang SR. Crystal Structures of the G Protein Giα1 Complexed with GDP and Mg2+: A Crystallographic Titration Experiment. Biochemistry. 1998;37:14376–14385. doi: 10.1021/bi9810306. [DOI] [PubMed] [Google Scholar]
- 8.Lambright DG, Noel JP, Hamm HE, Sigler PB. Structural determinants for activation of the α-subunit of a heterotrimeric G protein. Nature. 1994;369:621–628. doi: 10.1038/369621a0. [DOI] [PubMed] [Google Scholar]
- 9.Mixon MB, Lee E, Coleman DE, Berghuis AM, Gilman AG, Sprang SR. Tertiary and Quaternary Structural Changes in Giα1 Induced by GTP Hydrolysis. Science. 1995;270:954–960. doi: 10.1126/science.270.5238.954. [DOI] [PubMed] [Google Scholar]
- 10.Noel JP, Hamm HE, Sigler PB. The 2.2 Å crystal structure of transducin-α complexed with GTPγS. Nature. 1993;366:654–663. doi: 10.1038/366654a0. [DOI] [PubMed] [Google Scholar]
- 11.Sondek J, Lambright DG, Noel JP, Hamm HE, Sigler PB. GTPase mechanism of Gproteins from the 1.7-Å crystal structure of transducin α•GDP•AIF4−. Nature. 1994;372:276–279. doi: 10.1038/372276a0. [DOI] [PubMed] [Google Scholar]
- 12.Ferguson KM, Higashijima T, Smigel MD, Gilman AG. The influence of bound GDP on the kinetics of guanine nucleotide binding to G proteins. J Biol Chem. 1986;261:7393–7399. [PubMed] [Google Scholar]
- 13.Hamm HE, Deretic D, Arendt A, Hargrave PA, Koenig B, Hofmann KP. Site of G protein binding to rhodopsin mapped with synthetic peptides from the alpha subunit. Science. 1988;241:832–835. doi: 10.1126/science.3136547. [DOI] [PubMed] [Google Scholar]
- 14.Martin EL, Rens-Domiano S, Schatz PJ, Hamm HE. Potent peptide analogues of a G protein receptor-binding region obtained with a combinatorial library. J Biol Chem. 1996;271:361–366. doi: 10.1074/jbc.271.1.361. [DOI] [PubMed] [Google Scholar]
- 15.Rasenick MM, Watanabe M, Lazarevic MB, Hatta S, Hamm HE. Synthetic peptides as probes for G protein function. Carboxyl-terminal G alpha s peptides mimic Gs and evoke high affinity agonist binding to beta-adrenergic receptors. J Biol Chem. 1994;269:21519–21525. [PubMed] [Google Scholar]
- 16.Cai K, Itoh Y, Khorana HG. Mapping of contact sites in complex formation between transducin and light-activated rhodopsin by covalent crosslinking: use of a photoactivatable reagent. Proc Natl Acad Sci U S A. 2001;98:4877–4882. doi: 10.1073/pnas.051632898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Itoh Y, Cai K, Khorana HG. Mapping of contact sites in complex formation between light-activated rhodopsin and transducin by covalent crosslinking: use of a chemically preactivated reagent. Proc Natl Acad Sci U S A. 2001;98:4883–4887. doi: 10.1073/pnas.051632998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Nanoff C, Koppensteiner R, Yang Q, Fuerst E, Ahorn H, Freissmuth M. The carboxyl terminus of the Galpha-subunit is the latch for triggered activation of heterotrimeric G proteins. Mol Pharmacol. 2006;69:397–405. doi: 10.1124/mol.105.016725. [DOI] [PubMed] [Google Scholar]
- 19.Denker BM, Boutin PM, Neer EJ. Interactions between the amino- and carboxyl-terminal regions of G alpha subunits: analysis of mutated G alpha o/G alpha i2 chimeras. Biochemistry. 1995;34:5544–5553. doi: 10.1021/bi00016a028. [DOI] [PubMed] [Google Scholar]
- 20.Denker BM, Schmidt CJ, Neer EJ. Promotion of the GTP-liganded state of the Go alpha protein by deletion of the C terminus. J Biol Chem. 1992;267:9998–10002. [PubMed] [Google Scholar]
- 21.Kostenis E, Conklin BR, Wess J. Molecular Basis of Receptor/G Protein Coupling Selectivity Studied by Coexpression of Wild Type and Mutant m2 Muscarinic Receptors with Mutant Gαq Subunits. Biochemistry. 1997;36:1487–1495. doi: 10.1021/bi962554d. [DOI] [PubMed] [Google Scholar]
- 22.Conklin BR, Farfel Z, Lustig KD, Julius D, Bourne HR. Substitution of three amino acids switches receptor specificity of Gqα to that of Giα. Nature. 1993;363:274–276. doi: 10.1038/363274a0. [DOI] [PubMed] [Google Scholar]
- 23.Conklin BR, Herzmark P, Ishida S, Voyno-Yasenetskaya TA, Sun Y, Farfel Z, Bourne HR. Carboxyl-Terminal Mutations of Gqα and Gsα That Alter the Fidelity of Receptor Activation. Molecular Pharmacology. 1996;50:885–890. [PubMed] [Google Scholar]
- 24.Blahos J, II, Mary S, Perroy J, de Colle C, Brabet I, Bockaert J, Pin JP. Extreme C Terminus of G Protein α-Subunits Contains a Site that Discriminates between Gi-coupled Metabotropic Glutamate Receptors. Journal of Biological Chemistry. 1998;273:25765–25769. doi: 10.1074/jbc.273.40.25765. [DOI] [PubMed] [Google Scholar]
- 25.Kostenis E, Gomeza J, Lerche C, Wess J. Genetic Analysis of Receptor-Gαq Coupling Selectivity. Journal of Biological Chemistry. 1997;272:23675–23681. doi: 10.1074/jbc.272.38.23675. [DOI] [PubMed] [Google Scholar]
- 26.Natochin M, Muradov KG, McEntaffer RL, Artemyev NO. Rhodopsin Recognition by Mutant Gsα Containing C-terminal Residues of Transducin. Journal of Biological Chemistry. 2000;275:2669–2675. doi: 10.1074/jbc.275.4.2669. [DOI] [PubMed] [Google Scholar]
- 27.Ernst OP, Meyer CK, Marin EP, Henklein P, Fu WY, Sakmar TP, Hofmann KP. Mutation of the Fourth Cytoplasmic Loop of Rhodopsin Affects Binding of Transducin and Peptides Derived from the Carboxyl-terminal Sequences of Transducin α and γ Subunits. Journal of Biological Chemistry. 2000;275:1937–1943. doi: 10.1074/jbc.275.3.1937. [DOI] [PubMed] [Google Scholar]
- 28.Janz JM, Farrens DL. Rhodopsin Activation Exposes a Key Hydrophobic Binding Site for the Transducin α-Subunit C Terminus. Journal of Biological Chemistry. 2004;279:29767–29773. doi: 10.1074/jbc.M402567200. [DOI] [PubMed] [Google Scholar]
- 29.Scheerer P, Park JH, Hildebrand PW, Kim YJ, Krauss N, Choe HW, Hofmann KP, Ernst OP. Crystal structure of opsin in its G-protein-interacting conformation. Nature. 2008;455:497–502. doi: 10.1038/nature07330. [DOI] [PubMed] [Google Scholar]
- 30.Onrust R, Herzmark P, Chi P, Garcia PD, Lichtarge O, Kingsley C, Bourne HR. Receptor and βγ Binding Sites in the α Subunit of the Retinal G Protein Transducin. Science. 1997;275:381–384. doi: 10.1126/science.275.5298.381. [DOI] [PubMed] [Google Scholar]
- 31.Johnston CA, Siderovski DP. Structural basis for nucleotide exchange on G alpha i subunits and receptor coupling specificity. Proc Natl Acad Sci U S A. 2007;104:2001–2006. doi: 10.1073/pnas.0608599104. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 32.Herrmann R, Heck M, Henklein P, Hofmann KP, Ernst OP. Signal transfer from GPCRs to G proteins: role of the G alpha N-terminal region in rhodopsin-transducin coupling. J Biol Chem. 2006;281:30234–30241. doi: 10.1074/jbc.M600797200. [DOI] [PubMed] [Google Scholar]
- 33.Kostenis E, Zeng FY, Wess J. Functional Characterization of a Series of Mutant G Protein αq Subunits Displaying Promiscuous Receptor Coupling Properties. Journal of Biological Chemistry. 1998;273:17886–17892. doi: 10.1074/jbc.273.28.17886. [DOI] [PubMed] [Google Scholar]
- 34.Taylor JM, Jacob-Mosier GG, Lawton RG, Remmers AE, Neubig RR. Binding of an α2 Adrenergic Receptor Third Intracellular Loop Peptide to Gβ and the Amino Terminus of Gα. Journal of Biological Chemistry. 1994;269:27618–27624. [PubMed] [Google Scholar]
- 35.Preininger AM, Parello J, Meier SM, Liao G, Hamm HE. Receptor-Mediated Changes at the Myristoylated Amino Terminus of Galphail Proteins. Biochemistry. 2008;47:10281–10293. doi: 10.1021/bi800741r. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Lichtarge O, Bourne HR, Cohen FE. Evolutionarily conserved Gαβγ binding surfaces support a model of the G protein-receptor complex. Proceedings of the National Academy of Sciences of the United States of America. 1996;93:7507–7511. doi: 10.1073/pnas.93.15.7507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Oldham WM, Van Eps N, Preininger AM, Hubbell WL, Hamm HE. Mechanism of the receptor-catalyzed activation of heterotrimeric G proteins. Nature Structural and Molecular Biology. 2006;13:772–777. doi: 10.1038/nsmb1129. [DOI] [PubMed] [Google Scholar]
- 38.Marin EP, Krishna AG, Sakmar TP. Disruption of the α5 Helix of Transducin Impairs Rhodopsin-Catalyzed Nucleotide Exchange. Biochemistry. 2002;41:6988–6994. doi: 10.1021/bi025514k. [DOI] [PubMed] [Google Scholar]
- 39.Medkova M, Preininger AM, Yu NJ, Hubbell WL, Hamm HE. Conformational Changes in the Amino-Terminal Helix of the G Protein αi1 Following Dissociation from Gβγ Subunit and Activation. Biochemistry. 2002;41:9962–9972. doi: 10.1021/bi0255726. [DOI] [PubMed] [Google Scholar]
- 40.Dratz EA, Furstenau JE, Lambert CG, Thireault DL, Rarick H, Schepers T, Pakhlevaniants S, Hamm HE. NMR structure of a receptor-bound G-protein peptide. Nature. 1993;363:276–281. doi: 10.1038/363276a0. [DOI] [PubMed] [Google Scholar]
- 41.Otwinowski Z. In: CCP4 Study Weekend Data Collection and Processing. Sawyer L, Isaacs N, Bailey S, editors. SERC Daresbury Laboratory; UK: 1993. pp. 56–62. [Google Scholar]
- 42.Bailey S. The CCP4 suite - programs for protein crystallography. Acta Cryst. 1994;D50:760–763. doi: 10.1107/S0907444994003112. [DOI] [PubMed] [Google Scholar]
- 43.McCoy AJ, Grosse-Kunstleve RW, Storoni LC, Read RJ. Likelihood-enhanced fast translation functions. Acta Crystallogr D Biol Crystallogr. 2005;61:458–464. doi: 10.1107/S0907444905001617. [DOI] [PubMed] [Google Scholar]
- 44.de la Fortelle E, Bricogne G. Maximum-likelihood heavy-atom parameter refinement for multiple isomorphus replacement and multiwavelength anomalous diffraction methods. In: Carter CW, Sweet RM, editors. Methods in Enzymology Macromolecular Crystallography Part A. Academic Press; San Diego: 1997. pp. 472–494. [DOI] [PubMed] [Google Scholar]
- 45.Jones TA, Zou JY, Cowan SW, Kjeldgaard M. Improved methods for building protein models in electron density maps and the location of errors in these models. Acta Crystallogr. 1991;A47:110–119. doi: 10.1107/s0108767390010224. [DOI] [PubMed] [Google Scholar]
- 46.Brünger A, Adams P, Clore G, DeLano W, Gros P, Grosse-Kunstleve R, Jiang J, Kuszewski J, Nilges M, Pannu N, Read R, Rice L, Simonson T, Warren G. Crystallography & NMR System: A new software suite for macromolecular structure determination. Acta Cryst. 1998;D54:905–921. doi: 10.1107/s0907444998003254. [DOI] [PubMed] [Google Scholar]
- 47.Murshudov GN, Vagin AA, Dodson EJ. Refinement of macromolecular structures by the maximum likelihood method. Acta Crystallogr. 1997;D53:240–255. doi: 10.1107/S0907444996012255. [DOI] [PubMed] [Google Scholar]
- 48.Adams PD, Grosse-Kunstleve RW, Hung LW, Ioerger TR, McCoy AJ, Moriarty NW, Read RJ, Sacchettini JC, Sauter NK, Terwilliger TC. PHENIX: building new software for automated crystallographic structure determination. Acta Crystallogr D Biol Crystallogr. 2002;58:1948–1954. doi: 10.1107/s0907444902016657. [DOI] [PubMed] [Google Scholar]
- 49.Kleywegt GJ. Use of non-crystallographic symmetry in protein structure refinement. Acta Crystallogr D Biol Crystallogr. 1996;52:842–857. doi: 10.1107/S0907444995016477. [DOI] [PubMed] [Google Scholar]
- 50.Johnston CA, Willard FS, Jezyk MR, Fredericks Z, Bodor ET, Jones MB, Blaesius R, Watts VJ, Harden TK, Sondek J, Ramer JK, Siderovski DP. Structure of Gαi1 Bound to a GDP-Selective Peptide Provides Insight into Guanine Nucleotide Exchange. Structure. 2005;13:1069–1080. doi: 10.1016/j.str.2005.04.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Johnston CA, Lobanova ES, Shavkunov AS, Low J, Ramer JK, Blaesius R, Fredericks Z, Willard FS, Kuhlman B, Arshavsky VY, Siderovski DP. Minimal determinants for binding activated G alpha from the structure of a G alpha(i1)-peptide dimer. Biochemistry. 2006;45:11390–11400. doi: 10.1021/bi0613832. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Kanaho Y, Tsai SC, Adamik R, Hewlett EL, Moss J, Vaughan M. Rhodopsin-enhanced GTPase activity of the inhibitory GTP-binding protein of adenylate cyclase. J Biol Chem. 1984;259:7378–7381. [PubMed] [Google Scholar]
- 53.Van Eps N, Oldham WM, Hamm HE, Hubbell WL. Structural and dynamical changes in an alpha-subunit of a heterotrimeric G protein along the activation pathway. Proc Natl Acad Sci U S A. 2006;103:16194–16199. doi: 10.1073/pnas.0607972103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Shoemaker KR, Kim PS, York EJ, Stewart JM, Baldwin RL. Tests of the helix dipole model for stabilization of alpha-helices. Nature. 1987;326:563–567. doi: 10.1038/326563a0. [DOI] [PubMed] [Google Scholar]
- 55.Heydorn A, Ward RJ, Jorgensen R, Rosenkilde MM, Frimurer TM, Milligan G, Kostenis E. Identification of a Novel Site within G Protein α Subunits Important for Specificity of Receptor-G Protein Interaction. Molecular Pharmacology. 2004;66:250–259. doi: 10.1124/mol.66.2.250. [DOI] [PubMed] [Google Scholar]
- 56.Ramachandran S, Cerione RA. Stabilization of an intermediate activation state for transducin by a fluorescent GTP analogue. Biochemistry. 2004;43:8778–8786. doi: 10.1021/bi0362774. [DOI] [PubMed] [Google Scholar]
- 57.Abdulaev NG, Ngo T, Ramon E, Brabazon DM, Marino JP, Ridge KD. The receptor-bound “empty pocket” state of the heterotrimeric G-protein alpha-subunit is conformationally dynamic. Biochemistry. 2006;45:12986–12997. doi: 10.1021/bi061088h. [DOI] [PubMed] [Google Scholar]
- 58.Thomas C, Fricke I, Scrima A, Berken A, Wittinghofer A. Structural evidence for a common intermediate in small G protein-GEF reactions. Mol Cell. 2007;25:141–149. doi: 10.1016/j.molcel.2006.11.023. [DOI] [PubMed] [Google Scholar]
- 59.Fisher AJ, Smith CA, Thoden J, Smith R, Sutoh K, Holden HM, Rayment I. Structural studies of myosin:nucleotide complexes: a revised model for the molecular basis of muscle contraction. Biophys J. 1995;68:19S–26S. discussion 27S–28S. [PMC free article] [PubMed] [Google Scholar]
- 60.Rondard P, Iiri T, Srinivasan S, Meng E, Fujita T, Bourne HR. Mutant G protein α subunit activated by Gβγ: A model for receptor activation? Proceedings of the National Academy of Sciences of the United States of America. 2001;98:6150–6155. doi: 10.1073/pnas.101136198. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.