

Abstract

A concise total synthesis of S-14-methoxy-epothilone D has been accomplished. S-14-Methoxy-epothilone D represents a conceptually novel example of polyketide analogue design based on an alternative biogenetic pattern of extender units. The significant biological activity observed for this compound provides a foundation to support studies designed to prepare derivatives of this type through fermentation of genetically engineered organisms expressing the epothilone PKS gene cluster.
Polyketide natural products are typically produced by microorganisms to create an environmental advantage for the producing organism through antibiotic and antifungal activity. Their structures evolve during the ancestral history of the producing organism through modification in their PKS gene clusters and the development or sequestration of genes encoding post-PKS processing enzymes.1 The polyketide backbone is generated through the sequential incorporation of two-carbon extender units derived most commonly from malonyl-CoA or methylmalonyl-CoA. Advances in the genetic engineering of the gene clusters responsible for the production of biological active polyketides represents a valuable new source of chemical diversity essential to identification of the next generation of chemotherapeutic agents.2 Unfortunately, the biological implications of even minor structural modifications are typically difficult to predict. Our unique approach to this classic structure-activity relationship (SAR) problem was based on the initial appreciation that the main role for the functionality that adorns a polyketide is to control the overall conformation. Through extensive computer-based conformational analysis coupled with solution NMR studies on the naturally-ocurring epothilones as well as designed analogues, we were able to develop a conformationalactivity relationship profile which was used to propose a binding conformation for the class.3,4,5,6 With this information in hand it is possible to design biologically active analogues of the epothilones, such as epothilones C and D, Figure 1, which may be accessible through genetic engineering.
Figure 1.
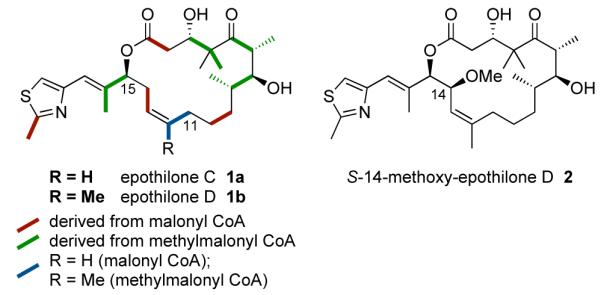
The structures and biogenetic pattern for epothilones C and D; the structure of (S)-14-methoxy-epothilone D.
Herein we report the total synthesis and biological evaluation of S-14-methoxy-epothilone D 2, a novel epothilone analogue. The stereochemistry of the C14-substitution was chosen to provide further support for the proposed binding conformation in the C11-C15 region.7 Moreover, the incorporation of a methoxy group at this position represents a target potentially available through modification to the acyltransferase domain of module 3 in the epothilone PKS gene cluster8 and the incorporation of an extender unit derived from methoxymalonyl CoA.9
The significant efforts required to develop a fermentation based route could be justified if the biological activity of the target were significant. Thus, a synthetic strategy for the production of S-14-methoxy-epothilone D was developed based upon modificatons to our previously reported route to epothilones B and D.10 This route relied on a sequential Nozaki-Hiyama-Kishi (NHK)11 couplingthionyl chloride induced allylic rearrangement12 to stereoselectively generate the C12-C13 trisubstituted olefin.
The starting thiazole aldehyde fragment 3 was prepared by previously described methods and detailed in Scheme 1.13 The introduction of the C14 methoxy substituent was accomplished via an asymmetric Brown allylation14 with lithiated allyl methyl ether in the presence of BF3•OEt2 to afford the alcohol 4 as a single diastereomer in >95% ee15 and 88% yield, Scheme 1. The undersired syn-stereochemistry was easily converted to the anti-diastereomer via a two step sequence. First, oxidation of the alcohol was achieved with Dess-Martin periodinane16 followed by a highly diastereoselective Luche reduction of the α, β-unsaturated ketone.17 This reduction yielded the optically pure anti-diastereomer 5 as a 93:7 mixture of diastereomers in 96% yield. The free hydroxyl group was protected with t-butoxydiphenylsilyl chloride (TBODPSCl) and subsequent oxidative cleavage of the terminal olefin under standard conditions afforded the desired aldehyde fragment 6.
Scheme 1.

Installation of C14-Methoxy Substituent.
As shown in Scheme 2, fragment coupling of aldehyde 6 with known vinyl iodide fragment 7, prepared according to a previously reported route,10 was achieved via a NHK coupling reaction. This key step afforded the desired allylic alcohol as a 3:1 mixture of unassigned and inconsequential diastereomers in 78% yield. Exposure of this mixture to thionyl chloride in ether-pentane provided the desired primary chloride 8 in 80% yield as a single olefin isomer.
Scheme 2.
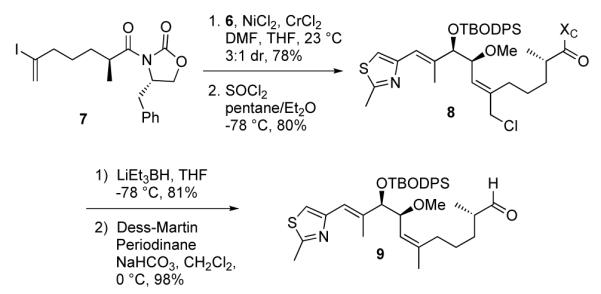
Sequential NHK Coupling/Thionyl Chloride Rearrangement for Trisubstituted Olefin Generation.
Removal of the chiral auxiliary and cleavage of the primary chloride was achieved in a single synthetic operation utilizing LiEt3BH, which was subsequently oxidized with Dess-Martin periodinane to afford the aldehyde 9. The synthesis of the C1-C6 fragment, Scheme 3, commenced with the readily available silyl ketene acetal 1018 and ketoaldehyde 11.19 Treatment with Kiyooka’s chiral boron reagent20 (100 mol%) provided the TMS-protected aldol adduct 12 in 69% yield and >95% ee. The TMS group was readily removed with TFA and the resultant secondary alcohol was protected with TBSOTf in the presence of 2,6-lutidine to afford the C1-C6 fragment 13 in 91% yield.
Scheme 3.
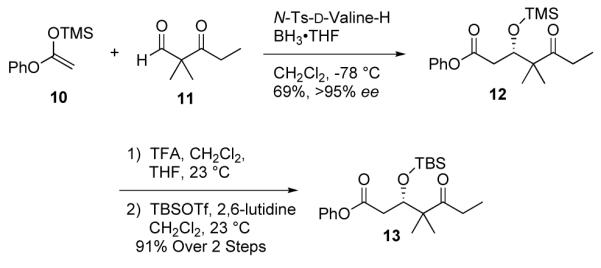
Synthesis of C1-C6 Fragment.
Aldol coupling between ketone fragment 13 and aldehyde 9 was accomplished by the previously reported, highly diastereoselective Ti-mediated coupling conditions.21 In our system, when 13 was treated with TiCl4 and i-Pr2NEt and then subsequently 9, the syn,anti-aldol product 14 was obtained in 80% yield and as a single diastereomer. The resultant secondary alcohol was protected as the TBS ether with TBSOTf and 2,6-lutidine in 95% yield. Hydrolysis of the phenyl ester under standard conditions (LiOH in THF/H2O) provided a small amount of the desired acid, as well as a large amount of C3 elimination product. After screening various conditions, this problem was overcome by the use of milder hydrolysis conditions (NaHCO3 and H2O2 in THF), which afforded the desired acid in 85% yield without any formation of a C3-elimination product. Subsequent selective removal of the C15 TBODPS ether was accomplished by careful treatment with TBAF at 0 °C to afford the seco-acid 15 in 75% yield. Classic Yamaguchi macrolactonization conditions22 using trichlorobenzoyl chloride (TCBCl) provided the 16-membered lactone in good yield when heated to 40 °C. Finally, global deprotection was carried out using HF•py to afford S-14-methoxyepothilone D 2 in 88% yield.
The biological activity of S-14-methoxyepothilone D 2 was evaluated against a two human tumor cell lines (Table 1) in direct comparison to natural epothilone B, ixabepilone, and paclitaxel. The presence of a C12,C13 epoxide as in epothilone B is known to enhance activity by an order of magnitude. Ixabepilone is Bristol-Myers Squibb’s semi-synthetic analogue of epothilone B recently approved by the FDA for advanced breast cancer therapy.23 S-14-Methoxyepothilone D retains significant cytotoxic activity on par with both naturally occurring as well as synthetically derived analogues of the epothilones.
Table 1.
Cytotoxicity of Epothilone Analogs (IC50 nM)
| compound | MCF-7 | H460 |
|---|---|---|
| S-14-methoxyepothilone D 3 | 3.7 | 4.9 |
| epothilone B | 0.5 | 0.3 |
| ixabepilone (Ixempra®) | 2.1 | 2.3 |
| paclitaxel (Taxol®) | 4.7 | 1.5 |
S-14-Methoxyepothilone D represents a conceptually novel example of polyketide analogue design based on an alternative biogenetic pattern of extender units. The significant biological activity observed for this compound provides a foundation to support studies designed to prepare derivatives of this type through fermentation of genetic engineered organisms expressing the modified forms of the epothilone PKS gene cluster and/or precurser-directed biosynthesis. Results along these lines will be reported in due course.
Supplementary Material
Scheme 4.

Selective Aldol and Completion of Synthesis.
Acknowledgment
Dr. Bandaru Sreenivasulu (Notre Dame) is acknowledged for preliminary studies. Support provided by the National Institutes of General Medical Sciences (GM077683) is greatfully acknowledged. JDF thanks the Walther Cancer Research Center (Notre Dame) for fellowship support. This work was supported in part by intramural funds of the Eunice Kennedy Shriver National Institute for Child Health and Human Development.
Footnotes
Supporting Information Available: Full experimental and characterization data for all new compounds. This material is available free of charge via the Internet at http://pubs.acs.org.
References
- 1.Ridley CP, Lee HY, Khosla C. Proc. Nat. Acad. Sci. 2008;105:4595–4600. doi: 10.1073/pnas.0710107105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Kealey JT. Front. Biosci. 2003;8:C1–C13. doi: 10.2741/885. [DOI] [PubMed] [Google Scholar]
- 3.Taylor RE, Chen Y, Galvin GM, Pabba PK. Org. Biomol. Chem. 2004;2:127–132. doi: 10.1039/b312213c. [DOI] [PubMed] [Google Scholar]
- 4.Carlomagno T, Blommers MJJ, Meiler J, Jahnke W, Schupp T, Peterson F, Schinzer D, Altmann K-H, Griesinger C. Angew. Chem., Int. Ed. 2003;42:2511–2515. doi: 10.1002/anie.200351276. [DOI] [PubMed] [Google Scholar]
- 5.An alternative binding conformation has been proposed based on electron crystallography: Nettles JH, Li H, Cornett B, Krahn JM, Snyder JP, Downing KH. Science. 2004;305:866–869. doi: 10.1126/science.1099190.
- 6.A critical review of published efforts towards identification of the binding conformation of the epothilones has been published: Heinz DW, Schubert W-D, Höfle G. Angew. Chem., Int. Ed. 2005;44:1298–1301. doi: 10.1002/anie.200462241.
- 7.Taylor RE, Chen Y, Beatty A, Myles DC, Zhou Y. J. Am. Chem. Soc. 2003;125:26–27. doi: 10.1021/ja028196l. [DOI] [PubMed] [Google Scholar]
- 8.Tang L, Shah S, Chung L, Carney J, Katz L, Khosla C, Julien B. Science. 2000;199:640–642. doi: 10.1126/science.287.5453.640. [DOI] [PubMed] [Google Scholar]
- 9.Chan YA, Boyne MT, II, Podevels AM, Klimowicz AK, Handelsman J, Kelleher NL, Thomas MG. Proc. Nat. Acad. Sci. 2006;103:14349–14354. doi: 10.1073/pnas.0603748103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Taylor RE, Chen Y. Org. Lett. 2001;3:2221–2224. doi: 10.1021/ol010094x. [DOI] [PubMed] [Google Scholar]
- 11 (a).Takai K, Tagashira M, Kuroda T, Oshima K, Utimoto K, Nozaki H. J. Am. Chem. Soc. 1986;108:6048–6050. doi: 10.1021/ja00279a068. [DOI] [PubMed] [Google Scholar]; (b) Jin H, Uenishi J-I, Christ WJ, Kishi Y. J. Am. Chem. Soc. 1986;108:5644–5646. [Google Scholar]
- 12.Caserio FF, Dennis GE, DeWolfe RH, Young WG. J. Am. Chem. Soc. 1955;77:4182–4183. [Google Scholar]
- 13.Taylor RE, Haley JD. Tetrahedron Lett. 1997;38:2061–2064. [Google Scholar]
- 14 a).Brown HC, Jadhav PK, Bhat KS. J. Am. Chem. Soc. 1988;110:1535–1538. [Google Scholar]; b) Hoffmann RW, Kemper B, Metternich R, Liebigs Lehmeier, T. Ann. Chem. 1985:2246–2260. [Google Scholar]
- 15.Dale JA, Mosher HS. J. Am. Chem. Soc. 1973;95:512–519. [Google Scholar]
- 16 a).Frigerio M, Santagostino M, Sputore S. J. Org. Chem. 1999;64:4537–4538. [Google Scholar]; b) Boeckman RK, Jr., Shao P, Mullins JJ. Org. Syn. 2004;77:141–146. [Google Scholar]
- 17.Luche J-L, Rodriguez-Hahn L, Crabbe P. J. Chem. Soc., Chem. Comm. 1978:601–602. [Google Scholar]
- 18.Slougui N, Rousseau G. Syn. Comm. 1987;17:1–11. [Google Scholar]
- 19.Inukai T, Yoshizawa R. J. Org. Chem. 1967;32:404–407. [Google Scholar]
- 20.Kiyooka S-I, Kira H, Hena MA. Tetrahedron Lett. 1996;37:2597–2600. [Google Scholar]
- 21.Koch G, Loiseleur O, Fuentes D, Jantsch A, Altmann K-H. Org. Lett. 2002;4:3811–3814. doi: 10.1021/ol026480b. [DOI] [PubMed] [Google Scholar]
- 22.Inanaga J, Hirata K, Saeki H, Katsuki T, Yamaguchi M. Bull. Chem. Soc. Jpn. 1979;52:1989–1993. [Google Scholar]
- 23. http://www.ixempra.com/
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.