

Abstract
Background
Obesity causes hypertension and sympathoactivation, a process proposed to be mediated by leptin. Protein tyrosine phosphatase 1B (PTP1B), a major new pharmaceutical target to treat obesity and type II diabetes, constrains leptin’s metabolic actions, but the extent to which PTP1B regulates leptin’s cardiovascular effects is unclear. This study examined the hypothesis that PTP1B is a negative regulator of the cardiovascular effects of leptin.
Methods and Results
PTP1B KO mice had lower body fat but higher mean arterial pressure (MAP: 116±5 vs. 105±5 mmHg, p<0.05) than controls. Leptin infusion produced a greater anorexic effect in PTP1B KO mice and a marked increase in MAP (135±5 mmHg) in PTP1B KO mice only. Decreased MAP to ganglionic blockade was higher in PTP1B KO mice (−38±3% vs. −29±3%, p<0.05) suggesting increased sympathetic tone. PTP1B deletion blunted MAP responses to phenylephrine (PE) injection (55±10% vs. 93±7%, p<0.05). PE-induced aortic contraction was reduced in PTP1B KO mice (57.7±9 vs. 96.3±12% of KCl, p<0.05), consistent with desensitization to chronically elevated sympathetic tone. Furthermore, PTP1B deletion significantly reduced gene expression of three alpha-1 adrenergic receptor sub-types, consistent with blunted constriction to PE.
Conclusion
these data indicate that PTP1B is a key regulator of the cardiovascular effects of leptin and that reduced vascular adrenergic reactivity provides a compensatory limit to leptin’s effects on MAP.
Keywords: PTP1B, leptin, blood pressure, alpha adrenergic receptor, sympathetic tone
Introduction
Obesity, a worldwide metabolic disorder affecting 60% of the population, is defined as an increased mass of adipose tissue and confers a higher risk for hypertension 1. While this risk is well-documented, underlying mechanisms are unknown. Obesity is clearly associated with sympathoactivation in humans and animal models 2, and increases in sympathetic activity appear to play a key role in obesity-induced hypertension 3. However, pathways responsible for the obesity-induced sympathetic overactivation have yet to be completely elucidated.
Leptin, an adipocyte derived hormone, is increased in most forms of obesity 4. This hormone communicates to the central nervous system the availability of energy stores, restrains food intake and induces energy expenditure 5. Besides its effects on appetite and metabolism, leptin binding its receptor (OB-Rb) acts in the hypothalamus to increase blood pressure through the activation of the sympathetic nervous system (SNS) 6, 7 via Janus kinases/Signal Transducers and Activators of Transcription (JAK/STAT) signaling pathways. Activated receptors induce the transphosphorylation of JAKs, and the phosphorylation of tyrosine residues on the leptin receptor, specifically Tyr1138, facilitates the binding of STAT proteins. Among the regulators of this signaling pathway is the protein tyrosine phosphatase 1B (PTP1B). PTP1B controls this pathway by dephosphorylating JAK2 8. Cheng9 and Zabolotny10 reported that genetic deletion of PTP1B increases leptin sensitivity, protecting mice against the development of obesity and type II diabetes.
In addition to its metabolic actions, leptin is involved in the control of cardiovascular function via regulation of sympathetic tone 7, 11, 12. The molecular mechanisms underlying leptin’s regulation of cardiovascular function are largely undetermined. Moreover, how the vasculature responds to chronic activation of leptin signaling is unknown. The availability of lean mice harboring a genetic deletion of PTP1B allows the examination of leptin actions on cardiovascular function without the confounding components of obesity. Using these mice, the hypothesis of the current study is that PTP1B is a specific negative regulator of leptin’s actions in the cardiovascular system. To test this hypothesis the regulation of blood pressure and vascular function in mice harboring a genetic deletion of PTP1B has been examined. Taken together, these studies critically test the circumstances under which leptin activation is a potential mechanism for hypertension.
Material and methods
Animals
PTP1B null mice were generated on a Balb/c background at Goodman Cancer Center of McGill University 9, 13. Balb/c mice, used as a control group, were purchased from the Jackson Laboratory (Bar Harbor, ME, USA). All animals were fed standard mouse chow and tap water was provided ad libitum. Mice were housed in an American Association of Laboratory Animal Care-approved animal care facility at the Medical College of Georgia, and the Institutional Animal Care and Use Committee approved all protocols.
Metabolic Measurements
Fasting blood glucose was assessed using a glucometer (Precision XL). Plasma total cholesterol and triglycerides were assessed with colorimetric assays (Wako, Richmond, VA, USA). Plasma insulin and leptin were respectively determined using the Mercodia Insulin ELISA assay and the mouse leptin EIA kit (ALPCO Diagnostics, Salem, NH, USA).
In Vivo Blood Pressure Measurement
Mice were instrumented with telemetry transmitters to record arterial pressure and heart rate (PA-C20, Data Sciences, St. Paul, MN, U.S.A.). Transmitters were implanted as previously described 14. Following 1 week of recovery from surgery, baseline data were recorded for 7 days prior to implantation of micro-osmotic pumps (ALZET®, 1007D, 0.5μL/hour) to infuse leptin (10 μg/day) 15, 16 subcutaneously. In a second set of mice, similar micro-osmotic pumps were implanted subcutaneously, in the PTP1B KO mice only, to determine the cardiovascular consequences of insulin treatment (2Units/kg/ day)17. To avoid any drop in blood glucose, mice were supplemented with a 10% glucose solution in drinking water 18. A different set of mice was used to determine the effects of a hypocaloric diet, mimicking leptin effects, on blood pressure. After 7 days of baseline blood pressure recording, mice were submitted to a 20% food restriction calculated from the average food intake during the baseline data collection period. Data were recorded throughout the infusion period. Mice were then sacrificed and tissues and plasma were collected for later analysis. In a different set of mice, the carotid artery and jugular vein were catheterized under isoflurane anesthesia (1.5%) for the measurement of mean arterial pressure (MAP) and drug delivery, respectively. To eliminate endogenous sympathetic vasomotor tone and baroreceptor-reflex-mediated responses, animals were given the ganglionic blocker mecamylamine (2 mg/kg, IV.). Effective blockade was confirmed by the absence of reflex bradycardia after constrictor administration. Changes in MAP were determined after injection of randomized boluses of PE (0.01 to 1 mg/kg) and expressed as percent of the baseline blood pressure 19.
Cardiovascular response to stress
Whether PTP1B deletion affects the cardiovascular response to stress was determined using the cage switch stress model that consists of measuring blood pressure variations by telemetry when a male mouse is placed in a cage previously occupied by another male mouse as described previously 14.
Vascular reactivity
Thoracic aortae from PTP1B KO and Balb/c mice were surgically dissected and mounted on a wire-myograph (DMT, Aarhus, Denmark) with 1g basal tension. Briefly, 2 tungsten wires were inserted into the lumen of the arteries and fixed to a force transducer and a micrometer. Arteries were bathed in a physiological salt solution as previously described 20. Arterial viability was determined using a potassium rich solution (40 mmol/L). Vascular contractility was assessed with cumulative concentration-response curves (CRC) to norepinephrine (NE, 1 pmol/L to 10 μmol/L), phenylephrine (PE, 1 pmol/L to 10 μmol/L) and serotonin (5HT, 1 pmol/L to 10 μmol/L). CRC to PE were repeated in the presence of the nitric oxide (NO) synthase inhibitor N(omega)-nitro-L-arginine methyl ester (L-NAME, 100 μmol/L, 20 minute incubation). Constriction to NE, PE, and 5HT were expressed as percent of KCl-induced constriction. The endothelial function was assessed with CRC to acetylcholine (ACh), and the involvement of NO was determined by inhibition of endothelial NO synthase (eNOS) with L-NAME (100 μmol/L, 20 min). Endothelium-independent relaxation was analyzed with a CRC to sodium nitroprusside (SNP, 1 pmol/L to 10 μmol/L). Relaxation curves were performed on preconstricted vessels (5HT, 1 μmol/L) and the relaxation expressed as percent of the precontraction.
Relation between sympathetic activity and vascular adrenergic reactivity
A last set of mice was treated with the specific α1-adrenergic receptor inhibitor, prazosin (2mg/kg/day, IP.) 21, 22 for 7 days, in association or not with leptin (10 μg/day) 15, 16. The cardiovascular consequences of the sympatho-inhibition were determine at the vascular level by assessing vascular reactivity and the gene expression of the three subtypes of the α1-adrenergic receptor by quantitative real-time RT-PCR, as described below.
Histological analysis
Mouse aortae were dissected under a microscope, fixed in formalin and embedded in resin. Sections (10 μm) stained with hematoxylin and eosin were observed under microscope (Carl Zeiss AX10, Thronwood, NY, USA) and the media cross sectional area calculated (AxioVision LE, AxioVs40).
Real-Time RT-PCR
Total aortic mRNA was extracted (Trizol® Plus, Invitrogen), purified using RNeasy spin columns and eluted from the column in 30μL of DEPC water, and the concentration was established using a NanoDrop ND-1000 (NanoDrop Technologies). cDNA was generated by RT-PCR using SuperScript III (Invitrogen) from 400 ng of total RNA using random hexamers. Reverse transcription was performed at 50°C for 50 min; the enzyme was heat inactivated at 85°C for 5 min and real-time quantitative RT-PCR was performed using the SYBR-Green Supermix (BioRad). The sequence of the primers used is presented in Table 1.
Table 1.
Basic Physiological and metabolic parameters of Balb/c and PTP1B KO mice.
| Balb/c | PTP1B | p value | |
|---|---|---|---|
| Body Weight (g) | 25.7 ± 0.4 | 27.4 ± 0.5 | 0.011 |
| Epididymal Fat Mass (mg) | 389 ± 25 | 163 ± 20 | 0.000 |
| Fat/body weight (mg/g) | 15.2 ± 0.9 | 5.32 ± 0.6 | 0.000 |
| Heart Weight (mg) | 122 ± 2 | 149 ± 5 | 0.003 |
| Heart / body weight (mg/g) | 4.7 ± 0.1 | 4.81 ± 0.1 | 0.372 |
| Leptin (ng/mL) | 1.11 ± 0.44 | 1.16 ± 0.33 | 0.3 |
| Insulin (ng/L) | 57.86 ± 17.01 | 54.13 ± 15.78 | 0.437 |
| Cholesterol (mg/dL) | 29.2 ± 3.8 | 35.5 ± 4.2 | 0.269 |
| Triglycerides (mg/dL) | 27.3 ± 5.5 | 16.8 ± 3.3 | 0.111 |
| Glycemia (mg/dL) | 70.9 ± 6.1 | 73.8 ± 8.7 | 0.782 |
| Hb1Ac (%) | 5.1 ± 0.1 | 5.3 ± 0.1 | 0.196 |
Statistics
All data are presented as mean ± SE. Differences in means among groups for non-repeated variables were compared by one-way ANOVA. Differences in means among groups and treatments, with repeated variables, were compared by two or three way ANOVA with repeated measures, when appropriate. Bonferroni and Fisher LSD tests were used as the post hoc test (SigmaStat).
Results
Baseline phenotypes
The effects of PTP1B deletion on the mouse phenotype were determined by measuring, body weight, organ weight and baseline plasma chemistry. As summarized in Table 1, PTP1B deletion induced a slight increase in body weight but a three fold decrease in epidydimal fat pad mass. Cardiac mass was similar when normalized for body weight. An increase in bone and muscle mass respectively triggered by the increased leptin and insulin sensitivity may explain the slight increase in body weight despite a decrease in fat content. As reported previously by Elchebly, in the same mice, PTP1B deletion had no effect on the fasting plasma level of insulin or leptin 13 and did not affect plasma concentrations of glucose, cholesterol or triglycerides.
Measurement of leptin sensitivity
The effects of PTP1B deletion on metabolic leptin sensitivity were determined by measuring changes in body weight during a 7-day subcutaneous leptin infusion. As shown in Figure 1, PTP1B KO mice presented a larger body weight reduction in response to leptin infusion compared to the Balb/c mice. (4.73±0.46 vs. 6.93±0.57% Balb/c vs. PTP1B KO mice, p<0.05).
Figure 1. PTP1B deletion increased the anorexigenic effects of leptin.
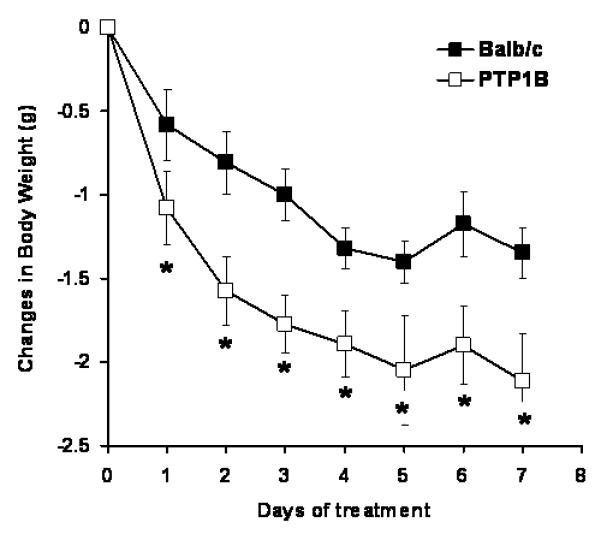
Changes in body weight induced by subcutaneous leptin infusion (10μg/day, 0.5μL/hour). Data are presented as mean ± sem. Effects of the treatment on body weight was determined by two-way ANOVA with repeated measures. * = p<0.05 vs. Balb/c, n=10-15 per group.
Effects of PTP1B deletion on blood pressure
The cardiovascular consequences of PTP1B deletion were first assessed by determining blood pressure in vivo via radiotelemetry. As shown in Figure 2A, the deletion of PTP1B resulted in a 20 mmHg rise in the baseline blood pressure (Balb/c: 99.4±4.6 vs. PTP1B: 120.4±3.1 mmHg, p<0.05). Treatment with leptin resulted in a significant increase in blood pressure only in PTP1B KO mice with no change in blood pressure in Balb/c mice (Balb/c: 100.3±1.5 vs. PTP1B: 130.8±2.1 mmHg, p<0.05). Treatment with insulin in PTP1B KO mice under glucose supplementation did not affect the mean arterial pressure (Figure 2C), nor did it affect the heart rate (Figure 2D) or the non-fasted glycemia (Glucose: 153±8 vs. Insulin + Glucose: 162±10 mg/dL). To confirm that the lack of response to leptin observed in Balb/c mice is not masked by the reduction in blood pressure associated with body weight loss, we determined changes in blood pressure induced by a hypocaloric diet, mimicking leptin effects. The reduction in food intake induced a similar drop in body weight in both Balb/c and PTP1B KO mice (Balb/c: −6.1±0.9 vs. PTP1B: 5.3±0.7%, NS, Figure 2E). As shown in Figure 2F, body weight reduction did not affect blood pressure in either Balb/c or PTP1B KO mice (Balb/c: 100±2 vs. PTP1B: 118±2 mmHg, p<0.05). As reported in Figure 2B, neither PTP1B deletion nor leptin treatment significantly affected heart rate in any group of mice.
Figure 2. PTP1B deletion increased baseline blood pressure and hemodynamic response to leptin infusion.
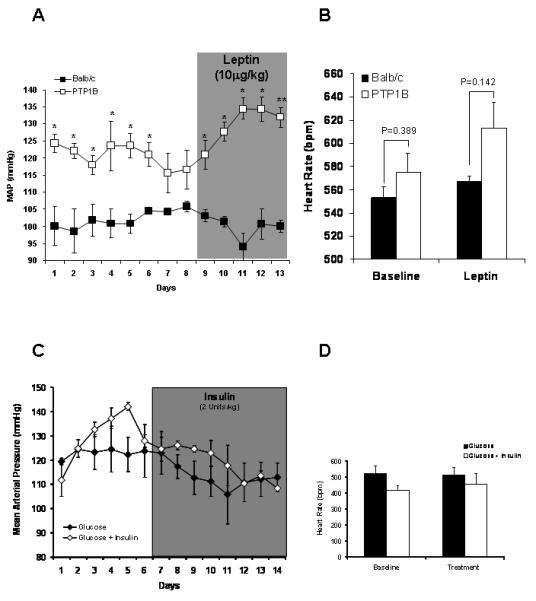
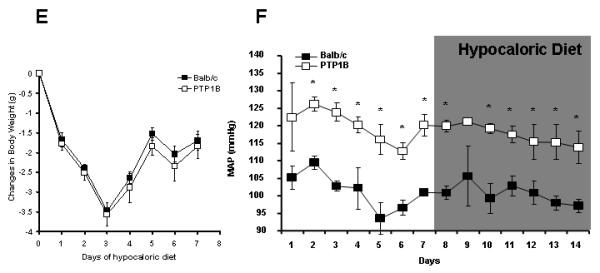
A) Baseline and mean arterial pressure (MAP) response to leptin infusion (10μg/day, 0.5μL/hour), determined by telemetry, in Balb/c and PTP1B KO mice (n=5 per group). B) Baseline and heart rate response to leptin infusion, determined by telemetry and presented as the average of the 7 days of recording, in Balb/c and PTP1B KO mice (n=5 per group). C) Baseline and mean arterial pressure (MAP) response to insulin infusion (2Units/kg/day, 0.5μL/hour), determined by telemetry in PTP1B KO mice supplemented with glucose. D) Baseline and heart rate response to glucose or glucose + insulin infusion determined by telemetry and presented as the average of the 7 days of recording, in PTP1B KO mice. E) Changes in body weight induced by 20% reduction in food intake. F) Mean arterial pressure response to the hypocaloric diet mimicking the anorexigenic effects of leptin (n=5 per group). Data are presented as mean ± sem. Effects of the treatment on continuous blood pressure recording and body weight, were analyzed by two-way ANOVA for repeated measurements (A,C,E,F).
Effects of PTP1B deletion on cardiovascular response to stress
To determine whether the effects of PTP1B deletion on arterial pressure were specific to leptin, male mice were switched to the cage of one of their congeners to measure the cardiovascular response to this territorial stress. Cage switch generated a 30 mmHg BP increase in both Balb/c and PTP1B KO mice (Figure 3A). The total pressor response was derived by calculating the integrated area under the curve, a value that was similar in both groups (Figure 3B).
Figure 3. PTP1B deletion does not affect acute response to stress.
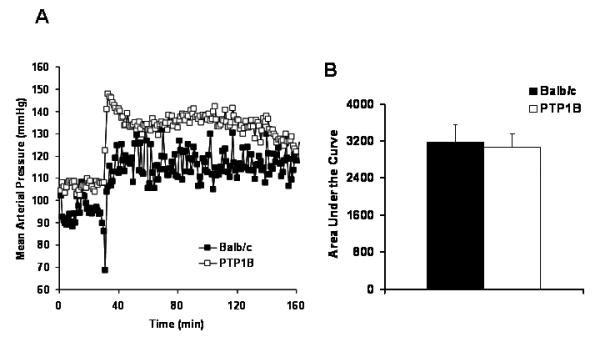
A) Representative changes in MAP induced by the cage switch. B) Integrated area under the MAP curve of the cage switch stress. Data are presented as mean ± sem. * = p<0.05 vs. Balb/c.
Effects of PTP1B deletion on sympathetic activity and adrenergic tone in vivo
To assess the effects of PTP1B deletion on the sympathetic activity and adrenergic sensitivity, mice were anesthetized and catheterized. An index of the sympathetic activity was obtained by determining decreases in blood pressure induced by ganglionic blockade. 23. As shown in Figure 4A, the drop in blood pressure induced by the ganglionic blocker mecamylamine was significantly larger in PTP1B KO mice compared to Balb/c (Balb/c: −29±3 vs. PTP1B: −38±3%, p<0.05), indicating greater sympathetic contribution to arterial pressure in PTP1B KO mice. Parallel increases in sympathetic tone were observed in Balb/c mice treated with leptin, suggesting consistency of mechanism between leptin-treated and leptin-sensitized mice. No further increase in the sympathetic tone was observed in the PTP1B KO mice under leptin treatment, suggesting a saturation of the sympathetic mechanism with deletion of PTP1B.
Figure 4. PTP1B deletion and leptin infusion enhanced sympathetic tone and reduced adrenergic reactivity.
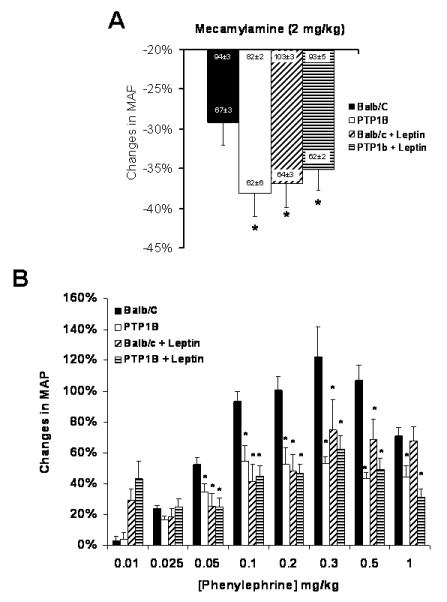
A) Drop in MAP pressure induced by ganglionic blockade (mecamylamine 2mg/kg). B) Changes in MAP induced by phenylephrine injection in mice under mecamylamine blockade. Data are presented as mean ± sem. Effect of the group and the treatment, were assessed with a two-way ANOVA (A) while the effects of the group, the treatment and the dose were analyzed with a three-way ANOVA (B). * = p<0.05 vs. Balb/c. n=5-9 per group.
Cumulative doses of PE were injected intravenously in mice under mecamylamine blockade to determine vascular adrenergic reactivity in vivo. As shown in Figure 4B, PTP1B deletion significantly blunted the rise in blood pressure induced by α1-adrenergic receptor stimulation (Maximum response: 63±5 vs. 50±4 mmHg, n>5, p<0.05), reflecting a decreased vascular adrenergic reactivity. A similar decrease in vascular adrenergic reactivity was noticed in the Balb/c mice treated with leptin, linking this hormone to the sympathetic overflow and the reduced adrenergic reactivity (Maximum response: 63±5 vs. 43±7 mmHg, n>5, p<0.05). No further reduction in the adrenergic sensitivity was observed in the PTP1B KO mice treated with leptin, consistent with the saturation of sympathetic drive by the combination of excess leptin stimulation and excess leptin sensitivity.
Effects of PTP1B deletion on vascular reactivity
To confirm whether the decreased response to PE observed in vivo reflects a decreased adrenergic reactivity of the vasculature, vasoconstrictor reactivity of aortic rings was assessed in vitro on a wire myograph. As shown in Figure 5, PTP1B deletion results in a decreased contraction to both NE (Fig. 5A) and PE (Fig. 5B). Specificity of this response to adrenergic stimulation was assessed by measuring the constriction induced by a depolarizing agent (KCl) and by 5HT as well as with the relaxation induced by the exogenous NO donor sodium nitroprusside (SNP). As shown in figure 5C, 5D and 5E, PTP1B deletion did not affect the constriction induced by KCl or 5HT, nor did it affect the relaxation induced by SNP, ruling out generalized vascular smooth muscle cell dysfunction as an explanation for the changes in adrenergic sensitivity.
Figure 5. PTP1B deletion reduced vascular adrenergic reactivity.
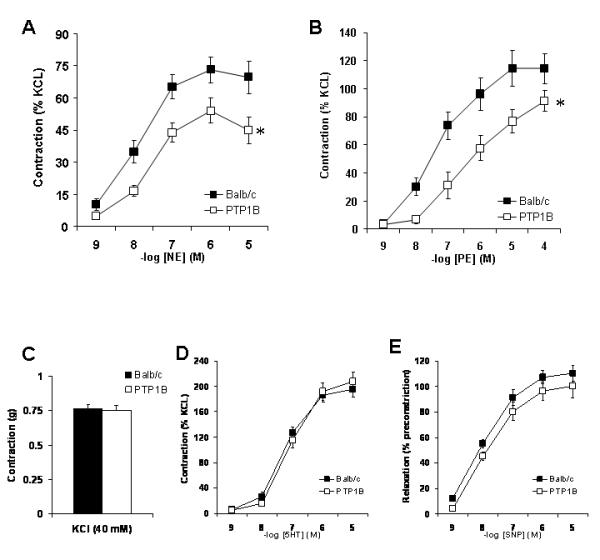
A) Cumulative concentration response curves to (A) Norepinephrine, (B) Phenylephrine, and (D) Serotonin, performed on aortic rings and expressed as percent of KCl-induced constriction (C). D) Endothelium-independent relaxation (SNP) expressed as percent of 5HT-induced constriction. Data are presented as mean ± sem. Comparison between groups were assessed with a two-way ANOVA for repeated measurements. * = p<0.05 vs. Balb/c. n= 8-10 per group.
Effects of PTP1B deletion on vascular structure
Since PTP1B has been reported to be involved in cell proliferation and migration 24, 25 and because changes in vascular architecture affect the generation of force 26, we determined whether the deletion of PTP1B affects vascular structure. Analysis of aortic cross sections and measurement of the media thickness and wall to lumen ratio indicated no significant difference in aortic structure between PTP1B KO and Balb/c mice (Figure I online-only Data Supplement).
Effects of PTP1B deletion on vascular endothelium
Because endothelial NO modulates adrenergic constriction 27, we examined endothelial function by two measures: relaxation induced by the endothelium-dependent vasodilator ACh and the effect of the eNOS inhibitor, L-NAME, on ACh and PE-induced constriction. ACh induced a similar relaxation of aortic rings in both PTP1B KO and Balb/c mice. Similarly, L-NAME completely inhibited the relaxation in both groups (Figure II online-only Data Supplement), indicating NO-dependency of this response. Furthermore, as shown in Figure II-B (online-only Data Supplement), eNOS inhibition had no effect on PE-induced contraction in either Balb/c or PTP1B KO mice, ruling out endothelium involvement in the reduced adrenergic reactivity observed in PTP1B KO mice.
Effects of PTP1B deletion on adrenergic receptor expression
To determine the origin of the reduced adrenergic reactivity observed in PTP1B KO mice, the gene expression of the three α1—adrenergic receptor subtypes (α1A, α1B, α1D) was measured by quantitative real-time RT-PCR, in the aorta. As shown in Figure 6, a significant decrease in gene expression of the three subtypes of adrenergic receptor was observed in the PTP1B KO mice compared to Balb/c.
Figure 6. PTP1B deletion and leptin infusion reduced α1-adrenergic receptor gene expression.
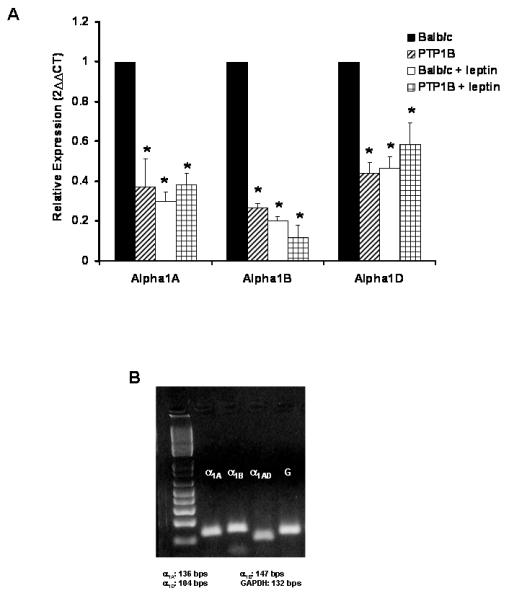
A) Quantification of α1-adrenergic receptor subtypes gene expression by quantitative real time RT-PCR performed on aortae taken from Balb/c and PTP1B KO mice. B) Representative gel used to confirm the specificity of our primers. Data are presented as mean ± sem. Effects of the group and the treatment within each gene, were determined with a two-way ANOVA. * = p<0.05 vs. Balb/c. n= 5 per group.
Effects of chronic treatment with leptin on vascular function
Vascular effects of chronic leptin treatment were determined by analyzing the reactivity of the aorta taken from the leptin treated mice. The 7-day leptin treatment had no effect on the endothelial function, as reflected by the lack of change in the ACh-mediated relaxation (Figure 7A). However, leptin induced a significant reduction in the PE-induced constriction in both the Balb/c and PTP1B KO mice. The PE-induced constriction, already reduced in the PTP1B KO mice was further reduced (Figure 7A). As shown by the results of the quantitative real time RT-PCR, the 7-day leptin treatment significantly reduced the expression of the 3 subunits of the alpha adrenergic receptor only in the Balb/c mice. Leptin treatment did not further decrease gene expression of these receptor subunits in the PTP1B KO mice (Figure 6). These data indicate that stimulation with leptin and sensitizations to leptin alter vascular function by parallel mechanisms.
Figure 7. Leptin infusion reduced vascular adrenergic reactivity without affecting endothelial function.
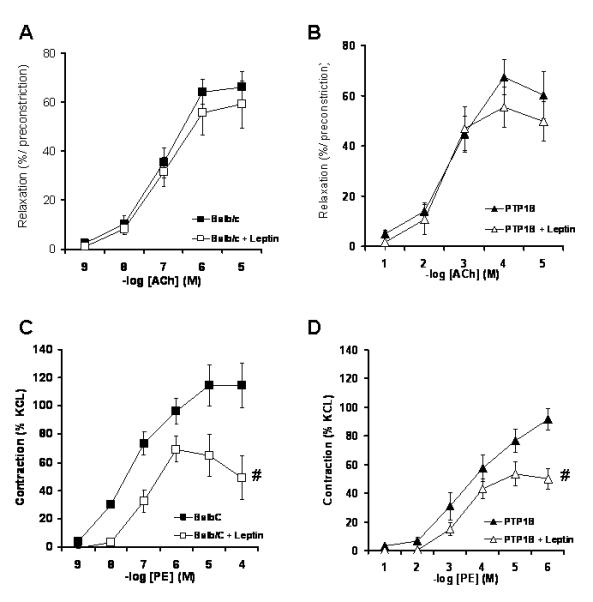
Cumulative concentration response curve to ACh (A: Balb/c, B: PTP1B) and PE (C: Balb/c, D: PTP1B) performed in the mice treated or not with leptin. Data are presented as mean ± sem. Effects of the group and the treatment were determined with a two-way ANOVA.# = p<0.05 vs. Ctrl without leptin. n= 8 per group.
Effects of chronic treatment with leptin on vascular function
To determine the extent to which vascular adrenergic changes were caused by sympathoactivation, vascular reactivity and adrenergic receptor expression were assessed by analyzing the vascular reactivity of mice treated chronically with prazosin in the presence or not of leptin. As reported in Figure 8A and 8B, α1-adrenergic receptor inhibition completely blunted the adrenergic desensitization induced by leptin and also restored the adrenergic reactivity in PTP1B KO mice. As reported by the results of the real-time RT-PCR (Figure 8C), blockade of adrenergic receptors with prazosin restored the vascular adrenergic reactivity by blunting the reduction in α1B and α1D-adrenergic receptors gene expression induced by both leptin and PTP1B deletion.
Figure 8. Chronic treatment with Prazosin restores vascular adrenergic reactivity in PTP1B KO mice.
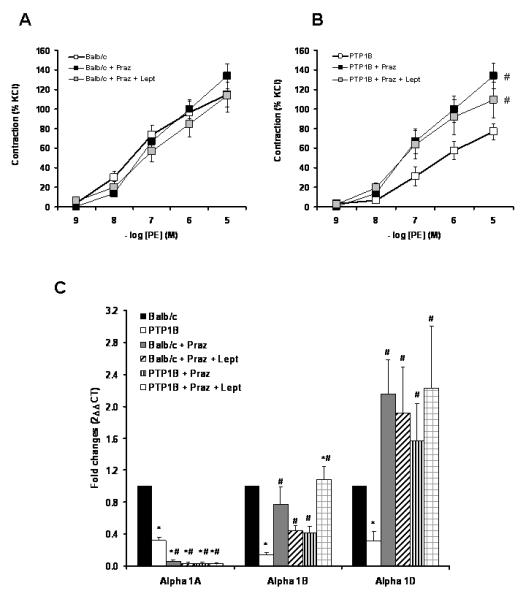
Cumulative concentration response curve to PE performed in Balb/c (A) and PTP1B KO mice (B) treated or not with Prazosin and Prazosin + leptin. C) Quantification of α1-adrenergic receptor subtypes gene expression by quantitative real time RT-PCR performed on aortae taken from Balb/c and PTP1B KO mice, treated or not with Prazosin or Prazosin + leptin. Data are presented as mean ± sem. Effects of the group and the treatment were analyzed by two-way ANOVA (C) and two-way ANOVA for repeated measurements (A and B). # = p<0.05 vs. Ctrl. n= 5 per group.
Discussion
The goal of the current study was to determine the mechanisms by which leptin, independent of obesity, modulates cardiovascular function. The key findings are 1) that increasing leptin sensitivity by PTP1B deletion enhances mean arterial pressure, 2) that the deletion of PTP1B increases the autonomic and presumably sympathetic contribution to blood pressure and 3) that blood pressure responses to leptin-induced sympathoactivation are associated with reductions in adrenergic reactivity.
PTP1B, Leptin and Mean Arterial Pressure
In this study, we demonstrate for the first time that disruption of the PTP1B gene increases mean arterial pressure, supporting the clinical association between PTP1B gene variants and systolic blood pressure 28. More interestingly, this study establishes a novel regulatory factor moderating the relationship between blood pressure and leptin. Specifically, subcutaneous leptin infusion at a subpressor dose to raise plasma concentrations into physiological ranges (Balb/c: 2.7±0.4 vs. PTP1B: 3.3±0.5 ng/mL, NS) failed to produce a pressor response in control Balb/c mice while producing a 20 mmHg increase in pressure in mice lacking PTP1B. These data provide strong evidence that PTP1B, in addition to limiting the metabolic actions of leptin, also acts as a brake on leptin’s hemodynamic actions.
It is noteworthy in these studies that relatively high levels of leptin did not produce an increase in mean arterial pressure in control mice. While several studies reported a mild increase in blood pressure with either an intracerebroventricular (+ 4.1 mmHg) 11, 29 or a subcutaneous leptin infusion (+ 7mmHg) 6, 30, others report no change in blood pressure 7, 29 despite an up to 50-fold increase in plasma leptin 7. A potential explanation for this observation is the report that body weight reduction reduces blood pressure 31. To test this hypothesis, blood pressure was determined during a 20% calorie restriction, mimicking leptin effects on body weight. Contrary to the data reported by Correeia 29 and Williams 31, the body weight reduction induced by the hypocaloric diet was not associated with changes in blood pressure, ruling out this hypothesis to explain the lack of response to leptin.
Finally, we observed that baseline blood pressure was also increased in PTP1B KO mice when compared to controls. While it is tempting to speculate that deletion of PTP1B causes a leptin-mediated hypertension at baseline, caution is warranted as PTP1B may affect other pro-hypertensive pathways. While a role of PTP1B in leptin-mediated hypertension is clear, further studies are needed to determine the mechanisms of baseline hypertension in PTP1B KO mice.
PTP1B, Leptin and the SNS
The deletion of PTP1B not only increased blood pressure, but, as indicated by the greater pressure drop during ganglionic blockade, also resulted in an increased contribution of the autonomic nervous system in blood pressure regulation that is presumably the consequence of a greater sympathetic tone. The ability of leptin to induce sympathetic overdrive was confirmed by the exaggeration of the mecamylamine-induced drop in blood pressure after leptin infusion in control mice. These results are in accordance with several studies establishing a clear relationship between leptin concentration and sympathetic activity 7, 11, 12, 32, 33 and thus, the increased sympathetic tone observed in the PTP1B KO mice might provide an explanation for the elevated blood pressure. Moreover, the deletion of PTP1B has an element of specificity in its pro-hypertensive effects: sympathetically-mediated increases in blood pressure via behavioral stress are similar with and without PTP1B. It is important to note, however, that PTP1B may act via leptin-independent mechanisms to increase sympathetic nerve activity. Indeed, PTP1B deletion not only results in increased leptin sensitivity but also enhanced insulin sensitivity 13. And while the effects of insulin on blood pressure have been largely discarded 17, other data support its role in sympathetic control 34. It is, therefore, possible that PTP1B-induced increased insulin sensitivity might account for at least part of the observed increase in the sympathetic control of arterial pressure. Nevertheless, the lack of change in mean arterial pressure, heart rate (Figure 2C, 2D) and vascular reactivity (Figure III, online-only Data Supplement) in mice chronically treated with insulin, minimizes the contribution of insulin in the development of the alterations observed.
Vascular adaptation to chronic sympathetic overdrive
While many studies of obesity-induced hypertension have documented increased sympathoactivation and speculated this as a mechanism of increased blood pressure, the literature is confounded by examples of sympathoactivation without hypertension. The missing piece of the puzzle to date has been how the vasculature adapts to chronic sympathetic activity. Through in vivo injection of PE (Figure 4) and ex vivo analysis of aortic ring reactivity (Figure 5), we determined that PTP1B deletion blunted increases in blood pressure and vasoconstriction in response to PE stimulation. Similar results were also observed in leptin treated Balb/c mice, attributing the adrenergic desensitization to leptin and its associated sympathoactiviation and suggesting that the reduced vascular adrenergic reactivity is the consequence of the leptin-induced sympathetic overflow. This was further supported by data from mice treated with prazosin which blunted the adrenergic desensitization induced by both leptin and the deletion of PTP1B. Moreover, it suggests that downregulation of adrenergic receptors may be a protective counter-regulation because leptin treated Balb/c mice did not exhibit elevated blood pressure despite increased sympathetic tone. Indeed, rodent models of obesity with relatively mild blood pressure increases show reduced adrenergic reactivity in the splanchnic circulation, consistent with the concept of adrenergic desensitization in the context of obesity-induced sympathoactivation 20, 35.
Finally, we have demonstrated that under leptin treatment, PTP1B KO mice develop an increased blood pressure with no further changes in adrenergic sensitivity. These data suggest that it is possible to saturate the ability to desensitize adrenergic reactivity, and the inability to further decrease adrenergic reactivity in response to sympathetic overdrive might result in a rise in blood pressure. More importantly, these data suggest that the failure to desensitize adrenergic reactivity under conditions of sympathoactiviation, such as obesity, may indeed be a major factor in determining whether frank hypertension develops.
Potential mechanisms for adrenergic desensitization
While increased sympathetic activity leads to α-adrenergic desensitization 36, 37 the mechanisms involved in this desensitization remain largely unknown. Due to the stimulatory effects of leptin on eNOS activity 38, 39 and to the protective effects of PTP1B deletion on flow-mediated dilation 40, we speculated that adrenergic desensitization observed in the PTP1B KO mice could be due to an improved endothelial function. Both ACh-mediated relaxation curves and PE-induced constriction curves, performed in the presence of L-NAME, ruled out this hypothesis. Furthermore, both the morphological analysis of the vessel structure and vascular responses to KCl, 5HT and SNP refuted a smooth muscle cell dysfunction and demonstrated that PTP1B deletion specifically targets the α1-adrenergic signaling pathway. In further support of this specificity, the quantification of the α1-adrenergic receptor gene expression by real time RT-PCR demonstrated that PTP1B deletion significantly reduced the expression of the three α1-adrenergic receptors in the aorta, and that leptin treatment had similar effects. This clearly suggests that the adrenergic desensitization, induced by an increase in leptin sensitivity or in leptin concentration, is the result of a decreased α1-adrenergic receptor expression. This receptor subtype most relevant is the α1D, since, although all receptors are downregulated with leptin/PTP1B-induced sympathoactivation, blockade of leptin-mediated actions by prasozin only restored α1D and completely restored vasoconstrictor reactivity.
In summary, by genetic disruption of PTP1B in lean mice, we have been able to identify PTP1B as a new modulator of blood pressure through its ability to regulate sympathetic tone and adrenergic reactivity. Through the experiments performed with subcutaneous leptin infusion, in a physiological range, we clearly established a role for leptin in the regulation of blood pressure, a role suggested by several authors but never clearly demonstrated. Finally, our data suggest that adrenergic desensitization may be a protective mechanism against hypertension, with a failure to properly desensitize the adrenergic signaling pathway leading to the development of hypertension.
Clinical Perspectives: Protein Tyrosine Phosphatase 1B (PTP1B) controls insulin as well as leptin sensitivity through its ability to directly inactivate catalytically phosphorylated tyrosines. Since deletion of PTP1B has been reported to improve insulin and leptin sensitivity and to protect rodents against the development of obesity and type II diabetes, PTP1B inhibitors are under development to treat metabolic disorders. The present study analyzed the cardiovascular function of PTP1B KO mice and reports that PTP1B deletion increased sympathetic tone and enhanced mean arterial pressure, highlighting the possible deleterious consequences of global PTP1B inhibitors in obese and type II diabetic patients already prone to the development of hypertension. This study further reports that the control mice, Balb/c, did not develop an increase in blood pressure in response to leptin infusion despite an increased sympathetic tone. The significant decreased vascular adrenergic reactivity reported both in vivo and ex vivo (isolated artery) and supported by the reduction in vascular α1-adrenergic receptors expression, suggest that adrenergic desensitization, in response to sympathetic overdrive, may be a protective mechanisms against the development of hypertension. Taken together, these data suggest that caution is warranted in consideration of PTP1B inhibitors as anti-diabetic drugs and further suggest that mechanisms that interfere with adrenergic desensitization may contribute to the hypertension observed in obese individuals with high levels of leptin.
Supplementary Material
Table I: Sequence of the primers used for the quantitative real time RT-PCR.
Figure I: PTP1B deletion did not affect vascular structure. A) Representative pictures of aortic sections. B) Wall to lumen ration and C) quantification of the media cross sectional area in the Balb/c (n=5) and PTP1B KO mice (n=6). Data are presented as mean ± sem.
Figure II: PTP1B deletion did not impair endothelial function. A) Cumulative concentration response curve to Acetylcholine (ACh) performed in the presence or absence of eNOS inhibitor (L-NAME, 10 mM). B) Cumulative concentration response curve to phenylephrine (PE) performed in the presence or absence of eNOS inhibitor (L-NAME, 10 mM). Data are presented as mean ± sem. n= 8-10 per group.
Figure III: Chronic insulin treatment in PTP1B KO mice did not affect the vascular reactivity. Cumulative concentration response curve to (A) PE, (B) 5HT and (C) SNP, performed in PTP1B KO mice treated with glucose or with glucose + leptin. Data are presented as mean ± sem.
Figure IV: Chronic sympatho-inhibition did not affect vascular reactivity to serotonin and sodium Nitroprusside. Cumulative concentration response curve to 5HT (A, in Balb/c and B, in PTP1B KO mice) and SNP (C, in Balb/c and D, in PTP1B KO mice), in mice treated with prazosin and prazosin + leptin. Data are presented as mean ± sem.
Acknowledgements
The authors wish to thank Drs. Brenda Lilly and Edward Inscho for technical assistance and the useful exchange of ideas in the generation of this manuscript. The authors acknowledge the helpful editorial assistance of Jessica Osmond in the preparation of the written document.
Funding sources The authors acknowledge research support from NIH R01 HL067533 to D.W.S. and an AHA Post-Doctoral Fellowship to EJBC.
Footnotes
Disclosures None
References
- 1.Kannel W, Brand M, Skinner J, Dawber T, McNamara P. The relation of adiposity to blood pressure and development of hypertension: the Framingham study. Ann Intern Med. 1967;67:48–59. doi: 10.7326/0003-4819-67-1-48. [DOI] [PubMed] [Google Scholar]
- 2.Reaven GM. Banting lecture 1988. Role of insulin resistance in human disease. Diabetes. 1988;37:1595–1607. doi: 10.2337/diab.37.12.1595. [DOI] [PubMed] [Google Scholar]
- 3.Rahmouni K, Correia ML, Haynes WG, Mark AL. Obesity-associated hypertension: new insights into mechanisms. Hypertension. 2005;45:9–14. doi: 10.1161/01.HYP.0000151325.83008.b4. [DOI] [PubMed] [Google Scholar]
- 4.Maffei M, Halaas J, Ravussin E, Pratley RE, Lee GH, Zhang Y, Fei H, Kim S, Lallone R, Ranganathan S, Kern PA, Friedman, JM. Leptin levels in human and rodent: measurement of plasma leptin and ob RNA in obese and weight-reduced subjects. Nature medicine. 1995;1:1155–1161. doi: 10.1038/nm1195-1155. [DOI] [PubMed] [Google Scholar]
- 5.Friedman JM, Halaas JL. Leptin and the regulation of body weight in mammals. Nature. 1998;395:763–770. doi: 10.1038/27376. [DOI] [PubMed] [Google Scholar]
- 6.Carlyle M, Jones OB, Kuo JJ, Hall JE. Chronic cardiovascular and renal actions of leptin: role of adrenergic activity. Hypertension. 2002;39:496–501. doi: 10.1161/hy0202.104398. [DOI] [PubMed] [Google Scholar]
- 7.Haynes WG, Morgan DA, Walsh SA, Mark AL, Sivitz WI. Receptor-mediated regional sympathetic nerve activation by leptin. J Clin Invest. 1997;100:270–278. doi: 10.1172/JCI119532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Hegyi K, Fülöp K, Kovács K, Tóth S, Falus A. Leptin-induced signal transduction pathways. Cell Biology International. 2004;28:159–169. doi: 10.1016/j.cellbi.2003.12.003. [DOI] [PubMed] [Google Scholar]
- 9.Cheng A, Uetani N, Simoncic PD, Chaubey VP, Lee-Loy A, McGlade CJ, Kennedy BP, Tremblay ML. Attenuation of leptin action and regulation of obesity by protein tyrosine phosphatase 1B. Dev Cell. 2002;2:497–503. doi: 10.1016/s1534-5807(02)00149-1. [DOI] [PubMed] [Google Scholar]
- 10.Zabolotny JM, Bence-Hanulec KK, Stricker-Krongrad A, Haj F, Wang Y, Minokoshi Y, Kim YB, Elmquist JK, Tartaglia LA, Kahn BB, Neel BG. PTP1B regulates leptin signal transduction in vivo. Dev Cell. 2002;2:489–495. doi: 10.1016/s1534-5807(02)00148-x. [DOI] [PubMed] [Google Scholar]
- 11.Dunbar JC, Hu Y, Lu H. Intracerebroventricular leptin increases lumbar and renal sympathetic nerve activity and blood pressure in normal rats. Diabetes. 1997;46:2040–2043. doi: 10.2337/diab.46.12.2040. [DOI] [PubMed] [Google Scholar]
- 12.Eikelis N, Schlaich M, Aggarwal A, Kaye D, Esler M. Interactions Between Leptin and the Human Sympathetic Nervous System. Hypertension. 2003;41:1072–1079. doi: 10.1161/01.HYP.0000066289.17754.49. [DOI] [PubMed] [Google Scholar]
- 13.Elchebly M, Payette P, Michaliszyn E, Cromlish W, Collins S, Loy AL, Normandin D, Cheng A, Himms-Hagen J, Chan CC, Ramachandran C, Gresser MJ, Tremblay ML, Kennedy BP. Increased insulin sensitivity and obesity resistance in mice lacking the protein tyrosine phosphatase-1B gene. Science. 1999;283:1544–1548. doi: 10.1126/science.283.5407.1544. [DOI] [PubMed] [Google Scholar]
- 14.Lee DL, Leite R, Fleming C, Pollock JS, Webb RC, Brands MW. Hypertensive response to acute stress is attenuated in interleukin-6 knockout mice. Hypertension. 2004;44:259–263. doi: 10.1161/01.HYP.0000139913.56461.fb. [DOI] [PubMed] [Google Scholar]
- 15.Della-Fera MA, Choi YH, Hartzell DL, Duan J, Hamrick M, Baile CA. Sensitivity of ob/ob mice to leptin-induced adipose tissue apoptosis. Obes Res. 2005;13:1540–1547. doi: 10.1038/oby.2005.189. [DOI] [PubMed] [Google Scholar]
- 16.Luque RM, Huang ZH, Shah B, Mazzone T, Kineman RD. Effects of leptin replacement on hypothalamic-pituitary growth hormone axis function and circulating ghrelin levels in ob/ob mice. Am J Physiol Endocrinol Metab. 2007;292:E891–899. doi: 10.1152/ajpendo.00258.2006. [DOI] [PubMed] [Google Scholar]
- 17.Hall JE, Brands MW, Zappe DH, Dixon WN, Mizelle HL, Reinhart GA, Hildebrandt DA. Hemodynamic and Renal Responses to Chronic Hyperinsulinemia in Obese, Insulin-Resistant Dogs. Hypertension. 1995;25:994–1002. doi: 10.1161/01.hyp.25.5.994. [DOI] [PubMed] [Google Scholar]
- 18.Johansson ME, Andersson IJ, Alexanderson C, Skott O, Holmang A, Bergstrom G. Hyperinsulinemic rats are normotensive but sensitized to angiotensin II. Am J Physiol Regul Integr Comp Physiol. 2008;294:R1240–1247. doi: 10.1152/ajpregu.00493.2007. [DOI] [PubMed] [Google Scholar]
- 19.D’Angelo G, Mintz JD, Tidwell JE, Schreihofer AM, Pollock DM, Stepp DW. Exaggerated Cardiovascular Stress Responses and Impaired {beta}-Adrenergic-Mediated Pressor Recovery in Obese Zucker Rats. Hypertension. 2006;48:1109–1115. doi: 10.1161/01.HYP.0000247306.53547.d4. [DOI] [PubMed] [Google Scholar]
- 20.Romanko OP, Stepp DW. Reduced constrictor reactivity balances impaired vasodilation in the mesenteric circulation of the obese Zucker rat. Am J Physiol Heart Circ Physiol. 2005;289:H2097–2102. doi: 10.1152/ajpheart.00213.2005. [DOI] [PubMed] [Google Scholar]
- 21.Jeffries WB, Tam LT, Wang Y, Smyth DD, Pettinger WA. Prazosin-induced alterations in renal alpha-adrenergic receptor function. Hypertension. 1987;9:III125–129. doi: 10.1161/01.hyp.9.6_pt_2.iii125. [DOI] [PubMed] [Google Scholar]
- 22.Zhang L, Taniguchi T, Tanaka T, Shinozuka K, Kunitomo M, Nishiyama M, Kamata K, Muramatsu I. Alpha-1 adrenoceptor up-regulation induced by prazosin but not KMD-3213 or reserpine in rats. British journal of pharmacology. 2002;135:1757–1764. doi: 10.1038/sj.bjp.0704639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Woods JR, Jr., Dandavino A, Murayama K, Brinkman CRd, Assali NS. Autonomic control of cardiovascular functions during neonatal development and in adult sheep. Circ Res. 1977;40:401–407. doi: 10.1161/01.res.40.4.401. [DOI] [PubMed] [Google Scholar]
- 24.Dube N, Cheng A, Tremblay ML. The role of protein tyrosine phosphatase 1B in Ras signaling. Proc Natl Acad Sci U S A. 2004;101:1834–1839. doi: 10.1073/pnas.0304242101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Nakamura Y, Patrushev N, Inomata H, Mehta D, Urao N, Kim HW, Razvi M, Kini V, Mahadev K, Goldstein BJ, McKinney R, Fukai T, Ushio-Fukai M. Role of Protein Tyrosine Phosphatase 1B in Vascular Endothelial Growth Factor Signaling and Cell-Cell Adhesions in Endothelial Cells. Circ Res. 2008;102:1182–1191. doi: 10.1161/CIRCRESAHA.107.167080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Driss AB, Devaux C, Henrion D, Duriez M, Thuillez C, Levy BI, Michel J-B. Hemodynamic Stresses Induce Endothelial Dysfunction and Remodeling of Pulmonary Artery in Experimental Compensated Heart Failure. Circulation. 2000;101:2764–2770. doi: 10.1161/01.cir.101.23.2764. [DOI] [PubMed] [Google Scholar]
- 27.Lembo G, Iaccarino G, Vecchione C, Barbato E, Izzo R, Fontana D, Trimarco B. Insulin modulation of an endothelial nitric oxide component present in the alpha2- and beta-adrenergic responses in human forearm. J Clin Invest. 1997;100:2007–2014. doi: 10.1172/JCI119732. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Cheyssac C, Lecoeur C, Dechaume A, Bibi A, Charpentier G, Balkau B, Marre M, Froguel P, Gibson F, Vaxillaire M. Analysis of common PTPN1 gene variants in type 2 diabetes, obesity and associated phenotypes in the French population. BMC Medical Genetics. 2006;7:44. doi: 10.1186/1471-2350-7-44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Correia MLG, Morgan DA, Sivitz WI, Mark AL, Haynes WG. Leptin Acts in the Central Nervous System to Produce Dose-Dependent Changes in Arterial Pressure. Hypertension. 2001;37:936–942. doi: 10.1161/01.hyp.37.3.936. [DOI] [PubMed] [Google Scholar]
- 30.Shek EW, Brands MW, Hall JE. Chronic Leptin Infusion Increases Arterial Pressure. Hypertension. 1998;31:409–414. doi: 10.1161/01.hyp.31.1.409. [DOI] [PubMed] [Google Scholar]
- 31.Williams TD, Chambers JB, Henderson RP, Rashotte ME, Overton JM. Cardiovascular responses to caloric restriction and thermoneutrality in C57BL/6J mice. Am J Physiol Regul Integr Comp Physiol. 2002;282:R1459–1467. doi: 10.1152/ajpregu.00612.2001. [DOI] [PubMed] [Google Scholar]
- 32.Shirasaka T, Takasaki M, Kannan H. Cardiovascular effects of leptin and orexins. Am J Physiol Regul Integr Comp Physiol. 2003;284:R639–651. doi: 10.1152/ajpregu.00359.2002. [DOI] [PubMed] [Google Scholar]
- 33.Aizawa-Abe M, Ogawa Y, Masuzaki H, Ebihara K, Satoh N, Iwai H, Matsuoka N, Hayashi T, Hosoda K, Inoue G, Yoshimasa Y, Nakao K. Pathophysiological role of leptin in obesity-related hypertension. J Clin Invest. 2000;105:1243–1252. doi: 10.1172/JCI8341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Rahmouni K, Morgan DA, Morgan GM, Liu X, Sigmund CD, Mark AL, Haynes WG. Hypothalamic PI3K and MAPK differentially mediate regional sympathetic activation to insulin. J Clin Invest. 2004;114:652–658. doi: 10.1172/JCI21737. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Schreihofer AM, Hair CD, Stepp DW. Reduced plasma volume and mesenteric vascular reactivity in obese Zucker rats. Am J Physiol Regul Integr Comp Physiol. 2005;288:R253–261. doi: 10.1152/ajpregu.00498.2004. [DOI] [PubMed] [Google Scholar]
- 36.Charkoudian N, Joyner MJ, Sokolnicki LA, Johnson CP, Eisenach JH, Dietz NM, Curry TB, Wallin BG. Vascular adrenergic responsiveness is inversely related to tonic activity of sympathetic vasoconstrictor nerves in humans. The Journal of physiology. 2006;572:821–827. doi: 10.1113/jphysiol.2005.104075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Smith EG, Voyles WF, Kirby BS, Markwald RR, Dinenno FA. Ageing and leg postjunctional {alpha}-adrenergic vasoconstrictor responsiveness in healthy men. The Journal of physiology. 2007;582:63–71. doi: 10.1113/jphysiol.2007.130591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Fruhbeck G. Pivotal role of nitric oxide in the control of blood pressure after leptin administration. Diabetes. 1999;48:903–908. doi: 10.2337/diabetes.48.4.903. [DOI] [PubMed] [Google Scholar]
- 39.Knudson JD, Dincer UD, Zhang C, Swafford AN, Jr., Koshida R, Picchi A, Focardi M, Dick GM, Tune JD. Leptin receptors are expressed in coronary arteries, and hyperleptinemia causes significant coronary endothelial dysfunction. Am J Physiol Heart Circ Physiol. 2005;289:H48–56. doi: 10.1152/ajpheart.01159.2004. [DOI] [PubMed] [Google Scholar]
- 40.Vercauteren M, Remy E, Devaux C, Dautreaux B, Henry J-P, Bauer F, Mulder P, van Huijsduijnen R Hooft, Bombrun A, Thuillez C, Richard V. Improvement of Peripheral Endothelial Dysfunction by Protein Tyrosine Phosphatase Inhibitors in Heart Failure. Circulation. 2006;114:2498–2507. doi: 10.1161/CIRCULATIONAHA.106.630129. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Table I: Sequence of the primers used for the quantitative real time RT-PCR.
Figure I: PTP1B deletion did not affect vascular structure. A) Representative pictures of aortic sections. B) Wall to lumen ration and C) quantification of the media cross sectional area in the Balb/c (n=5) and PTP1B KO mice (n=6). Data are presented as mean ± sem.
Figure II: PTP1B deletion did not impair endothelial function. A) Cumulative concentration response curve to Acetylcholine (ACh) performed in the presence or absence of eNOS inhibitor (L-NAME, 10 mM). B) Cumulative concentration response curve to phenylephrine (PE) performed in the presence or absence of eNOS inhibitor (L-NAME, 10 mM). Data are presented as mean ± sem. n= 8-10 per group.
Figure III: Chronic insulin treatment in PTP1B KO mice did not affect the vascular reactivity. Cumulative concentration response curve to (A) PE, (B) 5HT and (C) SNP, performed in PTP1B KO mice treated with glucose or with glucose + leptin. Data are presented as mean ± sem.
Figure IV: Chronic sympatho-inhibition did not affect vascular reactivity to serotonin and sodium Nitroprusside. Cumulative concentration response curve to 5HT (A, in Balb/c and B, in PTP1B KO mice) and SNP (C, in Balb/c and D, in PTP1B KO mice), in mice treated with prazosin and prazosin + leptin. Data are presented as mean ± sem.