

To the Editor
The availability of high-resolution microarrays has greatly improved the detection of genomic rearrangements in patients with multiple congenital anomalies, developmental delays, and mental retardation [Ming et al., 2006; Stankiewicz et al., 2007; Shaffer et al., 2007; Sharp et al., 2008]. These abnormal phenotypes are commonly due to an alteration of one or more dosage-sensitive gene(s) within the affected region. Alternatively, deletions may unmask an autosomal recessive mutation carried on the non-deleted allele, which has led to the discovery of several genetic and biochemical abnormalities inherited in an autosomal recessive fashion [Ludlow et al., 1996; Garshasbi et al., 2008; Bisgaard et al., 2008]. We describe here a male patient with multiple congenital anomalies who was diagnosed with Peters plus syndrome (PPS). He was found to have a heterozygous deletion by microarray analysis that included the B3GALTL gene. Sequencing of B3GALTL demonstrated a mutation on the non-deleted allele that has been seen previously in PPS patients [Lesnik Oberstein et al., 2006; Kapoor et al., 2008; Reis et al., 2008].
The patient was evaluated at one month of age for hydrocephalus thought to be due to aqueductal stenosis, thin corpus callosum, corneal clouding, dysmorphic facial features, bilateral cryptorchidism, and a sacral dimple with tethering of the spinal cord. His birth history was significant for a premature induced vaginal delivery at 33 weeks gestational age to a 23 yo G4P1→2 female (two prior first trimester spontaneous miscarriages) due to intrauterine growth restriction and poor scores on a biophysical profile. His birth weight and length were both at the 10th centile, and his head circumference was at the 80th centile. An echocardiogram performed due to the presence of a murmur was normal except for mild peripheral pulmonic stenosis. Radiological studies included a skeletal survey that revealed platybasia and toes of equal lengths, and a renal ultrasound showing duplication of the left renal collecting system. The family history was noncontributory. His parents were non-consanguineous and of German, Irish, and Lithuanian decent. A repeat genetics evaluation at 7 months demonstrated short stature (50th centile for 4 months), macrocephaly with a prominent forehead, short palpebral fissures (5th centile), cupped right helix, depressed nasal bridge, retrognathia, cryptorchidism, sacral dimple, rhizomelic shortening and bilateral clinodactyly of the upper extremities, and syndactyly between the 2nd and 3rd toes (Fig 1).
Figure 1. Photographs of the patient at age 7 months.
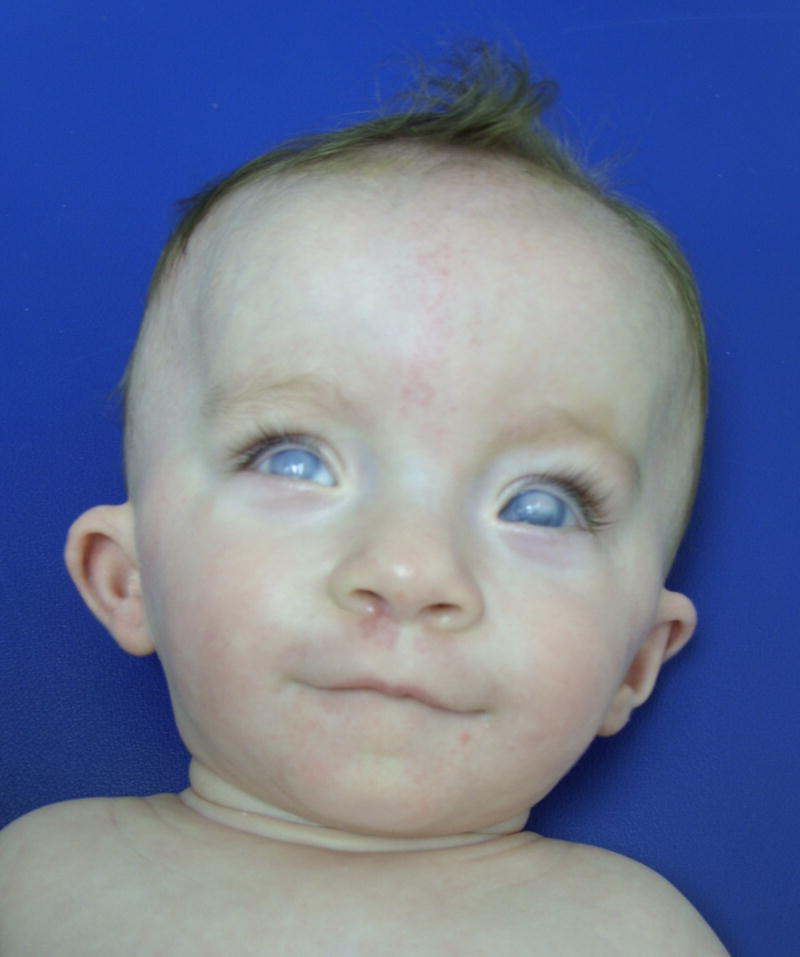

Dysmorphic features included frontal prominence with occipital flattening, corneal clouding, upslanting palpebral fissures, hypertelorism, small and low-set ears with thickened and protuberant helices, and a thin upper lip with an exaggerated Cupid's bow.
Due to his multiple congenital anomalies, a high-density oligonucleotide microarray was performed using the Affymetrix SNP6.0 platform (Affymetrix, Santa Clara, CA). A 781-kb heterozygous deletion was found on 13q12.3 (chr13:30,069,778-30,850,926, NCBI hg18 build), and later confirmed by fluorescence in situ hybridization with a probe located in the deleted region (RP11-173P16). The deletion involves five genes (Figure 2), including the B3GALTL gene that is implicated in PPS. We then sequenced the exons and their flanking regions and determined our patient to carry a c.660+1G>A mutation in addition to the deletion. Parental analysis confirmed them to be heterozygous carriers, with the deletion inherited from the mother and the point mutation inherited from the father.
Figure 2. Microarray results.
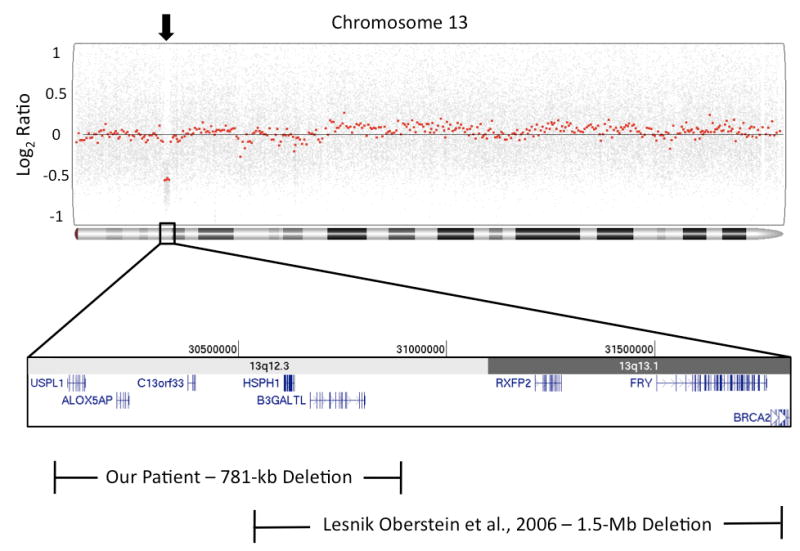
An Affymetrix SNP6.0 microarray detected a heterozygous 781-kb deletion of 13q12.3 (arrow). Analysis of the deletion coordinates in the UCSC Genome Browser (http://genome.ucsc.edu) showed five genes to be affected, including B3GALTL. Overlap of our patient's deletion is shown compared to one previously described in two brothers with Peters plus syndrome [Lesnick Oberstein et al., 2006].
Peters plus syndrome (OMIM 261540) is a multiple congenital anomaly syndrome where patients present with the Peters anomaly as well as several extra ocular characteristics [Maillette de Buy Wenninger-Prick and Hennekam, 2002]. The β1,3-galactosyltransferase-like (B3GALTL) gene on 13q12.3 was recently discovered to be the causative gene in this syndrome [Lesnik Oberstein et al., 2006]. The protein product is a glucosyltransferase, which adds a glucose molecule to an O-linked fucose located on thrombospondin type 1 repeats of several important proteins [Heinonen et al., 2003; Hess et al., 2008]. PPS is therefore considered a new congenital disorder of O-glycosylation [Heinonen et al., 2009]. Every PPS patient with molecular analysis of the B3GALTL gene harbors at least one common point mutation involving the donor splice site of exon 8, c.660+1G>A [Lesnick Oberstein et al., 2006; Kapoor et al., 2008; Reis et al., 2008]. Most PPS patients analyzed are homozygous for this mutation, but there have been additional mutations identified in patients with PPS [Lesnick Oberstein et al., 2006; Reis et al., 2008]. Although PPS is a clinically heterogeneous disorder, our patient appears to have many features previously described [Maillette de Buy Wenninger-Prick and Hennekam, 2002]. New findings in our patient include platybasia of the skull and tethering of the spinal cord. However, vertebral segmentation abnormalities and spina bifida occulta have been reported [Maillette de Buy Wenninger-Prick and Hennekam, 2002; Kapoor et al., 2008].
Two brothers were reported with heterozygous 1.5-Mb deletions involving B3GALTL and the common c.660+1G>A mutation on the other allele [Lesnick Oberstein et al., 2006]. Our patient's deletion is 781-kb, and is the smallest deletion in this region reported in the literature or online databases such as DECIPHER (http://decipher.sanger.ac.uk/). Comparing our deletion to the two brothers reported by Lesnick Oberstein et al. [2006], the common region of overlap includes a small area of 13q12.3 that contains 2 genes, HSPH1 and B3GALTL (Figure 2). HSPH1 encodes heat shock protein 105-kDa, a protein that is over expressed in certain cancers [Muchemwa et al., 2008] but has no association with a congenital anomaly syndrome. The other genes deleted only in our patient include USPL1, ALOX5AP, and C13orf33. USPL1 is an ubiquitin-specific peptidase with unclear function, and C13orf33 is a hypothetical protein based on DNA sequence analysis. ALOX5AP polymorphisms have been reported in atherosclerosis and atopic diseases [Halloway et al., 2008; Lemaitre et al., 2008], but no deletions of this gene have been implicated in human disease. Therefore, the heterozygous deletion and mutation of B3GALTL in our patient are the likely cause of his clinical features.
The deletion observed by Lesnick Oberstein et al. [2006] was more distal compared to our patient and contained additional genes such as LGR8/RXFP2, FRY, and BRCA2. It was suggested that the cryptorchidism observed in their patients may be due to haploinsufficiency of the nearby LGR8/RXFP2 gene, as this has been associated with cryptorchidism [Ferlin et al., 2008]. However, we did not find involvement of this gene in our patient (Figure 2). As cryptorchidism has been observed in PPS and other congenital disorders of glycosylation [Maillette de Buy Wenninger-Prick and Hennekam, 2002; Schollen et al., 2004], this specific finding in our patient is likely related to the underlying disorder rather than a positional effect of the deletion. There was also a family history of breast cancer in the family of the other deletion patients due to involvement of BRCA2. Our patient does not have BRCA2 deleted, nor is there a family history of breast or ovarian cancer, again suggesting there is no positional effect of the deletion on the function of this gene. The commonly observed clinical characteristics in both our patient and the two brothers is therefore most likely due to the involvement of B3GALTL, yet their variable phenotypes may be caused by the multisystemic disease process seen in glycosylation defects or to the different genes associated with their respective deletions.
In summary, we present here a patient with Peters plus syndrome, who was found by microarray analysis to have a contiguous gene deletion of 781-kb on 13q12.3. Sequencing analysis of the non-deleted allele of the B3GALTL gene identified a previously known pathogenic point mutation. This patient involves the second reported finding of a deletion of B3GALTL in PPS, and is the smallest known deletion involving this specific locus. Our report also highlights the utility of microarray-based copy number analysis in the diagnosis of patients with multiple congenital anomalies, especially when the copy number abnormalities involve genes that are associated with autosomal recessive disorders.
Acknowledgments
We would like to thank the patient's family for their participation. This work is partially supported by a grant from the NIH (GM081519) to T.H.S. C.H-E. is supported by a Medical Genetics Research Training Grant, 5-T32-GM-008638-11, to the University of Pennsylvania.
References
- Bisgaard AM, Kirchhoff M, Nielsen JE, Kibæk M, Lund A, Schwartz M, Christensen E. Chromosomal deletion unmasking a recessive disease: 22q13 deletion syndrome and metachromatic leukodystrophy. Clin Genet. 2008 Nov 21; doi: 10.1111/j.1399-0004.2008.01113.x. Epub ahead of print. [DOI] [PubMed] [Google Scholar]
- Ferlin A, Zuccarello D, Zuccarello B, Chirico MR, Zanon GF, Foresta C. Genetic alterations associated with cryptorchidism. JAMA. 2008;300:2271–2276. doi: 10.1001/jama.2008.668. [DOI] [PubMed] [Google Scholar]
- Garshasbi M, Hadavi V, Habibi H, Kahrizi K, Kariminejad R, Behjati F, Tzschach A, Najmabadi H, Ropers HH, Kuss AW. A defect in the TUSC3 gene is associated with autosomal recessive mental retardation. Am J Hum Genet. 2008;82:1158–1164. doi: 10.1016/j.ajhg.2008.03.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Halloway JW, Barton SJ, Holgate ST, Rose-Zerilli MJ, Sayers I. The role of LTA4H and ALOX5AP polymorphism in asthma and allergy susceptibility. Allergy. 2008;63:1046–1053. doi: 10.1111/j.1398-9995.2008.01667.x. [DOI] [PubMed] [Google Scholar]
- Heinonen TYK, Pasternack L, Lindfors K, Breton C, Gastinel LN, Maki M, Kainulainen H. A novel human glycosyltransferase: primary structure and characterization of the gene and transcripts. Biochem Biophys Res Commun. 2003;309:166–174. doi: 10.1016/s0006-291x(03)01540-7. [DOI] [PubMed] [Google Scholar]
- Heinonen TYK, Maki M. Peters'-plus syndrome is a congenital disorder of glycosylation caused by a defect in the 1,3-glucosyltransferase that modifies thrombospondin type 1 repeats. Ann Med. 2009;41:2–10. doi: 10.1080/07853890802301975. [DOI] [PubMed] [Google Scholar]
- Hess D, Keusch JJ, Lesnik Oberstein SA, Hennekam RC, Hofsteenge J. Peters plus syndrome is a new congenital disorder of glycosylation and involves defective O-glycosylation of thrombospondin type 1 repeats. J Biol Chem. 2008;283:7354–7360. doi: 10.1074/jbc.M710251200. [DOI] [PubMed] [Google Scholar]
- Kapoor S, Banerjee Mukherjee S, Arora R, Shroff D. Peters plus syndrome. Indian J Pediatr. 2008;75:635–637. doi: 10.1007/s12098-008-0122-6. [DOI] [PubMed] [Google Scholar]
- Lemaitre RN, Rice K, Marciante K, Bis JC, Lumley TS, Wiggins KL, Smith NL, Heckbert SR, Psaty BM. Variation in eicosanoid genes, non-fatal myocardial infarction and ischemic stroke. Atherosclerosis. 2008 Nov 1; doi: 10.1016/j.atherosclerosis.2008.10.011. Epub ahead of print. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lesnik Oberstein SA, Kriek M, White SJ, Kalf ME, Szuhai K, den Dunnen JT, Breuning MH, Hennekam RC. Peters Plus syndrome is caused by mutations in B3GALTL, a putative glycosyltransferase. Am J Hum Genet. 2006;79:562–566. doi: 10.1086/507567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ludlow LB, Schick BP, Budarf ML, Driscoll DA, Zackai EH, Cohen A, Konkle BA. Identification of a mutation in a GATA binding site of the platelet glycoprotein Ib-beta promoter resulting in the Bernard-Soulier syndrome. J Biol Chem. 1996;271:22076–22080. doi: 10.1074/jbc.271.36.22076. [DOI] [PubMed] [Google Scholar]
- Maillette de Buy Wenninger-Prick LJJ, Hennekam RCM. Peters' plus syndrome: a review. Ann Genet. 2002;45:97–103. doi: 10.1016/s0003-3995(02)01120-6. [DOI] [PubMed] [Google Scholar]
- Ming JE, Geiger E, James AC, Ciprero KL, Nimmakayalu M, Zhang Y, Huang A, Vaddi M, Rappaport E, Zackai EH, Shaikh TH. Rapid detection of submicroscopic chromosomal rearrangements in children with multiple congenital anomalies using high-density oligonucleotide arrays. Hum Mutat. 2006;27:467–473. doi: 10.1002/humu.20322. [DOI] [PubMed] [Google Scholar]
- Muchemwa FC, Nakatsura T, Fukushima S, Nishimura Y, Kageshita T, Ihn H. Differential expression of heat shock protein 105 in melanoma and melanocytic naevi. Melanoma Res. 2008;18:166–171. doi: 10.1097/CMR.0b013e3282fe9a16. [DOI] [PubMed] [Google Scholar]
- Reis LM, Tyler RC, Abdul-Rahman O, Trapane P, Wallerstein R, Broome D, Hoffman J, Khan A, Paradiso C, Ron N, Bergner A, Semina EV. Mutation analysis of B3GALTL in Peters plus syndrome. Am J Med Genet A. 2008;146:2603–2610. doi: 10.1002/ajmg.a.32498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schollen E, Frank CG, Keldermans L, Reyntjens R, Grubenmann CE, Clayton PT, Winchester BG, Smeitink J, Wevers RA, Aebi M, Hennet T, Matthijs G. Clinical and molecular features of three patients with congenital disorders of glycosylation type Ih (CDG-Ih) (ALG8 deficiency) J Med Genet. 2004;41:550–556. doi: 10.1136/jmg.2003.016923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shaffer LG, Theisen A, Bejjani BA, Ballif BC, Aylsworth AS, Lim C, McDonald M, Ellison JW, Kostiner D, Saitta S, Shaikh T. The discovery of microdeletion syndromes in the post-genomic era: review of the methodology and characterization of a new 1q41q42 microdeletion syndrome. Genet Med. 2007;9:607–616. doi: 10.1097/gim.0b013e3181484b49. [DOI] [PubMed] [Google Scholar]
- Sharp AJ, Mefford HC, Li K, Baker C, Skinner C, Stevenson RE, Schroer RJ, Novara F, De Gregori M, Ciccone R, Broomer A, Casuga I, Wang Y, Xiao C, Barbacioru C, Gimelli G, Bernardina BD, Torniero C, Giorda R, Regan R, Murday V, Mansour S, Fichera M, Castiglia L, Failla P, Ventura M, Jiang Z, Cooper GM, Knight SJ, Romano C, Zuffardi O, Chen C, Schwartz CE, Eichler EE. A recurrent 15q13.3 microdeletion syndrome associated with mental retardation and seizures. Nat Genet. 2008;40:322–328. doi: 10.1038/ng.93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stankiewicz P, Beaudet AL. Use of array CGH in the evaluation of dysmorphology, malformations, developmental delay, and idiopathic mental retardation. Curr Opin Genet Dev. 2007;17:182–192. doi: 10.1016/j.gde.2007.04.009. [DOI] [PubMed] [Google Scholar]