

Abstract
Quercetin is a flavonoid present in many vegetables, fruits and beverages. Due to its anti-oxidant, anti-tumor and anti-inflammatory activity, quercetin has been studied extensively as a chemoprevention agent in several cancer models. Since most of these studies used higher doses of quercetin than clinically achievable, we focused on the effectiveness of physiologically relevant doses of quercetin. A low dose of quercetin exerted cancer cell-specific inhibition of proliferation and this inhibition resulted from cell cycle arrest at the G1 phase. Quercetin induced p21 CDK inhibitor with a concomitant decrease of phosphorylation of pRb, which inhibits the G1/S cell cycle progression by trapping E2F1. A low dose of quercetin induced mild DNA damage and Chk2 activation, which is the main regulator of p21 expression by quercetin. In addition, quercetin down-regulated the cyclin B1 and CDK1, essential components of G2/M cell cycle progression. Inhibition of the recruitment of key transcription factor NF-Y to cyclin B1 gene promoter by quercetin led to transcriptional inhibition. This study proved that the chemopreventive efficacy of a physiologically relevant dose of quercetin can be achievable through the inhibition of cell cycle progression.
Keywords: quercetin, cell cycle, p21, cyclin B1, Chk2, DNA damage
Introduction
Flavonoids are polyphenolic compounds which occur ubiquitously in foods of plant origin and are categorized according to chemical structure into flavonols, flavones, flavanones, isoflavones, catechins, anthocyanidins and chalcones [Kühnau, 1976]. Over 4,000 flavonoids have been identified, many of which occur in fruits, vegetables and beverages. Flavonoids have aroused considerable interest recently because of their potential beneficial effects on human health--they have been reported to have antiviral, anti-allergic, antiplatelet, anti-inflammatory, antitumor and antioxidant activities [Lea et al., 1993; Plaumann et al., 1996; Di Carlo et al., 1999]. Epidemiological studies have shown that the consumption of sources of flavonoids (vegetables, fruits, and tea) is associated with a low risk of cancer [Block et al., 1992]. Many flavonoids exert potent antitumor activity through induction of apoptosis and cell cycle arrest in several cancer cell lines [Wenzel et al., 2000].
Quercetin (3,5,7,3′,4′-pentahydroxyflavone) is the major representative of the flavonoid subclass of flavonols. Due to its anti-oxidant, anti-tumor and anti-inflammatory activity, quercetin has been studied extensively as a chemoprevention agent in several cancer models [Hertog et al., 1993]. Quercetin has been shown to inhibit the proliferation of a wide range of cancers such as prostate, cervical, lung, breast, and colon. Recent studies have revealed that quercetin inhibits cell proliferation by causing apoptosis and/or cell cycle arrest [Yang et al., 2005; Lee et al., 2006]. It has been shown that quercetin treatment causes cell cycle arrests such as G2/M arrest or G1 arrest in different cell types [Choi et al., 2001; Ong et al., 2004; Beniston and Campo, 2003]. Moreover, quercetin-mediated apoptosis may result from the induction of stress proteins, disruption of microtubules and mitochondria, release of cytochrome c, and activation of caspases [Yoshizumi et al., 2001]. Quercetin is a strong antioxidant because it can chelate metals, scavenge oxygen free radicals [Kandaswami and Middleton, 1994; Bors et al., 1990] and inhibit xanthine oxidase and lipid peroxidation in vitro [Bors et al., 1994; da Silva et al., 1998; Vulcain et al., 2005].
Most of the studies which have shown the antitumor activity of quercetin were performed with a high concentration of quercetin, rangin from 25 μM to 200 μM. But pharmacokinetic research reveals that the peak concentration of quercetin in blood after food uptake reaches to a peak of around 10 μM [Gugler et al., 1975; Nielsen et al., 1997; Hollman et. al., 1997]. Therefore we focused on examining the effectiveness of physiologically relevant concentrations of quercetin, by administrating low amounts of quercetin every 24 h to breast cancer cells to mimic the conditions experienced by daily consumption. Here, we show that clinically relevant amounts of quercetin induce cell cycle arrest in the G0/G1 phase through hypo-phosphorylation of pRb, which is accompanied by the induction of CDK inhibitor p21. We further show that quercetin generates mild DNA damage and activates Chk2 kinase, which plays a crucial role in p21 induction. In addition, transcriptional repression of cyclin B1 is revealed as another mechanism of the anti-proliferation effect of a low dose of quercetin.
Materials and Methods
Cell culture
Human breast carcinoma SK-Br3, MDA-MB-453, and MDA-MB-231 cells were cultured in Dulbecco's Modified Eagle's Medium (DMEM) with 10% fetal bovine serum (HyClone, Logan, UT). MCF-10A cells were cultured in DMEM and Ham's F12 medium (1:1) supplemented with 5% horse serum, 20 ng/ml epidermal growth factor, 10 μg/ml insulin, 500 ng/ml hydrocortisone, and 100 ng/ml cholera enterotoxin [Soule et al., 1990]. Both media contained 100 units of penicillin and streptomycin (Invitrogen, Carlsbad, CA). The cells were maintained in a humidified atmosphere containing 5% CO2 and air at 37°C. All cell lines were obtained from American Type Culture Collection (Manassas, VA).
Drug treatment
Cells were seeded into 100 mm tissue culture plates and then left for 24 h before being treated with quercetin. Cells were treated for 24, 48, 72 or 96 h, with new medium being added with fresh quercetin every 24 h. Cell proliferation was assayed by cell counts using a hemocytometer or Z1 Coulter Counter (Beckman Coulter, Fullerton, CA). For trypan blue exclusion assay [Burow et al., 1998], trypsinized cells were pelleted and resuspended in 0.5 ml of medium, and 0.5 ml of 0.4% trypan blue solution. The samples were mixed thoroughly, incubated at room temperature for 15 min, and examined under a light microscope. At least 300 cells were counted for each survival determination.
Reagents
Quercetin (2-(3,4-Dihydroxyphenyl)-3,5,7-trihydroxy-4H-1-benzopyran-4-one dehydrate) and Chk2 inhibitor II were purchased from Sigma Chemical Co. (St. Louis, MO). The chemicals were dissolved in dimethyl sulfoxide (DMSO) and added directly to the culture medium.
Antibodies
Antibodies were obtained as follow. Anti-PARP was from Biomol Research Laboratory (Plymouth Meeting, PA), anti-actin monoclonal antibody was from ICN (Costa Mesa, CA), anti-phospho-pRb (S780 and S807/811), anti-pRb, anti-p27, anti-p21, anti-INK4B, anti-phospho-Chk2 (T68), anti-Chk2, anti-p-H2A.X (S139), anti-H2A.X, anti-cyclin D3, and anti-CDK4 were from Cell Signaling Technology (Danvers, MA), anti-p73 from Lab Vision (Fremont, CA), anti-p53, anti-cyclin E, anti-cyclin A, anti-cyclin B1, anti-CDK2, and anti-CDK1 were from Santa Cruz Biotechnology (Santa Cruz, CA).
Protein analysis
Cells were washed once with cold phosphate-buffered saline (pH 7.0) and collected by scraping. Harvested cells were lysed by resuspending in lysis buffer (50 mM Tris-HCl (pH 7.5), 0.5 mM EDTA, 200 mM NaCl, and 0.5% Nonidet P-40 (NP-40)) supplemented with protease inhibitors and phosphatase inhibitors (EMD Chemicals, Gibbstown, NJ). Cell lysates were clarified by centrifugation at 13,000 rpm (4°C) for 15 min, and protein concentration was determined by the Bradford method (Bio-Rad, Hercules, CA). After adding equal volume of 2× SDS sample buffer (100 mM Tris-HCl (pH 6.8), 4% SDS, 20% glycerol, and 0.02% bromophenol blue containing 2.56 M β-mercaptoethanol), cell lysates were boiled for 5 min. Equal amounts of cell lysate were separated by 10% to 15% SDS-polyacrylamide gels, transferred to nitrocellulose, immunoblotted, and detected by chemiluminescence using ECL detection reagents.
Cell cycle analysis
SK-Br3 cells were treated with 0, 5, or 10 μM of quercetin for 2 or 4 d. Cells were trypsinized and centrifuged at 1000 rpm for 5 min. The supernatant was then removed before cells were resuspended in 0.2 ml of PBS and fixed with 1 ml of cold absolute ethanol. Fixed cells were stained using 20 μg/ml propidium iodide (Invitrogen) and 200 μg/ml RNaseA, before being assayed on a Coulter Epics XL flow cytometer (Beckman-Coulter).
Comet assay
Control and quercetin treated cells were imbedded into an agarose gel and the proteins and lipids were removed by exposing the gel to an alkaline NaOH solution. DNA fragments were electrically separated from the nucleus (comet head) and visualized by staining with propidium iodide. DNA damage was seen in the form of a comet tail under a fluorescence microscope. For each comet image, PI intensities of the head and tail portions were obtained, and the percentage intensity of the tail portion was multiplied by the size of the tail (DNA migration) to yield a tail moment.
Real-time RT-PCR
Total RNA was prepared using RNeasy-mini kit (Qiagen, Valencia, CA) including on-column DNase digestion to eliminate genomic DNA contamination. cDNA was generated from 2 μg of RNA using SuperScript III™ reverse transcriptase (Invitrogen). Real-time RT-PCR was carried out using Platinum®SYBR® Green (Invitrogen) with ABI 7300 Real time PCR system. All samples were duplicated (or triplicated). Primer sequences are as follows: CCNB1 forward, 5′-TCTGGATAATGGTGAATGGACA-3′; CCNB1 reverse, 5′-CGATGTGGCATACTTGTTCTTG-3′; 18S-rRNA forward, 5′-GTAACCCGTTGAACCCCATT-3′; 18S-rRNA reverse, 5′-CCATCCAATCGGTAGTAGCG-3′ [Menssen and Hermeking, 2002].
Chromatin immunoprecipitation assays
SK-Br3 cells were seeded into 100 mm tissue culture plates and then left for 24 h before being treated with quercetin. Cells were treated for 48 h, with new medium being added with fresh quercetin (10 μM) every 24 h. Chromatin immunoprecipitation (ChIP) assay was done with the ChIP assay kit (Millipore, Temecula, CA). Briefly, cells were cross-linked with 1% formaldehyde for 10 min at room temperature. The cross-linked chromatin was then extracted, diluted with SDS lysis buffer, and sheared by sonication. After preclearing with Protein-A Agarose/Salmon Sperm DNA, the chromatin was immunoprecipitated with anti-NF-YA or anti-immunoglobulin G (negative control) polyclonal antibody (Santa Cruz Biotechnology). The immunoprecipitates were pelleted by centrifugation and incubated at 65°C to reverse the protein-DNA cross-linking. The DNA was extracted by phenol/chloroform extraction and then precipitated by ethanol. Purified DNA was subjected to PCR with primers specific for a region (-319 to -4) in the CCNB1 promoter spanning two NF-Y binding sites. The sequences of the PCR primers used are as follows: F2 5′-AGAGGCAGACCACGTGAGA-3′, R1 5′- TTCCTCTTCACCAGGCAGCA-3′.
Results
A low dose of quercetin inhibited the proliferation of breast cancer cells
Initially, in order to know whether quercetin has biochemopreventive effects in a physiologically relevant dose, human breast carcinoma SK-Br3 cells were treated with a clinically achievable dose of fresh quercetin every 24 h. In this experimental condition, quercetin inhibited SK-Br3 cell proliferation in a dose dependent manner (Fig. 1A). Growth inhibitory activity of quercetin was confirmed in another breast cancer cell line, MDA-MB-453 (Fig. 1B). Interestingly, the proliferation of MCF-10A cells, which have the characteristics of normal breast epithelium [Soule et al. 1990], was not affected by 10 μM of quercetin (Fig. 1C). These results suggest that quercetin has a cancer cell-specific anti-proliferation effect.
Fig. 1. Quercetin inhibited the proliferation of breast cancer cells.
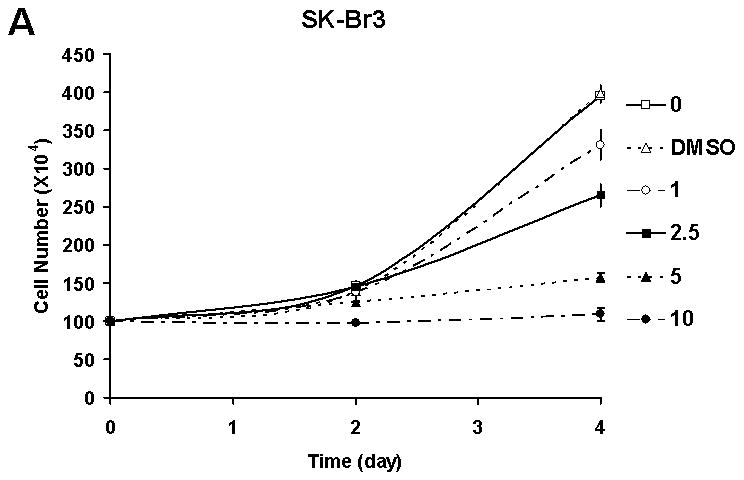
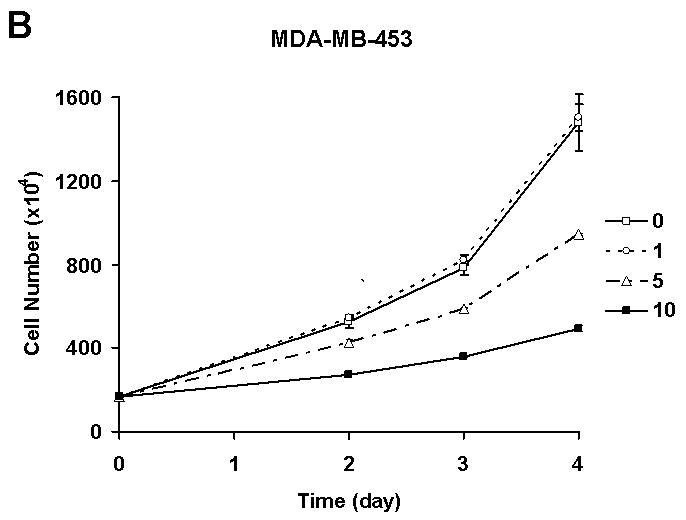
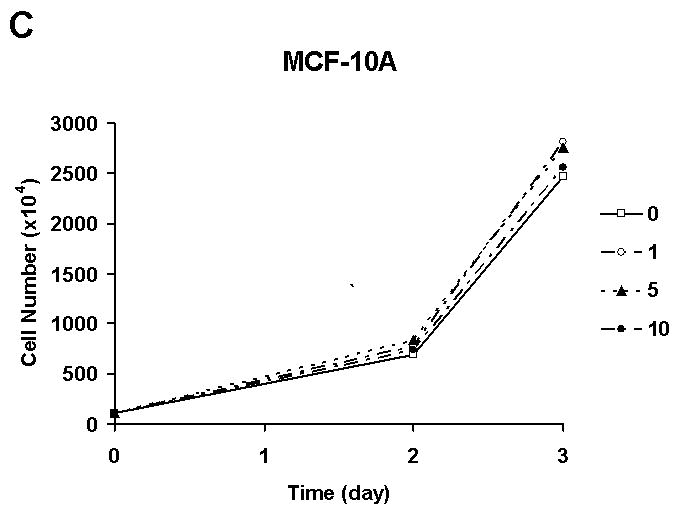
Breast cancer cell lines SK-Br3 (A) and MDA-MB-453 (B) were treated with an indicated amount, in μM, of quercetin every 24 h and cell proliferation was measured by counting the cells. C: The effect of quercetin on the proliferation of normal breast cell line MCF-10A was also analyzed and compared. The experiment was repeated three times and the average number of cell is shown with error bars (s.d.).
A low dose of quercetin showed a mild cytotoxic effect
Previous studies showed that quercetin is able to work as an anti-cancer agent by inducing apoptotic cell death [Yang et al., 2005; Lee et al., 2006]. In order to examine whether the anti-proliferation effect of quercetin is due to cell death, the viability of cells treated with quercetin was measured by tryphan blue exclusion assay (Fig. 2A). 94% of cells were viable even after 10 μM of quercetin was treated for 4 d. Coincident with this result, PARP protein cleavage, which is a representative marker of apoptotic cell death, was not detected (Fig. 2B). These results suggest that cytotoxic activity is not the main cause of the anti-proliferation effect of quercetin.
Fig. 2. A low dose of quercetin did not show significant cytotoxic effects.
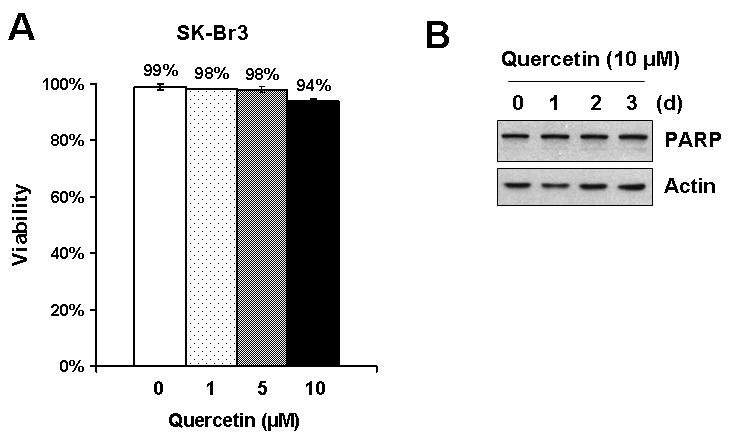
A: SK-Br3 cells were treated with an indicated amount of quercetin every 24 h for 4 d. Cell survival was determined by the tryphan blue exclusion assay. Error bars represent standard error from the mean (SEM) for three separate experiments. B: SK-Br3 cells were treated with 10 μM of quercetin and PARP status was analyzed as an indicator of apoptosis by Western blot analysis.
Quercetin induced cell cycle arrest in the G1 phase
From the finding that the low amount of quercetin used in this experimental condition did not induce cell death, we hypothesized that quercetin inhibits cell cycle progression. SK-Br3 cells were exposed to 0-10 μM of quercetin for 2 or 4 d. After the cells were fixed, cell cycle distribution was measured by measuring DNA content after staining with propidium iodide (PI). Figure 3 shows that SK-Br3 cells underwent a distinct increase in the G0/G1 population with a corresponding decrease in S phase. After 4 d of quercetin treatment, the G0/G1 population was increased from 61.8% to 70.7% (5 μM) or 75.9% (10 μM) and the S phase population was decreased from 30.2% to 21.4% (5 μM) or 18.2% (10 μM). The cell population in the G2/M phase was not changed significantly by quercetin treatment. The subG1 population increased to 6.4% and 9.2% after 2 and 4 d incubation with 10 μM of quercetin, which shows a similar range of cytotoxicity as the viability measurement. From these results, although a low dose of quercetin showed a mild cytotoxic effect, cell cycle arrest in the G1 phase is the main cause of anti-proliferation effect of quercetin.
Fig. 3. Cell cycle analysis by FACS.
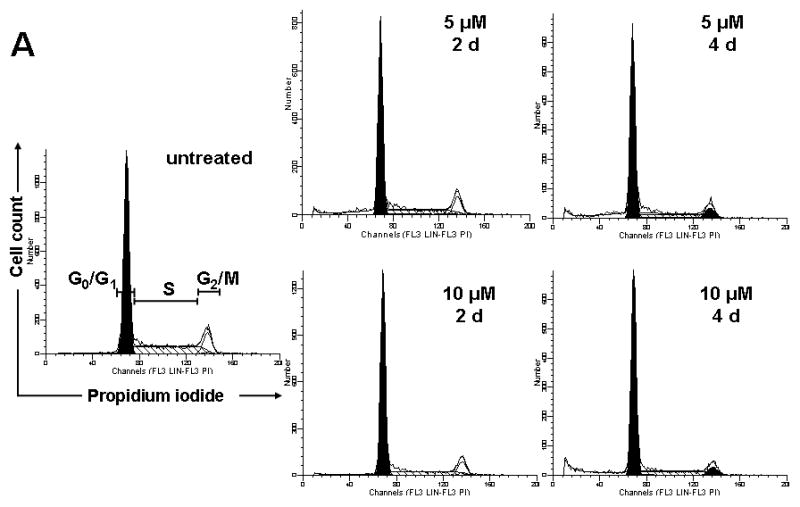
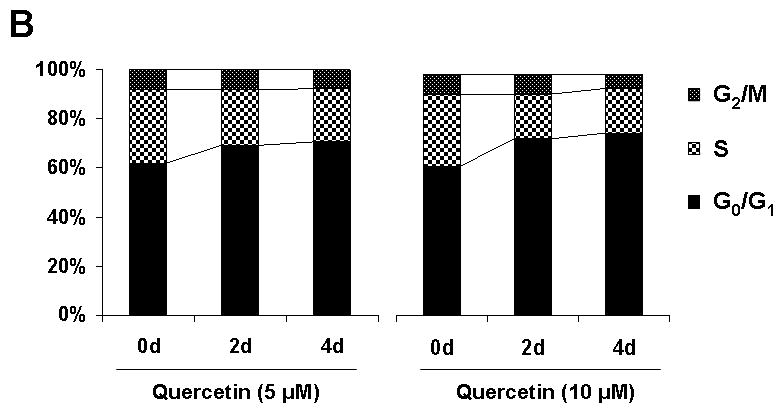
SK-Br3 cells were treated with 0, 5 or 10 μM of quercetin for 2 or 4 d. Cell cycle distribution was measured by measuring DNA content after staining with propidium iodide (PI) using flow cytometer. A: Representative histograms depicting cell cycle distribution are shown. B: Percent cells in G0/G1, S and G2/M phases are analyzed as a bar graph.
Quercetin induced cell cycle arrest in the G1 phase by hypo-phosphorylation of pRb
The retinoblastoma tumor suppressor protein, Rb, acts as a critical regulator for the G1 to S phase progression of the cell cycle by trapping E2F1, an essential transcriptional factor required for the expression of cell proliferation-associated genes. Hypophosphorylated Rb binds to and sequesters the transcription factor E2F1, resulting in cell cycle arrest at the G1 phase [Sherr, 1996]. So we checked the possibility that quercetin causes cell cycle arrest at the G1 phase through dephosphorylation of Rb. Quercetin decreased phosphorylation at Ser780 and Ser807/811 of Rb, which suggests that cell cycle progression from G1 to S phase is inhibited by quercetin (Fig. 4A). Eukaryotic cell cycle progression requires sequential activation of cyclin-dependent kinases (CDKs), which must be associated with corresponding regulatory cyclins in order to be activated [Schafer, 1998; Molinari, 2000]. Complexes formed by cyclin D and CDK4 or CDK6 and cyclin E and CDK2 phosphorylate Rb and enable the G1-S transition. To gain insight into the mechanism of hypo-phosphorylation of Rb, we first checked the protein level of these cyclins and CDKs. As shown in Fig. 4B and 4C, quercetin did not affect their expression. The CDK inhibitors (CKIs) play a crucial role in the regulation of the G1-S transition by binding to and inhibiting kinase activity of CDK/cyclin complexes [Molinari, 2000; Schafer, 1998]. Among the CDK inhibitors checked, p21 was increased dramatically by quercetin treatment (Fig. 4D). These results suggest that quercetin inhibited cyclin-CDK activity through the induction of p21, resulting in a decrease of phosphorylation of Rb. p21 induction by quercetin was confirmed in another breast cancer cell line, MDA-MB-453 (data not shown). Phosphorylation of Rb is also known to be negatively regulated by protein phosphatase 1α (PP1α). PP1α activity was checked by measuring its phosphorylation level, which showed that PP1α is dispensable for hypo-phosphorylation of Rb is (Fig. 4E). In contrast to the case of breast cancer SK-Br3 cells, neither hypo-phosphorylation nor p21 induction was observed in normal MCF-10A cells (Fig. 4F), which agrees with the cell proliferation result (Fig. 1C) and confirms the cancer cell-specific effects of quercetin.
Fig. 4. Quercetin inhibited cell cycle progression at the G1 phase by hypo-phosphorylation of pRb.
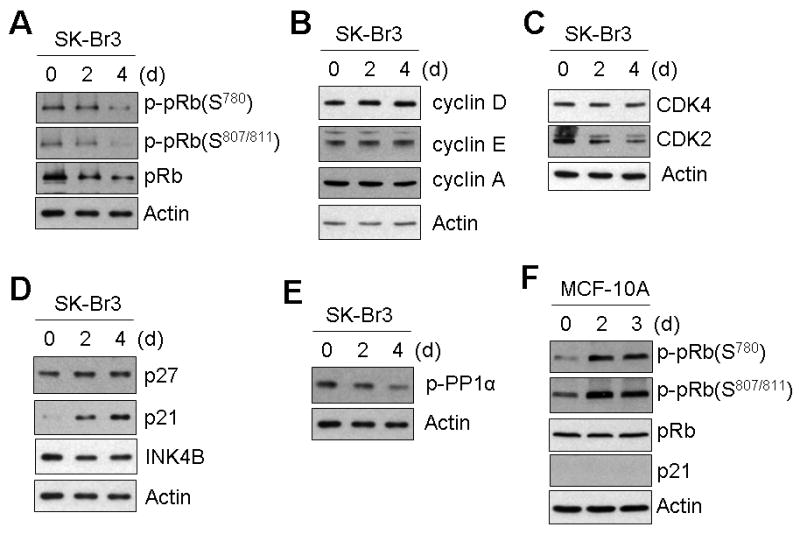
A: Quercetin (10 μM) was applied to SK-Br3 cells for an indicated time and harvested. Cell lysates containing equal amounts of protein (20 μg) were separated by SDS-PAGE and immunoblotted with anti-phospho-pRb or anti-pRb antibodies. In SK-Br3 cells, protein levels of cyclins (B) and CDKs (C) were compared by Western blot analysis. In SK-Br3 cells, the effect of quercetin treatment in the expression of CDK inhibitors (D) and phosphorylation of protein phosphatase 1α (PP1 α) (E) was examined by Western blot analysis. F: The phosphorylation of Rb and the expression level of p21, a CDK inhibitor, were examined in normal MCF-10A cells.
Chk2 played a crucial role in p21 induction by quercetin
The expression of p21 is known to be regulated by the tumor suppressor protein p53 [Taylor and Stark, 2001; Bode and Dong, 2004]. To address the question of whether quercetin-mediated induction of p21 protein expression was a p53-regulated response, p53 and p73, a p53 homologous protein, were checked by Western blot analysis. Both p53 and p73 proteins were not induced by quercetin treatment in SK-Br3 cells (Fig. 5A), leading to the conclusion that p21 induction by quercetin is mediated by a p53-independent pathway. A recent report shows that checkpoint kinase 2 (Chk2) mediates p21 induction through a p53-independent mechanism [Aliouat-Denis, 2005]. In order to check this possibility, we first checked whether a low dose of quercetin activates Chk2. Quercetin induced Chk2 phosphorylation at Thr68 in a time- and dose- dependent manner (Fig. 5B, C), which suggests that p21 induction may be mediated by activated Chk2. To confirm whether p21 induction is dependent on Chk2 activation, a Chk2 inhibitor was co-treated with quercetin. As shown in Fig. 5D, the Chk2 inhibitor significantly inhibited the induction of p21 by quercetin. From these results, we can conclude that quercetin induced the expression of p21 in a Chk2 dependent manner and caused hypo-phosphorylation of Rb, which led to inhibition of cell cycle progression at the G1 phase.
Fig. 5. The mechanism of p21 accumulation.
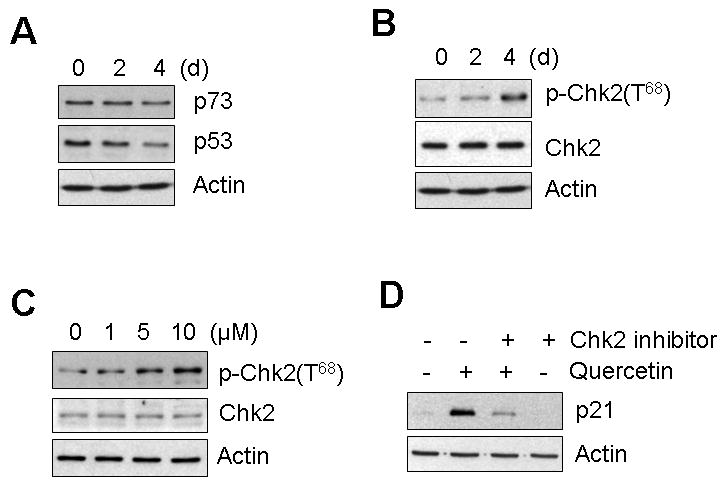
A: SK-Br3 cells were treated with 10 μM of quercetin for 2 and 4 d and then the protein level of the p53 family was examined by Western blot analysis. B: SK-Br3 cells were treated with 10 μM of quercetin for 2 and 4 d and then the phosphorylation of Thr68 of Chk2 kinase was examined by Western blot analysis. C: SK-Br3 cells were treated with an indicated amount of quercetin for 4 d. The phosphorylation of Thr68 of Chk2 kinase was examined by Western blot analysis. D: SK-Br3 cells were treated with quercetin (10 μM) and/or Chk2 inhibitor II (5 μM) for 4 d and the p21 level was examined by Western blot analysis.
A low dose of quercetin induced mild DNA damage
After DNA damage by ionizing radiation (IR), UV irradiation or hydroxyurea treatment, Chk2 becomes phosphorylated by ATM/ATR [Kastan and Lim, 2000; Matsuoka et al., 2000; Melchionna et al., 2000]. To gain insight into the mechanism of Chk2 activation by quercetin, we checked whether quercetin treatment causes DNA damage to cells. For quantitative analysis of DNA damage, alkaline comet assay was performed. As shown in Fig. 6A, DNA damage is seen in the form of a comet tail under a fluorescence microscope. For each comet image, PI intensities of the head and tail portions were obtained, and the percentage intensity of the tail portion was multiplied by the size of the tail (DNA migration) to yield a tail moment (Fig. 6B). Quercetin treatment (10 μM) for 1 h caused an increase in tail moments (greater than the value at the dotted line), indicating an increase in DNA damage, but the damage level was not as great as the damage exerted by 5 Gy of X-rays. DNA damage caused by ionizing radiation, UV-light, or radiomimetic agents results in rapid phosphorylation of the histone H2A family member H2A.X at Ser139 by ATM [Rogakou et al., 2000]. Within minutes following DNA damage, Ser139-phosphorylated H2A.X localizes to sites of DNA damage at subnuclear foci [Rogakou et al., 1999]. Coincident with the results of comet assay, histone H2A.X phosphorylation was increased by quercetin in a time- (Fig. 6C) and dose- (Fig. 6D) dependent manner. All these results indicate that quercetin induced mild DNA damage, which is the direct cause of Chk2 mediated p21 accumulation.
Fig. 6. Quercetin induced mild DNA damage.
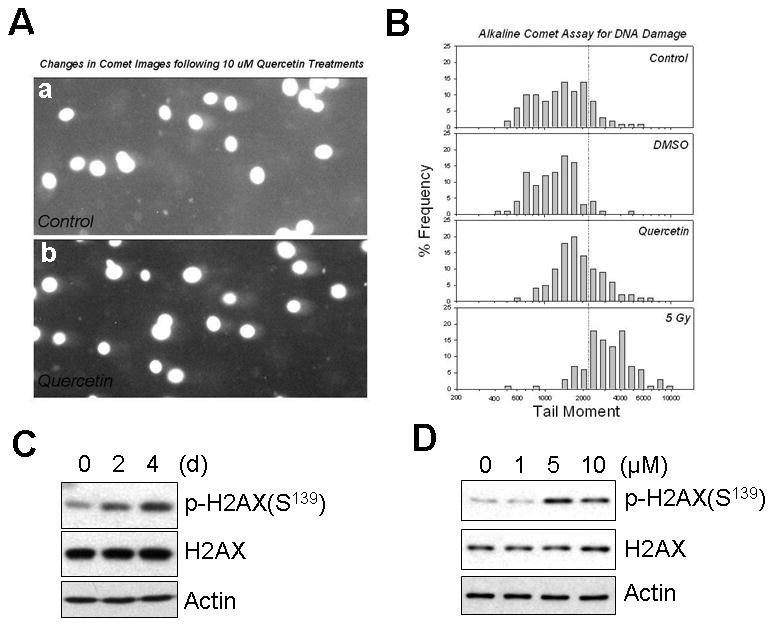
A: Alkaline Comet Images. Control (a) and quercetin treated cells (b) were embedded into an agarose gel and the proteins and lipids were removed by exposing the gel to an alkaline NaOH solution. DNA fragments were electrically separated from the nucleus (comet head) and visualized by staining with propidium iodide (PI). DNA damage is seen in the form of a comet tail under a fluorescence microscope. B: DNA Damage Quantification. For each comet image, PI intensities of the head and tail portions were obtained, and the percentage intensity of the tail portion was multiplied by the size of the tail (DNA migration) to yield a tail moment. Sample Size: Control (114), DMSO (67), Quercetin (125), and 5 Gy (72). SK-Br3 cells were treated with 10 μM of quercetin for 2 and 4 days (C) or with an indicated amount of quercetin for 4 d (D). The phosphorylation at Ser69 of histone H2A.X was examined by Western blot analysis.
Quercetin down-regulated cyclin B1 and CDK1 proteins
To address the question of whether the progression of cell cycles was affected by quercetin, protein levels of cyclin B and CDK1 in SK-Br3 cells were compared by Western blot analysis. Interestingly, these two proteins were downregulated by quercetin treatment (Fig. 7A), which suggests that G2-M phase cell cycle progression was also inhibited by quercetin. Downregulation of cyclin B1 and CDK1 was confirmed in another breast cancer cell line, MDA-MB-453 (Fig. 7B). Again this effect on cyclin B1 and CDK1 was cancer cell specific, and did not occur in normal MCF-10A cells (Fig. 7C).
Fig. 7. Quercetin inhibited cell cycle progression at the G2/M phase by cyclin B and CDK1 downregulation.
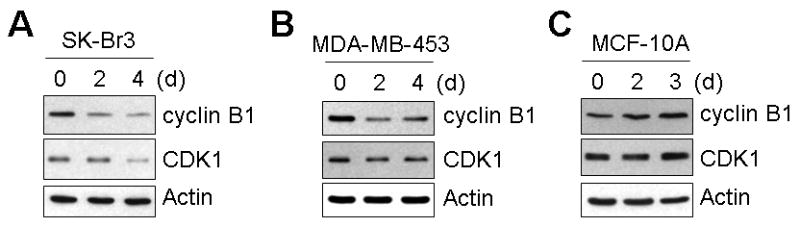
SK-Br3 (A), MDA-MB-453 (B) and MCF-10A (C) cells were treated with 10 μM of quercetin for an indicated time and then cyclin B1 and CDK1 protein levels were examined by Western blot analysis.
Cyclin B1 transcription was repressed by quercetin
Protein down-regulation can be mediated by decrease in transcription, inhibition of protein synthesis and/or increased protein degradation. Initially, mRNA level of the cyclin B1 gene (CCNB1) was analyzed using realtime RT-PCR. Transcription of the CCNB1 dramatically decreased by 50% after 2 d of 10 μM of quercetin treatment and was not recovered, suggesting that downregulation of cyclin B1 is mediated by transcriptional repression (Fig. 8A). To gain further insight into the mechanism of transcription repression of CCNB1 by quercetin, promoter structure was analyzed. In the promoter region of the CCNB1, there are two CCAAT motifs, which are binding sites for the transcription factor NF-Y (Fig. 8B). In order to know whether quercetin affects NF-Y binding on the CCNB1 promoter, chromatin immunoprecipitation (ChIP) assay was performed. SK-Br3 cells were untreated or treated with 10 μM of quercetin for 2d and then ChIP assay was performed by pulling-down with the antibody against NF-Y subunit A. The results clearly show that quercetin inhibited NF-Y binding on the CCNB1 promoter, which eventually led to transcriptional repression of cyclin B1 (Fig. 8C). We recently showed that quercetin can regulate gene expression epigenetically by decreasing the acetylation of histone H3; quercetin inhibits the binding of Sp1 to the survivin gene promoter and down-regulates survivin expression [Kim et al, 2008]. In order to check the possibility that epigenetic modification of the CCNB1 promoter region can inhibit the binding of NF-Y, ChIP assay was performed by pulling-down with the antibody against acetyl-histone H3 or H4 (Fig. 8D). In contrast to the regulation of survivin gene expression, quercetin did not change the histone acetylation in CCNB1 promoter region.
Fig. 8. The mechanism of cyclin B downregulation.
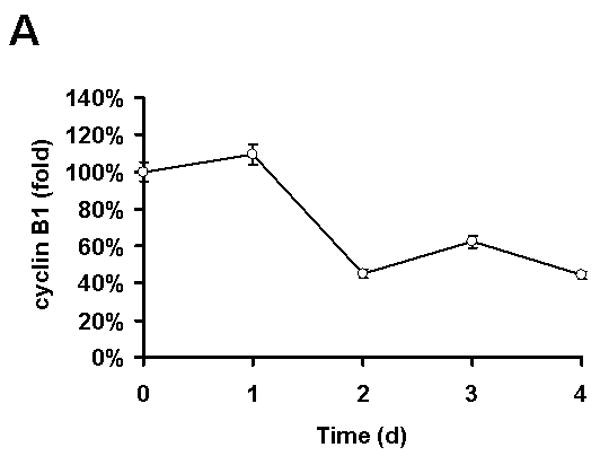
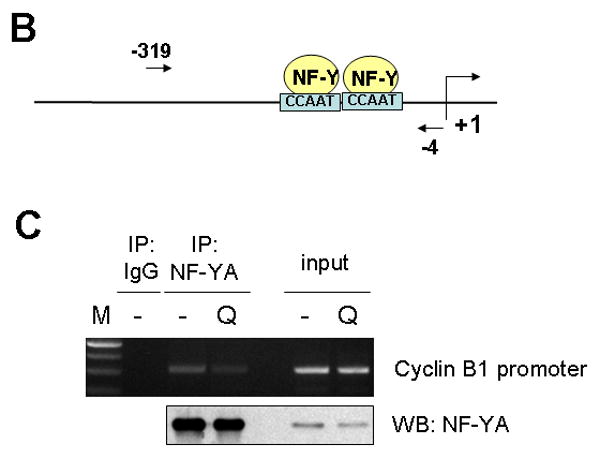
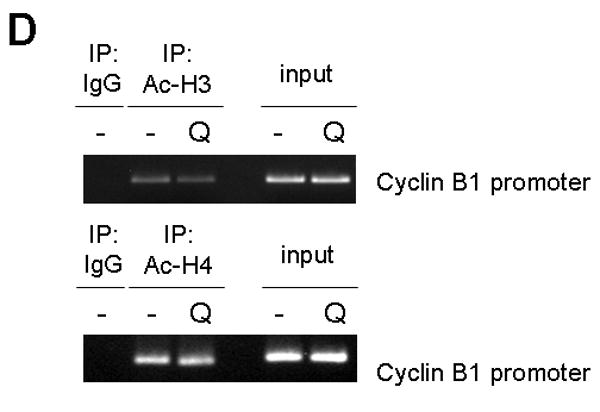
A: SK-Br3 cells were treated with 10 μM of quercetin for an indicated time and then RNA was extracted. Cyclin B1 mRNA level was measured by realtime RT-PCR assay. B: Diagram of cyclin B1 promoter region. Two NF-Y binding sites are indicated as boxes and the regions used for ChIP assay indicated as an arrow. C: SK-Br3 cells were untreated or treated with 10 μM of quercetin for 2d and then ChIP assay was performed by pulling-down with the antibody against NF-Y subunit A. The co-immunoprecipitated promoter DNA was amplified by PCR and is shown in the upper panel. Immunoprecipitated NF-Y subunit A was detected by Western blot analysis. D: ChIP assay was performed using anti-acetyl-Histone H3 and anti-acetyl-Histone H4 as in C.
Discussion
In this study, we demonstrated that physiologically relevant doses of quercetin inhibited the proliferation of cancer cells (Fig. 2A, B). Although a low dose of quercetin showed a small amount of cytotoxic activity (Fig. 2A), the anti-proliferation effect of quercetin mainly originated from the inhibition of cell cycle progression from G1 to S phase (Fig. 3A). This led to an examination of the molecular mechanisms of cell cycle regulation by quercetin. Quercetin induced mild DNA damage (Fig. 6) and subsequently activated Chk2 kinase (Fig. 5), which was the key regulator of p21 accumulation by quercetin. Increased p21 caused hypo-phosphorylation of Rb (Fig. 4A), which prevented the release of E2F1, an essential transcriptional factor required for the synthesis of many proteins involved in DNA synthesis [Sherr, 1996], and eventually led to G1 phase cell cycle arrest (Fig. 3). There are at least two possible mechanisms of quercetin-mediated DNA damage. One is that quercetin can induce DNA damage by inhibition of topo II activity. Previous report has shown that 10 of 20 bioflavonoids including quercetin induced in vivo DNA cleavage in the MLL BCR between nucleotides 6,800 and 7,000 by topo II inhibition similar to chemotherapeutic agents. The specificity of quercetin for topo II inhibition was directly confirmed by in vitro topo II catalytic and in nuclei topo II DNA cleavage experiments [Strick et al., 2000]. The other possibility is that DNA damage from quercetin occurs through reactive oxygen species (ROS) generation. This is a consequence of the peroxide activation of quercetin caused by peroxidase-catalyzed one-electron oxidation. Quercetin acts as an effective donor of electrons for scavenging reactive peroxyl radicals as indicated below.
| (Reaction 1) |
The Reactivity of quercetin radicals (Quercetin-O·) toward different biomolecules (Reaction 2) may be dependent upon redox states in cells.
| (Reaction 2) |
The resultant quercetin radicals enter the redox cycle to potentially increase ROS and subsequently cause oxidative DNA damage. Quercetin as a pro-oxidant was proven by showing that the semiquinone formed by lactoperoxidase/H2O2 in the presence of 0.25 M ZnCl2 (to stabilize the semiquinone) disappeared on addition of 1 mM GSH [Metodiewa et al., 1999]. A fundamental question, which remains unanswered, is the differential effect of quercetin on cancer cells in comparison with normal cells. Previous studies have shown that reduced amounts of antioxidant enzymes, especially MnSOD, are found in a variety of cancer cells [Oberley and Buettner, 1979]; also, lowered amounts of CuZnSOD have been found in many, but not all, tumors. MnSOD and CuZnSOD are essential primary enzymes that convert O2·- to H2O2 and O2 within the mitochondria and cytoplasm, respectively. These observations suggest a rationale as to why tumor cells are more sensitive than normal cells to quercetin-induced reactive oxygen species [Yu et al., 2007]. This possibility needs to be investigated in the future.
It is possible that peroxidase-catalyzed quercetin phenoxyl radicals oxidize protein SH groups, in particular redox-sensing molecules, directly or indirectly through ROS generation. Thioredoxin (TRX) and glutaredoxin (GRX), which are well known redox-sensing molecules, bind to apoptosis signal-regulating kinase 1 (ASK1) and suppress its activation [Saitoh et al., 1998; Song et al., 2002]. It is well known that both GRX and TRX contain two redox-active half-cystine residues, -Cys-Gly-Pro-Cys- or –Cys-Pro-Tyr-Cys-, in an active center [Holmgren, 1989; Padilla et al., 1995]. These molecules form an intramolecular disulfide bond between cysteine residues in the oxidizing environment. Formation of an intramolecular disulfide bond leads to a conformational change and subsequently results in dissociation of TRX and GRX from ASK1. TRX and GRX are negative regulators of ASK1; TRX or GRX binds to the N-terminal or C-terminal portion of ASK1, respectively [Saitoh et al., 1998; Song et al., 2002]. Their dissociation from ASK1 activates the ASK1-MEK-JNK/p38 MAPK signal transduction pathway [Saitoh et al., 1998; Song et al., 2002]. It is possible that these kinases affect some of the regulatory proteins of the cell cycle and cause the inhibition of cell cycle progression. Obviously, further studies are necessary to prove this possibility.
In addition, quercetin has down-regulated cyclin B1 protein (Fig. 7), which is essential in CDK1 kinase function and progression at the G2/M cell cycle phase. The synthesis of cyclin B1 was repressed at the transcriptional level (Fig. 8A). ChIP assay results suggested that the binding of a crucial transcriptional factor NF-Y to the CCNB1 promoter was inhibited by quercetin treatment (Fig. 8C). Recently, we showed that quercetin decreased the acetylation of histone H3 and down-regulated survivin expression by inhibiting the binding of Sp1 to the survivin gene promoter [Kim et al, 2008]. In contrast, quercetin did not regulate the CCNB1 transcription by reducing the histone acetylation (Fig. 8D). Another possibility is that quercetin regulates the CCNB1 gene expression through DNA methylation of the promoter region. A recent study showed that cell cycle-dependent transcription of cyclin B2 is influenced by DNA methylation [Tschöp and Engeland, 2007]. Computational analysis [Takai and Jones, 2003] of the CCNB1 promoter regions revealed one DNA methylation candidate region of CpG island (data not shown). From these findings, we speculate that quercetin induces DNA methylation in CCNB1 promoter and represses its transcription.
Acknowledgments
This study was supported by grants from the National Cancer Institute, National Institutes of Health (CA95191, CA96989, CA121395), DOD prostate program funds (PC020530, PC040833) and Susan G. Komen Breast Cancer Foundation fund (BCTR60306) (Y.J.L.).
Abbreviations
- CDK
cyclin-dependent kinase
- ChIP
Chromatin immunoprecipitation
- Chk2
checkpoint kinase 2
- CKI
CDK inhibitor
- DMEM
Dulbecco's modified Eagle's medium
- DTT
dithiothreitol
- HRP
horse radish peroxidase
- PBS
phosphate-buffered saline
- PI
propidium iodide
- PPIα
protein phosphatase 1α
- ROS
reactive oxygen species
References
- Aliouat-Denis CM, Dendouga N, Van den Wyngaert I, Goehlmann H, Steller U, van de Weyer I, Van Slycken N, Andries L, Kass S, Luyten W, Janicot M, Vialard JE. p53-independent regulation of p21Waf1/Cip1 expression and senescence by Chk2. Mol Cancer Res. 2005;3:627–34. doi: 10.1158/1541-7786.MCR-05-0121. [DOI] [PubMed] [Google Scholar]
- Beniston RG, Campo MS. Quercetin elevates p27(Kip1) and arrests both primary and HPV16 E6/E7 transformed human keratinocytes in G1. Oncogene. 2003;22:5504–14. doi: 10.1038/sj.onc.1206848. [DOI] [PubMed] [Google Scholar]
- Block G, Patterson B, Subar A. Fruit, vegetables, and cancer prevention: a review of the epidemiological evidence. Nutr Cancer. 1992;18:1–29. doi: 10.1080/01635589209514201. [DOI] [PubMed] [Google Scholar]
- Bors W, Heller W, Michel C, Saran M. Flavonoids as antioxidants: determination of radical-scavenging efficiencies. Methods Enzymol. 1990;186:343–55. doi: 10.1016/0076-6879(90)86128-i. [DOI] [PubMed] [Google Scholar]
- Bors W, Michel C, Saran M. Flavonoid antioxidants: rate constants for reactions with oxygen radicals. Methods Enzymol. 1994;234:420–9. doi: 10.1016/0076-6879(94)34112-5. [DOI] [PubMed] [Google Scholar]
- Burow ME, Weldon CB, Tang Y, Navar GL, Krajewski S, Reed JC, Hammond TG, Clejan S, Beckman BS. Differences in susceptibility to tumor necrosis factor alpha-induced apoptosis among MCF-7 breast cancer cell variants. Cancer Res. 1998;58:4940–6. [PubMed] [Google Scholar]
- Choi JA, Kim JY, Lee JY, Kang CM, Kwon HJ, Yoo YD, Kim TW, Lee YS, Lee SJ. Induction of cell cycle arrest and apoptosis in human breast cancer cells by quercetin. Int J Oncol. 2001;19:837–44. doi: 10.3892/ijo.19.4.837. [DOI] [PubMed] [Google Scholar]
- da Silva EL, Piskula MK, Yamamoto N, Moon JH, Terao J. Quercetin metabolites inhibit copper ion-induced lipid peroxidation in rat plasma. FEBS Lett. 1998;430:405–8. doi: 10.1016/s0014-5793(98)00709-1. [DOI] [PubMed] [Google Scholar]
- Di Carlo G, Mascolo N, Izzo AA, Capasso F. Flavonoids: old and new aspects of a class of natural therapeutic drugs. Life Sci. 1999;65:337–53. doi: 10.1016/s0024-3205(99)00120-4. [DOI] [PubMed] [Google Scholar]
- Gugler R, Leschik M, Dengler HJ. Disposition of quercetin in man after single oral and intravenous doses. Eur J Clin Pharmacol. 1975;9:229–34. doi: 10.1007/BF00614022. [DOI] [PubMed] [Google Scholar]
- Hertog MG, Hollman PC, Katan MB, Kromhout D. Intake of potentially anticarcinogenic flavonoids and their determinants in adults in The Netherlands. Nutr Cancer. 1993;20:21–9. doi: 10.1080/01635589309514267. [DOI] [PubMed] [Google Scholar]
- Hollman PC, van Trijp JM, Buysman MN, van der Gaag MS, Mengelers MJ, de Vries JH, Katan MB. Relative bioavailability of the antioxidant flavonoid quercetin from various foods in man. FEBS Lett. 1997;418:152–6. doi: 10.1016/s0014-5793(97)01367-7. [DOI] [PubMed] [Google Scholar]
- Holmgren A. Thioredoxin and glutaredoxin systems. J Biol Chem. 1989;264:13963–6. [PubMed] [Google Scholar]
- Kandaswami C, Middleton E., Jr Free radical scavenging and antioxidant activity of plant flavonoids. Adv Exp Med Biol. 1994;366:351–76. doi: 10.1007/978-1-4615-1833-4_25. [DOI] [PubMed] [Google Scholar]
- Kastan MB, Lim DS. The many substrates and functions of ATM. Nat Rev Mol Cell Biol. 2000;1:179–86. doi: 10.1038/35043058. [DOI] [PubMed] [Google Scholar]
- Kim YH, Lee DH, Jeong JH, Guo ZS, Lee YJ. Quercetin augments TRAIL-induced apoptotic death: involvement of the ERK signal transduction pathway. Biochem Pharmacol. 2008;75:1946–58. doi: 10.1016/j.bcp.2008.02.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuhnau J. The flavonoids. A class of semi-essential food components: their role in human nutrition. World Rev Nutr Diet. 1976;24:117–91. [PubMed] [Google Scholar]
- Lea MA, Xiao Q, Sadhukhan AK, Cottle S, Wang ZY, Yang CS. Inhibitory effects of tea extracts and (-)-epigallocatechin gallate on DNA synthesis and proliferation of hepatoma and erythroleukemia cells. Cancer Lett. 1993;68:231–6. doi: 10.1016/0304-3835(93)90151-x. [DOI] [PubMed] [Google Scholar]
- Lee TJ, Kim OH, Kim YH, Lim JH, Kim S, Park JW, Kwon TK. Quercetin arrests G2/M phase and induces caspase-dependent cell death in U937 cells. Cancer Lett. 2006;240:234–42. doi: 10.1016/j.canlet.2005.09.013. [DOI] [PubMed] [Google Scholar]
- Matsuoka S, Rotman G, Ogawa A, Shiloh Y, Tamai K, Elledge SJ. Ataxia telangiectasia-mutated phosphorylates Chk2 in vivo and in vitro. Proc Natl Acad Sci U S A. 2000;97:10389–94. doi: 10.1073/pnas.190030497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Melchionna R, Chen XB, Blasina A, McGowan CH. Threonine 68 is required for radiation-induced phosphorylation and activation of Cds1. Nat Cell Biol. 2000;2:762–5. doi: 10.1038/35036406. [DOI] [PubMed] [Google Scholar]
- Menssen A, Hermeking H. Characterization of the c-MYC-regulated transcriptome by SAGE: identification and analysis of c-MYC target genes. Proc Natl Acad Sci U S A. 2002;99:6274–9. doi: 10.1073/pnas.082005599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Metodiewa D, Jaiswal AK, Cenas N, Dickancaite E, Segura-Aguilar J. Quercetin may act as a cytotoxic prooxidant after its metabolic activation to semiquinone and quinoidal product. Free Radic Biol Med. 1999;26:107–16. doi: 10.1016/s0891-5849(98)00167-1. [DOI] [PubMed] [Google Scholar]
- Molinari M. Cell cycle checkpoints and their inactivation in human cancer. Cell Prolif. 2000;33:261–74. doi: 10.1046/j.1365-2184.2000.00191.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nielsen SE, Kall M, Justesen U, Schou A, Dragsted LO. Human absorption and excretion of flavonoids after broccoli consumption. Cancer Lett. 1997;114:173–4. doi: 10.1016/s0304-3835(97)04654-5. [DOI] [PubMed] [Google Scholar]
- Oberley LW, Buettner GR. Role of superoxide dismutase in cancer: a review. Cancer Res. 1979;39:1141–9. [PubMed] [Google Scholar]
- Ong CS, Tran E, Nguyen TT, Ong CK, Lee SK, Lee JJ, Ng CP, Leong C, Huynh H. Quercetin-induced growth inhibition and cell death in nasopharyngeal carcinoma cells are associated with increase in Bad and hypophosphorylated retinoblastoma expressions. Oncol Rep. 2004;11:727–33. [PubMed] [Google Scholar]
- Padilla CA, Martinez-Galiseo E, Barcena JA, Spyrou G, Holmgren A. Purification from placenta, amino acid sequence, structure comparisons and cDNA cloning of human glutaredoxin. Eur J Biochem. 1995;227:27–34. doi: 10.1111/j.1432-1033.1995.tb20356.x. [DOI] [PubMed] [Google Scholar]
- Plaumann B, Fritsche M, Rimpler H, Brandner G, Hess RD. Flavonoids activate wild-type p53. Oncogene. 1996;13:1605–14. [PubMed] [Google Scholar]
- Rogakou EP, Boon C, Redon C, Bonner WM. Megabase chromatin domains involved in DNA double-strand breaks in vivo. J Cell Biol. 1999;146:905–16. doi: 10.1083/jcb.146.5.905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rogakou EP, Nieves-Neira W, Boon C, Pommier Y, Bonner WM. Initiation of DNA fragmentation during apoptosis induces phosphorylation of H2A.X histone at serine 139. J Biol Chem. 2000;275:9390–5. doi: 10.1074/jbc.275.13.9390. [DOI] [PubMed] [Google Scholar]
- Saitoh M, Nishitoh H, Fujii M, Takeda K, Tobiume K, Sawada Y, Kawabata M, Miyazono K, Ichijo H. Mammalian thioredoxin is a direct inhibitor of apoptosis signal-regulating kinase (ASK) 1. EMBO J. 1998;17:2596–606. doi: 10.1093/emboj/17.9.2596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schafer KA. The cell cycle: a review. Vet Pathol. 1998;35:461–78. doi: 10.1177/030098589803500601. [DOI] [PubMed] [Google Scholar]
- Sherr CJ. Cancer cell cycles. Science. 1996;274:1672–7. doi: 10.1126/science.274.5293.1672. [DOI] [PubMed] [Google Scholar]
- Song JJ, Rhee JG, Suntharalingam M, Walsh SA, Spitz DR, Lee YJ. Role of glutaredoxin in metabolic oxidative stress. Glutaredoxin as a sensor of oxidative stress mediated by H2O2. J Biol Chem. 2002;277:46566–75. doi: 10.1074/jbc.M206826200. [DOI] [PubMed] [Google Scholar]
- Soule HD, Maloney TM, Wolman SR, Peterson WD, Jr, Brenz R, McGrath CM, Russo J, Pauley RJ, Jones RF, Brooks SC. Isolation and characterization of a spontaneously immortalized human breast epithelial cell line, MCF-10. Cancer Res. 1990;50:6075–86. [PubMed] [Google Scholar]
- Strick R, Strissel PL, Borgers S, Smith SL, Rowley JD. Dietary bioflavonoids induce cleavage in the MLL gene and may contribute to infant leukemia. Proc Natl Acad Sci U S A. 2000;97:4790–5. doi: 10.1073/pnas.070061297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takai D, Jones PA. The CpG island searcher: a new WWW resource. Silico Biol. 2003;3:235–40. [PubMed] [Google Scholar]
- Taylor WR, Stark GR. Regulation of the G2/M transition by p53. Oncogene. 2001;20:1803–15. doi: 10.1038/sj.onc.1204252. [DOI] [PubMed] [Google Scholar]
- Tschop K, Engeland K. Cell cycle-dependent transcription of cyclin B2 is influenced by DNA methylation but is independent of methylation in the CDE and CHR elements. Febs J. 2007;274:5235–49. doi: 10.1111/j.1742-4658.2007.06045.x. [DOI] [PubMed] [Google Scholar]
- Vulcain E, Goupy P, Caris-Veyrat C, Dangles O. Inhibition of the metmyoglobin-induced peroxidation of linoleic acid by dietary antioxidants: Action in the aqueous vs. lipid phase. Free Radic Res. 2005;39:547–63. doi: 10.1080/10715760500073865. [DOI] [PubMed] [Google Scholar]
- Wenzel U, Kuntz S, Brendel MD, Daniel H. Dietary flavone is a potent apoptosis inducer in human colon carcinoma cells. Cancer Res. 2000;60:3823–31. [PubMed] [Google Scholar]
- Yang JH, Hsia TC, Kuo HM, Chao PD, Chou CC, Wei YH, Chung JG. Inhibition of lung cancer cell growth by quercetin glucuronides via G2/M arrest and induction of apoptosis. Drug Metab Dispos. 2006;34:296–304. doi: 10.1124/dmd.105.005280. [DOI] [PubMed] [Google Scholar]
- Yoshizumi M, Tsuchiya K, Kirima K, Kyaw M, Suzaki Y, Tamaki T. Quercetin inhibits Shc- and phosphatidylinositol 3-kinase-mediated c-Jun N-terminal kinase activation by angiotensin II in cultured rat aortic smooth muscle cells. Mol Pharmacol. 2001;60:656–65. [PubMed] [Google Scholar]
- Yu JQ, Liu HB, Tian DZ, Liu YW, Lei JC, Zou GL. Changes in mitochondrial membrane potential and reactive oxygen species during wogonin-induced cell death in human hepatoma cells. Hepatol Res. 2007;37:68–76. doi: 10.1111/j.1872-034X.2007.00003.x. [DOI] [PubMed] [Google Scholar]