

Abstract
Apelin is the peptidic ligand for the G-protein-coupled receptor APJ. The apelin−APJ system is important in cardiovascular regulation, fluid homeostasis, and angiogenesis, among other roles. In this study, we investigate interactions between apelin and membrane-mimetic micelles of the detergents sodium dodecyl sulfate (SDS), dodecylphosphocholine (DPC), and 1-palmitoyl-2-hydroxy-sn-glycero-3-[phospho-rac-(1-glycerol)] (LPPG). Far-ultraviolet circular dichroism spectropolarimetry and diffusion-ordered spectroscopy indicate that apelin peptides bind to micelles of the anionic detergents SDS and LPPG much more favorably than to zwitterionic DPC micelles. Nuclear magnetic resonance spectroscopy allowed full characterization of the interactions of apelin-17 with SDS micelles. Titration with paramagnetic agents and structural determination of apelin-17 in SDS indicate that R6−K12 is highly structured, with R6−L9 directly interacting with headgroups of the micelle. Type I β-turns are initiated between R6 and L9, and a well-defined type IV β-turn is initiated at S10. Furthermore, binding of apelin-17 to SDS micelles causes structuring of M15-F17, with no evidence for direct binding of this region to the micelles. These results are placed into the context of the membrane catalysis hypothesis for peptide−receptor binding, and a hypothetical mechanism of APJ binding and activation by apelin is advanced.
Introduction
The apelin peptides activate the class A rhodopsin-like G-protein-coupled receptor (GPCR) named APJ.(1) Apelin−APJ signaling has been demonstrated within the cardiovascular system, central nervous system, and adipoinsular axis (as previously reviewed(2)), among other physiological settings. Apelin is an extremely potent inotropic agent, increasing muscle contractility with a half-maximal effective concentration of ∼33 pmol/L in rat heart.(3) Upregulation of apelin is observed in conjunction with an elevated body mass index,(4) and plasma levels of apelin are increased during heart failure.(5) Furthermore, apelin inhibits human immunodeficiency virus fusion to host cells,(6) and apelin has been shown to induce angiogenesis during tumor formation.7,8 As a whole, these functions have made the apelin−APJ system popular as a proposed therapeutic target.2,9,10
Apelin is produced as a 77 amino acid preproprotein that is cleaved into a variety of bioactive forms from 13 to 36 residues in length, each of which retain the C-terminus of the precursor.(11) The 12 C-terminal residues of apelin constitute the core region essential for apelin function.12−14 Our recent NMR studies on apelin-17 suggest that this core region is only moderately structured in solution at physiological temperature, but that binding to APJ may lead to more defined structuring.(15)
Sargent and Schwyzer(16) first formulated the “membrane catalysis” hypothesis, stating that a small peptide first interacts with the target cell plasma membrane before binding to and activating its target receptor. The membrane is proposed to act as a receptor−ligand catalyst by increasing the local concentration of the ligand, improving the probability of diffusional collision between the receptor and ligand, and/or by inducing a conformational change in the peptide to increase the receptor-binding energetics. This is supported by the fact that many peptide hormones bind to membranes or micelles17−21 and in some cases have been shown to adopt a specific structure when bound to the membrane.22−24 A striking example is calcitonin, a peptide hormone involved in calcium homeostasis.(22) In the presence of sodium dodecyl sulfate (SDS) micelles, calcitonin assumes an α-helical structure while being unstructured in solution. Furthermore, deletion of the critical F16 residue disrupts in parallel both the induction of the α-helical structure and the function of calcitonin.
Results of this nature, in the context of the membrane catalysis model, provide strong incentive to investigate the potential interactions of apelin with lipids. Herein, we use circular dichroism (CD) spectropolarimetry and pulsed field gradient NMR diffusion measurements to probe interactions of apelin-17 and apelin-12 with SDS micelles, dodecylphosphocholine (DPC) micelles, and 1-palmitoyl-2-hydroxy-sn-glycero-3-[phospho-rac-(1-glycerol)] (LPPG) micelles. An NMR spectroscopy based structure of apelin-17 bound to SDS micelles is also presented, and paramagnetic titration gives an overview of the topology of the apelin-17/SDS micelle complex. Finally, the significance of these findings in relation to the membrane catalysis model is discussed.
Materials and Methods
Materials
LPPG was purchased from Avanti Polar lipids (Alabaster, AL). Deuterium oxide (D2O; 99.8 atom % D), SDS-d25, DPC, DPC-d38, and D2O containing 1% (w/w) sodium 2,2-dimethyl-2-silapentane-5-sulfonate (DSS) were obtained from C/D/N Isotopes (Pointe-Claire, QC, Canada). Fmoc-protected amino acids and coupling reagents were obtained from AAPPTec (Louisville, KY). All other chemicals were obtained at biotechnology, high-performance liquid chromatography, or reagent grade, as appropriate, from Sigma-Aldrich (Oakville, ON, Canada).
Peptide Synthesis and Purification
Apelin-17 (H2N-KFRRQRPRLSHKGPMPF-COO−) and apelin-12 (H2N-RPRLSHKGPMPF-COO−) were produced and purified in multimilligram quantities using previous procedures.(15) Peptide identities were confirmed using matrix-assisted laser desorption ionization mass spectrometry and quantitative amino acid analysis (Hospital for Sick Children, Toronto, ON, Canada).
CD Spectropolarimetry
Far-ultraviolet CD spectra of apelin-17 and apelin-12 were recorded at 35 °C using a Jasco J-810 spectropolarimeter (Easton, MD). Solutions of apelin peptides (apelin-12, 55.2 ± 0.7 μM; apelin-17, 64.5 ± 5.2 μM; exact concentration determined by quantitative amino acid analysis; Hospital for Sick Children) were prepared from a single stock solution. For each apelin peptide, samples were prepared with no lipid added and 16 mM SDS, 19 mM DPC, or 38 mM LPPG in 20 mM sodium phosphate adjusted to pH 7.00 ± 0.05. Spectra were acquired from 260 nm downward (1 nm steps), with reliable ellipticity values observed at >190 nm based on spectropolarimeter photomultiplier tube voltages, in 0.5 mm path length quartz cuvettes (Hellma, Müllheim, Germany). All measurements were collected in triplicate. Machine data were converted to the mean residue ellipticity, [θ], averaged over all trials, blank subtracted, and subjected to sliding-window averaging over 3 nm stretches so that [θ] reported at a given wavelength (λ) is
Finally, each spectrum was baseline adjusted by subtracting the average ellipticity value over 250−260 nm.
Paramagnetic Relaxation Measurements
Three identical NMR samples were prepared with 1 mM apelin-17 in a 90% H2O/10% D2O mixture with 20 mM Na+CD3COO−, 1 mM NaN3, 90 mM SDS, and 1 mM DSS. Each sample was titrated with MnCl2, 5-doxylstearic acid (5-DSA), or 16-doxylstearic acid (16-DSA) up to a maximum of 8 mM paramagnetic agent at 35 °C. Titrations were halted when a ∼50% reduction in the intensities of ∼50% of the peaks was observed using 1H−1H total correlation spectroscopy (TOCSY) relative to a reference spectrum. For each residue, the intensities of one or two nonoverlapped cross-peaks were measured (depending on the availability of nonoverlapped peaks). Titration of 16-DSA was at 800 MHz using a Varian INOVA spectrometer (Palo Alto, CA) at the National High Field Nuclear Magnetic Resonance Centre (NANUC; Edmonton, AB, Canada), while both Mn2+ and 5-DSA were titrated at 700 MHz on a Bruker Avance III spectrometer (Milton, ON, Canada) at the National Research Council Institute for Marine Biosciences (NRC-IMB; Halifax, NS, Canada). Both spectrometers are equipped with cryogenically cooled triple-resonance probes. NMR data were processed using NMRPipe(25) and peak intensities measured with a Gaussian line shape in Sparky 3 (T. D. Goddard and D. G. Kneller, University of California, San Francisco).
Diffusion-Ordered Spectroscopy
Four samples of apelin-17 were prepared for DOSY experiments with 1 mM apelin-17, 20 mM Na+CD3COO−, 1 mM NaN3, and 1 mM DSS in 90% H2O/10% D2O, with three samples containing 81 mM SDS-d25, 94 mM DPC-d25, or 187 mM LPPG. Identical samples were also prepared without apelin. Detergent concentrations were chosen to give ∼1.25:1 micelle:peptide ratios. Sample pH values were adjusted to 5.00 ± 0.05 using DCl and NaOD. DOSY spectra were acquired at 35 °C using a stimulated echo sequence with bipolar gradients(26) and a longitudinal eddy current decay of 5 ms at 500 MHz on a Bruker Avance II spectrometer with a TXI probe at the Nuclear Magnetic Resonance Research Resource (NMR3; Halifax, NS). Susceptibility-matched tubes (Shegemi, Allison Park, PA) were used to minimize diffusion artifacts due to gradient nonlinearity or convection currents. Signal attenuation at the maximum gradient strength was adjusted to be 95%, with 32 increments collected in the diffusion dimension with the gradient strength ranging from 1.7 to 32.0 G/cm. DOSY experiments were performed in triplicate.
Diffusion coefficients for apelin-17 in each condition alongside those of each micelle in the absence of apelin were obtained using the relaxation module of Topspin 2.1 (Bruker). Topspin uses an iterative procedure to fit the equation
to the data, where I and I0 are the peak intensities when the experiment is performed with and without a gradient, respectively, D is the translational diffusion coefficient, γ is the gyromagnetic ratio, and δ and Δ are the gradient duration and diffusion time, respectively. Due to what appeared to be a consistent artifact, the first (lowest gradient strength) increment of each experiment was discarded, and 31 diffusion time points were used for fitting. For apelin-17 and each micelle, the translational diffusion coefficient (D) was calculated using three separate 1H NMR peaks for each spectral data set (i.e., nine measurements per condition), and the values were averaged to give a final value of D with associated standard error.
To determine the fraction of apelin-17 bound to each micelle, a fast two-site exchange model(27) was used:
where Dobsd is the observed diffusion coefficient of apelin-17 in the presence of micelles, fb and Db are the fraction of apelin-17 bound to micelles and the diffusion coefficient of the micelles, respectively, and Df is the translational diffusion coefficient of free apelin. We made the assumption that the diffusion coefficient of a micelle, Db, does not differ significantly with the presence or absence of apelin-17. The viscosity of each solution was not measured. Instead, the expected effect of the presence of micelles on the value of Df was estimated via the following expression for small molecules obstructed by spherical particles:(28)
where D0f is the measured diffusion coefficient of apelin-17 in water and ϕ is the volume fraction of lipids in the sample, which can be estimated by using the weight fraction.(28) Using eqs 3 and 4 and the values of D obtained using Topspin, the fraction of apelin-17 bound to each micelle was estimated.
Sequential Assignment of Apelin-17 in SDS and DPC Micelles
Two 600 μL NMR samples were made with 20 mM Na+CD3COO−, 1 mM NaN3, 1 mM DSS, 0.5 mM apelin-17, and either 81 mM SDS-d25 or 94 mM DPC-d38. 1H−1H TOCSY (80 ms mixing time), 1H−1H nuclear Overhauser effect spectroscopy (NOESY; 350 ms mixing time), gradient-enhanced natural abundance 1H−13C heteronuclear single-quantum coherence (HSQC), and 1H−15N HSQC experiments were performed at 700 MHz (NRC-IMB Avance III) with a TCI cryoprobe at 35 °C. NMR data were processed using NMRPipe(25) and manually assigned in Sparky 3 (T. D. Goddard and D. G. Kneller, University of California, San Francisco). Secondary chemical shifts, Δδ = δ(obsd) − δ(random coil),29−31 of the Hα, Hβ, Cα, and Cβ nuclei were calculated using the random coil chemical shifts of Wishart and co-workers.(32)
Structural Calculations of Apelin-17 Bound to SDS Micelles
XPLOR-NIH version 2.18(33) was used for final structure determination of apelin-17 in the presence of SDS micelles using the methods of NOE intensity calculation and refinement detailed by Langelaan and co-workers.(15) An ensemble of 200 structures was generated, with the 80 lowest energy structures retained as the final ensemble. The LSQKAB software of the CCP4 suite(34) was used to iteratively superpose the ensemble over 2−17-residue stretches while an in-house tcl/tk script was used to calculate the root mean squared deviation (rmsd) of the backbone atoms within the superposed region (using a previously developed iterative superposition protocol35,36). Lastly, the ensemble was analyzed (i) using Procheck-NMR to determine the proportion of residues in favored, allowed, generously allowed, and disallowed regions of the Ramachandran plot,(37) (ii) with Promotif-NMR to determine the number, type, and position of β-turns present in the ensemble,(38) and (iii) using an in-house tcl/Tk script to calculate both the deviation and order parameter (S, as defined by Hyberts et al.(39)) of the ϕ and ψ dihedral angles. All chemical shift data for apelin-17 with SDS and DPC micelles as well as the final structural ensemble for apelin-17 with SDS micelles have been deposited to the Biological Magnetic Resonance Bank(40) using SMSDep (accession numbers 20082 and 16275 for apelin-17 with SDS and DPC micelles, respectively).
Results and Discussion
CD Spectropolarimetry of Apelin-12 and Apelin-17
The far-ultraviolet (far-UV) CD spectra from 260 to 190 nm of both apelin-12 and apelin-17 in buffer and in the presence of SDS, DPC, or LPPG micelles are shown in Figure 1. For any given condition, the CD spectra for both apelin-12 and apelin-17 have similar features. As we have previously described,(15) the spectra for both apelin-12 and apelin-17 in buffer at 35 °C are random coil(41) in nature. This primarily random coil spectrum is convoluted with positive bands at 195 and 218 nm, most likely attributable to the 1B and 1La transitions42,43 of the C-terminal phenylalanine.(15) For both apelin-12 and apelin-17, DPC micelles do not strongly perturb the CD spectrum relative to the buffer (Figure 1). In contrast, dramatic spectral changes occur with both apelin isoforms in the presence of either SDS or LPPG micelles. The CD spectra with these micelles present are convoluted by an α-helix-like band pattern (difference spectra in Figure 1), which may be attributed either to formation of the α-helical secondary structure(41) or to the formation of β-turns.44,45
Figure 1.

Far-ultraviolet circular dichroism spectra of (A) apelin-12 and (B) apelin-17 in buffer, SDS micelles, DPC micelles, and LPPG micelles alongside the difference spectrum between LPPG and the buffer. Spectropolarimetry (sliding-window (eq 1) averaged blank-subtracted averages of three replicates) was performed at 35 °C in 20 mM phosphate buffer at pH 7.00 ± 0.05.
Although the main trends observed in the CD spectra of apelin-12 and apelin-17 are identical, there are a few minor differences between isoforms. In the presence of DPC micelles, a slightly larger deviation from buffer conditions is observed for apelin-12 than for apelin-17. Also, the CD spectra of apelin-17 in the presence of LPPG and SDS are nearly identical, while for apelin-12 the CD spectrum in LPPG shows a larger deviation from buffer conditions in comparison to that in SDS (Figure 1). Despite these slight differences, the underlying trends in the CD spectra of apelin-12 and apelin-17 are the same, with DPC micelles causing little to no change in the spectra relative to the buffer versus SDS and LPPG, which produce increasing amounts of perturbation of the CD spectrum relative to the buffer. A similar trend was observed with DPC and SDS micelles for apelin-13, pyroglutamate−apelin-13, and apelin-36 (Bebbington and Rainey, unpublished), suggesting that all of these apelin peptides undergo similar structural changes in the presence of anionic micelles.
Diffusion Coefficient Determination
Translational diffusion coefficients (D) were measured for SDS, DPC, and LPPG micelles, for apelin-17 in buffer, and for apelin-17 in the presence of each micelle (Table 1). Reported literature diffusion values (DOSY determined) are 1.2 × 10−10 m2/s for DPC micelles,(46) 9.5 × 10−11 m2/s for SDS micelles,(47) and 6.62 × 10−11 m2/s for LPPG micelles.(48) Once adjusted for temperature and viscosity using literature viscosity values(49) to match our experimental conditions, these measurements become 1.35 × 10−10, 1.47 × 10−10, and 8.49 × 10−11 m2/s, respectively. Although our reported D values are slightly higher, the agreement with previous D values is reasonable. Discrepancies may be due to effects of the different temperature and buffer conditions in our study upon the aggregation number of the micelles. In particular, the aggregation number of the micelles decreases with increasing temperature,(50) and our study was at 35 °C, while the reported values of D for SDS and LPPG were at 25 °C and that for DPC was at 30 °C. Also, the observed D for a micelle is often an overestimate due to contributions from monomeric detergent remaining in solution. Under buffer conditions, however, the critical micelle concentrations of the detergents are lowered, and for each micelle employed herein, the contribution of monomeric detergent to D was minor since only ∼5%, ∼1%, and ∼0.03% of SDS, DPC, and LPPG are estimated to be present as monomers (based upon critical micelle concentration values of ∼4 mM,(51) 1.1 mM,(52) and 0.06 mM(52) for SDS, DPC, and LPPG, respectively).
Table 1. Fraction of Apelin-17 Bound (fb) to the Indicated Micelle Types as Determined through Diffusion-Ordered NMR Spectroscopy Methodsa.
| component | Δ (ms) | δ (ms) | Dobsd (m2/s) | fb |
|---|---|---|---|---|
| SDS micelles | 60 | 6.4 | 1.7 × 10−10 ± 1.6 × 10−12 | N/A |
| DPC micelles | 60 | 6.4 | 1.8 × 10−10 ± 1.6 × 10−12 | N/A |
| LPPG micelles | 85 | 7.0 | 9.6 × 10−11 ± 2.8 × 10−13 | N/A |
| apelin-17 | 55 | 4.4 | 3.6 × 10−10 ± 3.4 × 10−12 | N/A |
| apelin-17 with SDS micelles | 65 | 5.2 | 2.0 × 10−10 ± 1.2 × 10−11 | 82 ± 5% |
| apelin-17 with DPC micelles | 60 | 5.2 | 2.8 × 10−10 ± 3.4 × 10−12 | 46 ± 5% |
| apelin-17 with LPPG micelles | 70 | 6.6 | 1.0 × 10−10 ± 1.2 × 10−12 | 98 ± 5% |
Topspin (Bruker) was used to determine the diffusion coefficients (Dobsd) of three peaks of the component of interest for three replicate experiments to give an average Dobsd with associated average deviation. Assuming two-state fast exchange for apelin-17, fb was estimated using eqs 3 and 4. The experimental settings for the diffusion time (Δ) and gradient duration (δ) for each experiment are indicated; pulsed field gradients were varied from 1.7 to 32.0 G/cm.
Upon determination of D, the fb of apelin-17 with each micelle was calculated using eq 3, indicating relatively weak binding of apelin-17 to DPC micelles and strong binding with both SDS and LPPG micelles (Table 1), in good agreement with the CD spectropolarimetry results. This interaction is somewhat predictable due to the strongly cationic nature of apelin-17, which contains eight basic residues to facilitate its interaction with anionic headgroups. However, without experimental demonstration such as this, it is not necessarily predictable that a peptide as hydrophilic as apelin-17 will interact so strongly with detergent micelles in solution.
NMR Assignment and Secondary Chemical Shifts
Nearly complete assignment of 1H, 13C, and 15N was obtained for apelin-17 (Table S1 in the Supporting Information) in the presence of both SDS and DPC micelles using standard methods,(53) allowing comparison of Δδ32 for Hα, Cα, Hβ, and Cβ nuclei in the presence of SDS and DPC micelles to those in buffer that we previously reported(15) (Figure 2). For both Hα and Cβ, there is no clear relationship between the value of the secondary chemical shift and the condition used. However, these Δδ values are significantly different in the presence of SDS micelles compared to those in either the buffer or DPC conditions over K1−K12, while Δδ values in the C-terminal of apelin-17 show little perturbation in the presence of micelles. The large changes in Δδ values of Cα and Hβ atoms in the presence of SDS suggest that apelin-17 is binding to the SDS micelles rather than forming a different, canonical secondary structure since Hα and Cβ would also be expected to have a consistent Δδ perturbation with secondary structure formation. Determination of the effect of LPPG micelles on Δδ values was not possible as deuterated LPPG is not available, making spectral assignment infeasible for apelin without isotopic enrichment.
Figure 2.

Apelin-17 secondary chemical shifts (Δδ = δ(obsd) − δ(random coil)(29)) for Hα, Cα, Hβ, and Cβ (the prime symbol denotes a degenerate shift) at 35 °C in buffer(15) (20 mM CD3COO−, pH 5.00 ± 0.05), SDS micelles, and DPC micelles. Horizontal lines show the Δδ cutoff significant for secondary structuring(31) (note that Cα and Cβ for Pro have identified significance ranges of ±4 ppm).
In all cases, apelin-17 showed a single, predominant set of chemical shifts at 35 °C with D values between those of free apelin and those of each detergent micelle indicative of fast exchange on the NMR time scale between the free and bound states (i.e., submillisecond exchange(54)). Fast exchange may be less relevant in the case of LPPG, given the almost 100% binding stoichiometry; however, it is certainly feasible that similar transient binding is taking place under an equilibrium favored further toward the bound state than in the SDS and DPC cases. Conformational sampling of apelin-17 was also evident in the NMR data, as we previously observed in buffer,(15) with a minor conformation in the P14−F17 region <10% as populated as the major one. Only the major conformer was sequentially assigned as there were few NOE contacts in the minor conformer. CD spectropolarimetry, DOSY, and chemical shift analysis therefore clearly demonstrate that the apelin peptides bind weakly and transiently to zwitterionic DPC micelles but bind favorably to anionic SDS and LPPG micelles.
Paramagnetic Relaxation Enhancement
To determine the region of apelin-17 that is interacting with anionic micelles, paramagnetic relaxation enhancement NMR spectroscopy experiments were used. The 1H−1H TOCSY peak attenuation of apelin-17 in SDS micelles with various concentrations of 5-DSA, 16-DSA, or Mn2+ is summarized in Figure 3. These reagents would be expected to attenuate the NMR signals for nuclei in the tailgroup core of the micelle (16-DSA), just below the anionic headgroup (5-DSA), or readily accessible to solution (Mn2+). For both 5-DSA and 16-DSA, there is no clear trend of peak attenuation over apelin-17, even at relatively high concentrations of paramagnetic agent, suggesting that apelin-17 does not associate in the hydrophobic core of the micelles or just below the headgroups of SDS (Figure 3A). In contrast, a clear trend in peak attenuation is observed with the titration of apelin-17 by Mn2+. At 0.25 mM Mn2+, many residues of apelin-17 show large attenuation of peak intensity, notably at the C-terminal region (Figure 3B). After titration with 1 mM Mn2+, only K1 and R6−L9 have an observable signal, indicating that these residues of apelin-17 interact most strongly with SDS micelles since they are partially shielded from the effects of Mn2+. However, even these residues are largely attenuated by Mn2+, indicating that although they interact with the micelle, they are still partially solvent exposed and very near the micelle surface, as supported by the lack of peak attenuation induced by 5-DSA or 16-DSA.
Figure 3.

Retained peak intensity (the error bar is the average deviation) of apelin-17 in SDS micelles with the indicated amounts of (A) 5-DSA or 16-DSA or (B) Mn2+ calculated from the intensities of the 1H−1H TOCSY peaks relative to a reference spectrum.
Structure of Apelin-17 in SDS Micelles
In total, 522 unique NOESY contacts were observed (with each NOE being considered from the perspective of each nucleus of the spin pair). This is an almost 2-fold increase in observed NOE contacts in comparison to those of apelin-17 in buffer at 35 °C (285 restraints(15)), indicating a dramatic increase in structuring of apelin-17 bound to SDS micelles vs free in solution. Of these contacts, the majority which are longer than i + 1 occur C-terminal to R6 (Figure S1 in the Supporting Information), implying that this region of apelin-17 is most structured in the presence of SDS micelles. Eight rounds of structure calculation were performed with these NOE restraints, with 100 structures generated in rounds 1−7 and 200 in round 8. Of the 200 structures in the final round, the 80 structures with the lowest total energy were retained as the final ensemble with minimal NOE violations and excellent Ramachandran plot statistics (Table 2). Evaluation of ϕ and ψ dihedral angle variation in terms of both the average deviation and order parameter (S of Hyberts et al.(39)) demonstrates a stretch of seven residues from R6 to K12 with both low average deviation and high S (Figure 4A,B) indicative of structural convergence.(35)
Table 2. Summary of the Restraints Employed for the Final Ensemble of 80 Retained Structures from 200 Calculated Structuresa.
| structure calculation parameter | apelin-17 with SDS | apelin-17 in bufferb |
|---|---|---|
| total rounds of NOE refinement | 8 | 12 |
| unique NOE restraints | ||
| total | 522 | 285 |
| intraresidue | 248 | 146 |
| sequential | 188 | 108 |
| medium range (|i − j| ≤ 4) | 46 | 22 |
| long range (|i − j| > 4) | 0 | 0 |
| ambiguous | 40 | 9 |
| Ramachandran plot statistics | ||
| core | 33% | 26% |
| allowed | 51% | 52% |
| generously allowed | 10% | 10% |
| disallowed | 6% | 13% |
| number of type I β-turns | 45 | 9 |
| number of type IV β-turns | 274 | 123 |
| number of type VIII β-turns | 6 | 10 |
| XPLOR-NIH energies (kcal/mol) | ||
| total | 26.9 ± 2.1 | 45.14 ± 5.39 |
| NOE | 4.6 ± 1.0 | 0.60 ± 0.49 |
| violations | ||
| NOE violations >0.5 Å | 0 | 0 |
| NOE violations of 0.3−0.5 Å | 1 | 0 |
| NOE violations of 0.2−0.3 Å | 4 | 4 |
Classification of NOE restraints using CYANA, LA Systems, Tokyo, Japan. XPLOR-NIH energies and average deviations and violation occurrences calculated using an in-house tcl/Tk script. Structural statistics for apelin-17 in buffer at 35 °C are shown for comparison.
Previously reported data.(15)
Figure 4.

Analysis of the 80-member NMR-based structural ensemble of apelin-17 bound to SDS micelles: (A) ϕ and (B) ψ angle average deviation (bars) and order parameters(39) (lines). Root mean square deviation (rmsd) calculated iteratively for (C) backbone atom superpositions over seven-residue segments and (D) all heavy atoms over three-residue superpositions with each bar (the error bar is the average deviation) plotted at the first residue in a superposition.
Independent of this, the ensemble of apelin-17 structures was superimposed in seven-residue stretches, and the root-mean-square deviation (rmsd) of backbone atoms was calculated (Figure 4C). (At superposition lengths of eight residues or greater, no RMSDs below 0.88 ± 0.2 Å were observed.) For a seven-residue superposition, the backbone atom rmsd is the lowest over R6−K12, suggesting that this region is well converged and mirroring the observation of highly defined ϕ and ψ dihedral angles. RMSDs over seven residues indicate that both the N- and C-terminal regions of apelin-17 are relatively unstructured in the presence of SDS micelles. Using Promotif-NMR,(38) 45 type I and 374 type IV β-turns were detected in the final ensemble (Table 2), with 98% of the type I β-turns being initiated between R6 and L9. Out of 80 structures, 30 have a type I β-turn initiation at P7. Type IV β-turns appear to be distributed throughout apelin-17, although there is a very well-defined type IV β-turn initiated at S10 in 77/80 of the ensemble members (Figure S2 in the Supporting Information). Paramagnetic relaxation enhancement experiments (Figure 3) indicate that the structural convergence over R6−K12 is the result of apelin binding to the micelle. In the superposed structural ensemble, the cationic side-chain atoms of R6 and R8 also tend to lie on the same face of the peptide, providing a cationic surface that may interact with the anionic micelle headgroups (Figure 5B). Since the structure of apelin-17 induced by anionic micelle binding is consistent between SDS and LPPG, as observed with CD spectropolarimetry, the NMR ensemble generated for apelin-17 interacting with SDS is also representative of the apelin-17 structure when bound to LPPG micelles.
Figure 5.
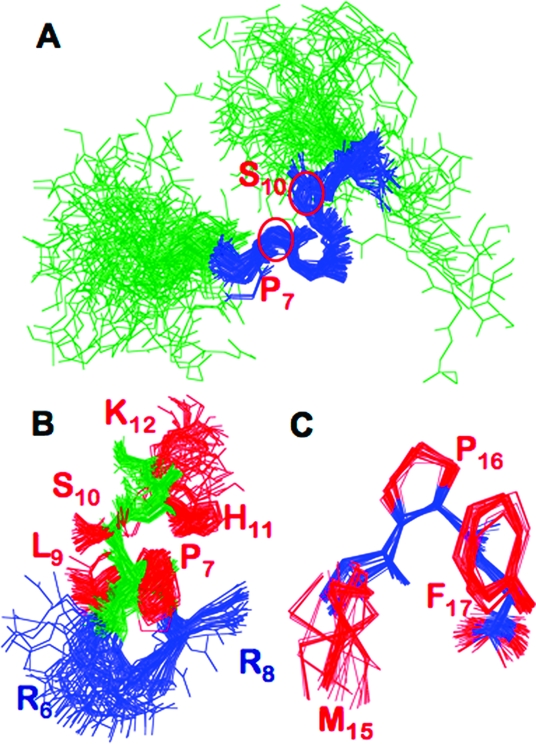
Structure of apelin-17 bound to an SDS micelle: (A) superposition of all 80 members of the final ensemble of structures from R6 to K12 (R6−K12 colored blue, remainder green) with P7 and S10, initiation points of type I and type IV β-turns, respectively, indicated; (B) zoom of the superposition in (A) (backbone atoms green) showing cationic side chains of R6 and R8 (blue; all other side chains red) falling on the same face of apelin-17; (C) superposition of all heavy atoms of M15−F17 for all 80 ensemble members, with the backbone shown in blue and side chains colored red.
Upon analysis of superpositions for shorter regions of apelin-17, an extremely well-converged region of apelin-17 was observed from M15 to F17 (Figure 5C), with a heavy atom rmsd of 0.37 ± 0.19 Å and well-converged ϕ and ψ dihedrals (Figure 4A,B,D). This region is just as converged as some of the residues between R6 and K12 in terms of both atom positions and dihedral angles but shows no canonical secondary structuring by Promotif-NMR analysis. Notably, this structural convergence was not observed in apelin-17 in solution at 35 °C,(15) indicating induction by apelin−micelle binding even though M15−F17 are not directly interacting with the micelle.
Implications for Membrane Catalysis of Apelin−APJ Binding
This study implies that apelin is likely to fit with the membrane catalysis model put forward by Sargent and Schwyzer.(16) Since apelin interacts almost equally well with two quite different anionic detergent headgroups, binding to biological membranes is likely. Furthermore, the structure observed with apelin bound to SDS micelles should represent the bound state with a biological membrane. Although a micelle surface is curved, in this system the curvature is negligible. The radius of an SDS micelle is ∼20 Å,(55) while the binding region of apelin-17 (R6−L9) spans ∼6.7 Å, or ∼5%, of the micelle circumference. Upon membrane interaction, the membrane catalysis model hypothesizes that a peptide will adopt a conformation that will accelerate binding to its receptor. The abundant type I and type IV β-turns which are induced by the anionic headgroups at the SDS and LPPG micelle surface may increase apelin’s affinity to the APJ receptor, in line with the suggestion of Tyndall et al. that β-turns are a ubiquitous motif recognized by GPCRs.(56) However, the high degree of structuring for M15−F17, although not β-turn in character, is a very striking micelle-induced structural element that may be even more likely to allow specific recognition of apelin by APJ since this portion of the peptide is not directly membrane-associated and therefore free to bind. Taken together, the structural changes that occur to apelin when bound to anionic micelles are likely essential for initiating interactions with APJ in a membrane. Since both regions of apelin-17 observed to undergo significant conformational restriction in comparison to the remainder of the peptide reside within the 12-residue functional core retained in all bioactive apelin isoforms, this binding hypothesis is immediately applicable to the other apelin peptides.
Concluding Remarks
The various active isoforms of apelin interact similarly with anionic SDS and LPPG micelles but not with the zwitterionic DPC micelle. Key to this interaction is the formation of a converged structure from R6 to K12 with significant type I and type IV β-turn initiation between R6 and L9 and at S10, respectively, while the residues N-terminal to this region are largely disordered. Part of this converged structure, R6−L9, interacts most strongly with anionic micelles. Binding of apelin-17 to SDS also induces structuring of M15−F17, even though this region is not in direct contact with the micelle surface. These results, obtained from disparate biophysical methods, suggest that APJ activation by apelin may follow the membrane catalysis model, requiring that apelin first bind to the plasma membrane, adopt the observed structure, and then bind to and activate APJ.
Acknowledgments
We thank Meghan Bebbington for preliminary CD studies, Bruce Stewart for technical support, Drs. Stephen Bearne and David Waisman for access to CD spectropolarimeters, Dr. Barbara Karten for access to a refrigerated centrifuge, Dr. Carmichael Wallace for access to his lyophilizer, Dr. Valerie Booth (Memorial University of Newfoundland) for lending us 5-DSA, and Drs. Ryan McKay (NANUC), Mike Lumsden (NMR3), and Ray Syvitski and Ian Burton (NRC-IMB) for assistance in the collection of NMR data. This research was supported by startup funding from Dalhousie University and a CIHR Regional Partnership Program Operating Grant matched through the Nova Scotia Health Research Foundation, the Dalhousie Cancer Research Program, and Dalhousie University’s Department of Biochemistry. Key infrastructure was funded by a Dalhousie University Faculty of Medicine Intramural Grant and a grant from the E. Gordon Young Endowment Fund. D.N.L. is supported through a Canada Graduate Scholarship from NSERC. Operation of NMR3 is funded by Dalhousie University and the Natural Sciences and Engineering Research Council of Canada (NSERC). NANUC receives funding from the Canadian Institutes for Health Research (CIHR), NSERC, and the University of Alberta. The 700 MHz cryoprobe at the NRC-IMB was the result of an Atlantic Canada Opportunities Agency Grant to Dalhousie University.
Supporting Information Available
Table S1 and Figures S1−S2. This information is available free of charge via the Internet at http://pubs.acs.org.
Supplementary Material
References
- O’Dowd B. F.; Heiber M.; Chan A.; Heng H. H.; Tsui L. C.; Kennedy J. L.; Shi X.; Petronis A.; George S. R.; Nguyen T. Gene 1993, 136, 355. [DOI] [PubMed] [Google Scholar]
- Masri B.; Knibiehler B.; Audigier Y. Cell. Signalling 2005, 17, 415. [DOI] [PubMed] [Google Scholar]
- Szokodi I.; Tavi P.; Foldes G.; Voutilainen-Myllyla S.; Ilves M.; Tokola H.; Pikkarainen S.; Piuhola J.; Rysa J.; Toth M.; Ruskoaho H. Circ. Res. 2002, 91, 434. [DOI] [PubMed] [Google Scholar]
- Heinonen M. V.; Purhonen A. K.; Miettinen P.; Paakkonen M.; Pirinen E.; Alhava E.; Akerman K.; Herzig K. H. Regul. Pept. 2005, 130, 7. [DOI] [PubMed] [Google Scholar]
- Foldes G.; Horkay F.; Szokodi I.; Vuolteenaho O.; Ilves M.; Lindstedt K. A.; Mayranpaa M.; Sarman B.; Seres L.; Skoumal R.; Lako-Futo Z.; deChatel R.; Ruskoaho H.; Toth M. Biochem. Biophys. Res. Commun. 2003, 308, 480. [DOI] [PubMed] [Google Scholar]
- Kleinz M. J.; Davenport A. P. Pharmacol. Ther. 2005, 107, 198. [DOI] [PubMed] [Google Scholar]
- Kalin R. E.; Kretz M. P.; Meyer A. M.; Kispert A.; Heppner F. L.; Brandli A. W. Dev. Biol. 2007, 305, 599. [DOI] [PubMed] [Google Scholar]
- Sorli S. C.; Le Gonidec S.; Knibiehler B.; Audigier Y. Oncogene 2007, 26, 7692. [DOI] [PubMed] [Google Scholar]
- Sorli S. C.; van den Berghe L.; Masri B.; Knibiehler B.; Audigier Y. Drug Discovery Today 2006, 11, 1100. [DOI] [PubMed] [Google Scholar]
- Principe A.; Melgar-Lesmes P.; Fernandez-Varo G.; del Arbol L. R.; Ros J.; Morales-Ruiz M.; Bernardi M.; Arroyo V.; Jimenez W. Hepatology 2008, 48, 1193. [DOI] [PubMed] [Google Scholar]
- Tatemoto K.; Hosoya M.; Habata Y.; Fujii R.; Kakegawa T.; Zou M. X.; Kawamata Y.; Fukusumi S.; Hinuma S.; Kitada C.; Kurokawa T.; Onda H.; Fujino M. Biochem. Biophys. Res. Commun. 1998, 251, 471. [DOI] [PubMed] [Google Scholar]
- Fan X.; Zhou N.; Zhang X.; Mukhtar M.; Lu Z.; Fang J.; DuBois G. C.; Pomerantz R. J. Biochemistry 2003, 42, 10163. [DOI] [PubMed] [Google Scholar]
- Medhurst A. D.; Jennings C. A.; Robbins M. J.; Davis R. P.; Ellis C.; Winborn K. Y.; Lawrie K. W.; Hervieu G.; Riley G.; Bolaky J. E.; Herrity N. C.; Murdock P.; Darker J. G. J. Neurochem. 2003, 84, 1162. [DOI] [PubMed] [Google Scholar]
- Lee D. K.; Saldivia V. R.; Nguyen T.; Cheng R.; George S. R.; O’Dowd B. F. Endocrinology 2005, 146, 231. [DOI] [PubMed] [Google Scholar]
- Langelaan D. N.; Bebbington E. M.; Reddy T.; Rainey J. K. Biochemistry 2009, 48, 537. [DOI] [PubMed] [Google Scholar]
- Sargent D. F.; Schwyzer R. Proc. Natl. Acad. Sci. U.S.A. 1986, 83, 5774. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mavromoustakos T.; Theodoropoulou E.; Dimitriou C.; Matsoukas J. M.; Panagiotopoulos D.; Makriyannis A. Lett. Pept. Sci. 1996, 3, 175. [Google Scholar]
- Deber C. M.; Behnam B. A. Proc. Natl. Acad. Sci. U.S.A. 1984, 81, 61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lopes S. C. D. N.; Fedorov A.; Castanho M. A. R. B. ChemBioChem 2005, 6, 697. [DOI] [PubMed] [Google Scholar]
- Lopes S. C. D. N.; Fedorov A.; Castanho M. A. R. B. Steroids 2004, 69, 825. [DOI] [PubMed] [Google Scholar]
- Young J. K.; Graham W. H.; Beard D. J.; Hicks R. P. Biopolymers 1992, 32, 1061. [DOI] [PubMed] [Google Scholar]
- Motta A.; Andreotti G.; Amodeo P.; Strazzullo G.; Castiglione Morelli M. A. Proteins 1998, 32, 314. [PubMed] [Google Scholar]
- Contreras L. M.; de Almeida R. F.; Villalain J.; Fedorov A.; Prieto M. Biophys. J. 2001, 80, 2273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Backlund B. M.; Wikander G.; Peeters T. L.; Graslund A. Biochim. Biophys. Acta 1994, 1190, 337. [DOI] [PubMed] [Google Scholar]
- Delaglio F.; Grzesiek S.; Vuister G. W.; Zhu G.; Pfeifer J.; Bax A. J. Biomol. NMR 1995, 6, 277. [DOI] [PubMed] [Google Scholar]
- Wu D. H.; Chen A. D.; Johnson C. S. J. Magn. Reson., A 1995, 115, 260. [Google Scholar]
- Stilbs P. J. Colloid Interface Sci. 1983, 87, 385. [Google Scholar]
- Jönsson B.; Wennerström H.; Nilsson P. G.; Linse P. Colloid Polym. Sci. 1986, 264, 77. [Google Scholar]
- Wishart D. S.; Sykes B. D.; Richards F. M. J. Mol. Biol. 1991, 222, 311. [DOI] [PubMed] [Google Scholar]
- Wishart D. S.; Sykes B. D. J. Biomol. NMR 1994, 4, 171. [DOI] [PubMed] [Google Scholar]
- Wishart D. S.; Sykes B. D.; Richards F. M. Biochemistry 1992, 31, 1647. [DOI] [PubMed] [Google Scholar]
- Wishart D. S.; Bigam C. G.; Holm A.; Hodges R. S.; Sykes B. D. J. Biomol. NMR 1995, 5, 67. [DOI] [PubMed] [Google Scholar]
- Schwieters C. D.; Kuszewski J. J.; Tjandra N.; Clore G. M. J. Magn. Reson. 2003, 160, 65. [DOI] [PubMed] [Google Scholar]
- Collaborative Computational Project N. Acta Crystallogr., D 1994, 50, 760.15299374 [Google Scholar]
- Rainey J. K.; Fliegel L.; Sykes B. D. Biochem. Cell Biol. 2006, 84, 918. [DOI] [PubMed] [Google Scholar]
- Reddy T.; Ding J.; Li X.; Sykes B. D.; Rainey J. K.; Fliegel L. J. Biol. Chem. 2008, 283, 22018. [DOI] [PubMed] [Google Scholar]
- Laskowski R. A.; Rullmannn J. A.; MacArthur M. W.; Kaptein R.; Thornton J. M. J. Biomol. NMR 1996, 8, 477. [DOI] [PubMed] [Google Scholar]
- Hutchinson E. G.; Thornton J. M. Protein Sci. 1996, 5, 212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hyberts S. G.; Goldberg M. S.; Havel T. F.; Wagner G. Protein Sci. 1992, 1, 736. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ulrich E. L.; Akutsu H.; Doreleijers J. F.; Harano Y.; Ioannidis Y. E.; Lin J.; Livny M.; Mading S.; Maziuk D.; Miller Z.; Nakatani E.; Schulte C. F.; Tolmie D. E.; Kent Wenger R.; Yao H.; Markley J. L. Nucleic Acids Res. 2008, 36, D402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Greenfield N. J. Methods Enzymol. 2004, 383, 282. [DOI] [PubMed] [Google Scholar]
- Sreerama N.; Manning M. C.; Powers M. E.; Zhang J. X.; Goldenberg D. P.; Woody R. W. Biochemistry 1999, 38, 10814. [DOI] [PubMed] [Google Scholar]
- Mehlis B.; Rueger M.; Becker M.; Bienert M.; Niedrich H.; Oehme P. Int. J. Pept. Protein Res. 1980, 15, 20. [PubMed] [Google Scholar]
- Rose G. D.; Gierasch L. M.; Smith J. A. Adv. Protein Chem. 1985, 37, 1. [DOI] [PubMed] [Google Scholar]
- Perczel A.; Hollosi M.. Turns. In Circular Dichroism and the Conformational Analysis of Biomolecules; Fasman G. D., Ed.; Plenum Press: New York, 1996; p 285. [Google Scholar]
- Shenkarev Z. O.; Balashova T. A.; Efremov R. G.; Yakimenko Z. A.; Ovchinnikova T. V.; Raap J.; Arseniev A. S. Biophys. J. 2002, 82, 762. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deaton K. R.; Feyen E. A.; Nkulabi H. J.; Morris K. F. Magn. Reson. Chem. 2001, 39, 276. [Google Scholar]
- Chou J. J.; Baber J. L.; Bax A. J. Biol. NMR 2004, 29, 299. [DOI] [PubMed] [Google Scholar]
- Cho C. H.; Urquidi J.; Singh S.; Robinson G. W. J. Phys. Chem. B 1999, 103, 1991. [Google Scholar]
- Malliaris A.; Le Moigne J.; Sturm J.; Zana R. J. Phys. Chem. 1985, 89, 2709. [Google Scholar]
- Paul B. C.; Islam S. S.; Ismail K. J. Phys. Chem. B 1998, 102, 7807. [Google Scholar]
- Stafford R. E.; Fanni T.; Dennis E. A. Biochemistry 1989, 28, 5113. [DOI] [PubMed] [Google Scholar]
- Wüthrich K.NMR of Proteins and Nucleic Acids; Wiley: New York, 1986. [Google Scholar]
- Evans J. N. S.Biomolecular NMR Spectroscopy; Oxford University Press: Oxford, U.K., 1995. [Google Scholar]
- Almgren M.; Gimel J. C.; Wang K.; Karlsson G.; Edwards K.; Brown W.; Mortensen K. J. Colloid Interface Sci. 1998, 202, 222. [Google Scholar]
- Tyndall J. D.; Pfeiffer B.; Abbenante G.; Fairlie D. P. Chem. Rev. 2005, 105, 793. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.