

Abstract
Recent studies demonstrate that activation of Ca2+-permeable N-methyl-D-aspartate (NMDA) receptors upregulates phosphorylation of mitogen-activated protein kinases (MAPKs) in heterologous cells and neurons. In cultured rat striatal neurons, the present work systematically evaluated the role of a number of protein kinases in forming a signaling cascade transducing NMDA receptor signals to MAPKs. It was found that a brief NMDA application consistently induced rapid and transient phosphorylation of the extracellular signal-regulated kinase 1/2 (ERK1/2), a best characterized subclass of MAPKs. This ERK1/2 phosphorylation was resistant to the inhibition of protein kinase C, p38 MAPK, cyclin-dependent kinase 5, receptor tyrosine kinase (epidermal growth factor receptors), or non-receptor tyrosine kinases (including Src) by their selective inhibitors. However, the increase in ERK1/2 phosphorylation was partially blocked by a protein kinase A (PKA) inhibitor. The inhibitors for Ca2+/calmodulin-dependent protein kinase (CaMK) or phosphatidylinositol 3-kinase (PI3-kinase) completely blocked the NMDA-stimulated ERK1/2 phosphorylation. In an attempt to characterize the sequential role of CaMK and PI3-kinase, we found that NMDA increased PI3-kinase phosphorylation on Tyr508, which kinetically corresponded to the ERK1/2 phosphorylation and was blocked by the CaMK inhibitor. These results indicate that the protein kinases are differentially involved in linking NMDA receptors to ERK1/2 in striatal neurons.
Keywords: glutamate, CaMK, PI3-kinase, PKA, PKC, tyrosine kinase, striatum
1. Introduction
Mitogen-activated protein kinases (MAPKs) are densely expressed in the postmitotic neuronal cells of adult mammalian brain and are involved in the regulation of multiple cellular activities [3]. Inducible phosphorylation of MAPKs by an upstream kinase, MAPK kinase (MEK), has been demonstrated in many cell lines in response to a wide range of extracellular stimuli [31]. The excitatory neurotransmitter L-glutamate (glutamate) is among effective extracellular signals that readily activate MAPK cascades [40]. Stimulation of the corticostriatal glutamatergic pathway increased phosphorylation of extracellular signal-regulated kinase 1/2 (ERK1/2 or p44/p42 MAPKs), a best characterized subclass of MAPKs, on their Thr202 and Tyr204 sites in the rat striatum in vivo [34,38]. The glutamate-sensitive ERK phosphorylation was also seen in cultured rat cortical [11,42], hippocampal [16], and striatal [13,30,39] neurons. In an attempt of characterizing the ERK1/2 phosphorylation by different subtypes of ionotropic glutamate receptors, we found that N-methyl-D-aspartate (NMDA) produced a rapid and transient phosphorylation of ERK1/2 in striatal neurons, which was blocked by the antagonists selective for NMDA, but not AMPA/kainate, receptors [23]. Moreover, the Ca2+ influx through Ca2+-permeable NMDA receptors mediates the NMDA effect because NMDA no longer phosphorylated ERK1/2 in an extracellular Ca2+-free medium [23].
Early studies evaluated roles of protein kinases in mediating the stimulus-induced ERK1/2 phosphorylation. In PC12 cells, the L-type Ca2+ channel-mediated Ca2+ influx activated the epidermal growth factor (EGF) receptor, a receptor tyrosine kinase, to phosphorylate MAPKs [32]. Like the receptor tyrosine kinase, the non-receptor tyrosine kinases Src and PYK2 have also been suggested to form a Ca2+-sensitive pathway to the Ras-MAPK cascade [17,33]. In cultured striatal neurons, the inhibitors relatively selective for Ca2+/calmodulin-dependent protein kinases (CaMK) or phosphatidylinositol (phosphoinositide) 3-kinase (PI3-kinase) attenuated NMDA-stimulated ERK1/2 phosphorylation [30], indicating a significant role of these two kinases in transducing NMDA receptor signals to ERK1/2. However, the role of other protein kinases and the interrelationship between CaMKs and PI3-kinase in transducing NMDA receptor signals to ERK1/2 in striatal neurons are poorly understood at present.
This study therefore systematically screened the involvement of a large number of protein kinases in coupling NMDA receptors to ERK1/2 in cultured rat striatal neurons. Using selective inhibitors, we evaluated the importance of protein kinase A (PKA), protein kinase C (PKC), p38 MAPK, cyclin-dependent kinase 5 (CDK5), EGF receptor tyrosine kinase, and non-receptor tyrosine kinase in comparison with CaMKs and PI3-kinase. Second, given the demonstrated role of CaMKs and PI3-kinase [30], we expanded our study to the investigation of a sequential relationship between the two kinases in mediating NMDA receptor signals to ERK1/2.
2. Materials and Methods
2.1. Primary striatal neuronal cultures
The standardized procedure preparing primary striatal neuronal cultures from the neonatal 1-day-old rat pups (Charles River, New York, NY) was employed in this study [24,43]. Predominant GABAergic neuronal cultures were obtained using this procedure as evidenced by the fact that > 90% of total cells were immunoreactive to glutamic acid decarboxylase-65/67 (GAD), GABA, or the specific marker for neurons (microtubule-associated protein-2a+2b, MAP2), but not for glia (glial fibrillary acidic protein, GFAP). Cells were usually cultured for 10-14 days before use.
2.2. Immunocytochemistry and quantitative analysis
The indirect ABC immunocytochemistry on slides was performed as described previously [25,26] to detect pERK1/2 immunoreactivity at the cellular level. Briefly, cultures were fixed in 4% paraformaldehyde (10 min), followed by incubation in 4% normal goat serum and 1% bovine serum album (20 min) to block nonspecific staining. The cells were treated with a primary antibody overnight at 4°C. The primary antibodies include rabbit polyclonal antibodies against ERK1/2 (Cell Signaling Technology, Beverly, MA; 1:2000), pERK1/2(Thr202/Tyr204) (Cell Signaling Technology; 1:2000), pPI3-kinase p85α(Tyr508) (Santa Cruz; 1:1000), NMDA receptor NR1-NT (recognizing the N-terminal domain; Upstate, Charlottesville, VA; 1:1000), or NR1-CT (recognizing the C-terminal cytoplasmic domain; Upstate; 1:1000), and mouse monoclonal antibodies against PI3-kinase p85α (Santa Cruz; 1:1000). The cells were incubated with biotinylated secondary antibody (goat anti-rabbit or anti-mouse IgG; 1:200; Vector) for 1 h, before incubation with avidin-biotin-horseradish peroxidase complex for 1 h. Finally, 3,3′-diaminobenzidine (0.25 mg/ml/0.01% H2O2/0.04% NiCl in 50 mM tris-HCl buffer, pH 7.4) was used as a chromagen (4-6 min). Omission of the primary antibodies served as negative controls.
The same strategy and procedure employed in our previous work [25,26] were performed in this study for quantitative analysis. The pERK1/2 immunocytochemical images were acquired through a video camera coupled to a Nikon E800 microscope. Cell counting was performed in each well by a person unaware of the treatment protocol. Both positive and negative cells were counted on the basis of a clearly visible pERK1/2-labeled (obviously different from the background) and unlabeled nucleus, respectively. Cells with ambiguous labeling or unidentifiable nuclei were excluded from analysis. Phenotypes of neuronal and astrocytic cells were easily identified according to their morphological characteristics. Neurons showed small (8-12 μm) to medium-sized (13-19 μm), phase-bright cell bodies with branching processes whereas astrocytes were large and flat with phase-dark, large pale nuclei (25-35 μm) and abundantly and widely spread cytoplasms [25,26]. Five optic fields per well (1 at the center and 4 at ∼1.5 mm from the four edges of the well; 800 × 800 μm each) were selected for cell counting. The pERK1/2-positive cells from five optic fields were calculated as the percentage of total cells from five fields in a well and treated as n = 1.
2.3. Western blot
Cultures were harvested in boiling 1X LDS sample buffer (Invitrogen, Carlsbad, CA). Cell lysates were sonicated with an ultrasonic dismembrator. Protein concentrations were determined with a Pierce BCA protein assay kit. The equal amount of protein (20 μg in 20 μl/well) was loaded on NuPAGE Novex 4-12% Bis-Tris precast mini-gels (Invitrogen) for separation of proteins. Proteins were transferred to polyvinylidene fluoride membrane (Immobilon-P; 0.45 mm; Millipore, Bedford, MA) and blocked in blocking buffer (5% nonfat dry milk in PBS and 0.1% Tween 20) for 1 h. The blots were incubated in primary rabbit polyclonal antibodies against ERK1/2, pERK1/2(Thr202/Tyr204), PI3-kinase p85α, or pPI3-kinase p85α(Tyr508) at 1:1,000 overnight at 4°C. This was followed by 1 h incubation in donkey anti-rabbit horseradish peroxidase-linked secondary antibodies (Jackson Immunoresearch Laboratory) at 1:10,000. Immunoblots were developed with the enhanced chemiluminescence reagents (ECL; Amersham Pharmacia Biotech, Piscataway, NJ), and exposed onto Kodak Biomax MS-1 films for 0.5-1 min. Kaleidoscope-prestained standards (Bio-Rad, Hercules, CA) were used for protein size determination. The integrated density (area × density) of immunoblots was analyzed using the NIH Image (1.60). Band density measurements were averaged over 2-3 control samples for each gel, and all bands were normalized as percentage of control values [19,20].
2.4. Drug treatments
The culture medium was replaced by HEPES-buffered salt solution (in mM: HEPES 20, NaCl 140, KCl 5, CaCl2 1.2, glycine 0.01, glucose 5.5, pH 7.4), and after 2 h incubation, cells were treated with drugs. All drugs were freshly made on the day of experiment. NMDA, 4-[5-(4-fluorophenyl)-2-[4-(methylsulphonyl)phenyl]-1H-imidazol-4-yl]pyridine hydrochloride (SB203580), 2-(4-morpholinyl)-8-phenyl-4H-1-benzopyran-4-one hydrochloride (LY294002), and wortmannin were purchased from Tocris Cookson Inc. (Ballwin, MO). H89, Ro-31-8220, roscovitine, KN93, KN92, AG1478, AG825, herbimycin A, genistein, and PP2 were purchased from Calbiochem (San Diego, CA). Gö6983, 8-bromoadenosine-3′,5′-cyclophosphoric acid (8-br-cAMP), phorbol 12-myristate 13-acetate (PMA), and human epidermal growth factor (hEGF) were purchased from Sigma. Drugs were dissolved in 1X PBS with or without an aid of dimethyl sulfoxide (DMSO). Whenever DMSO was used, PBS containing the same concentration of DMSO was used as the control vehicle.
2.5. Statistics
The results are presented as mean ± SEM. The percentages of pERK1/2-positive cells were evaluated using a one- or two-way analysis of variance, as appropriate, followed by a Bonferroni (Dunn) comparison of groups using least squares-adjusted means. Probability levels of < 0.05 were considered statistically significant.
3. Results
3.1. NMDA-induced ERK1/2 phosphorylation partially depends on PKA, but not PKC
Direct activation of PKA or PKC increased ERK1/2 phosphorylation in many cell lines [2,8]. Active PKA and PKC also mediate ERK1/2 phosphorylation induced by stimulation of G-protein-coupled receptors [5,15]. Thus, the first study was carried out to determine whether activation of either kinase contributes to transducing NMDA receptor signals to ERK1/2 in cultured striatal neurons. Using a PKA inhibitor H89 that concentration-dependently blocked the ERK1/2 phosphorylation induced by the PKA activator 8-br-cAMP (Fig. 1A and 1C), we found that the number of pERK1/2-labeled neurons seen after H89 (10 μM) + NMDA (100 μM) treatment was approximately a half of pERK1/2-positive neurons seen after NMDA treatment alone (Fig. 1A and 1C). Thus, in some striatal neurons, PKA contributes to the NMDA action. In contrast to PKA, PKC is not a kinase imperative for NMDA phosphorylation of ERK1/2 because the two PKC inhibitors Ro-31-8220 and Gö6983 at concentrations effective for blocking the ERK1/2 phosphorylation induced by PKC activator PMA did not alter the increases in pERK1/2 cells induced by NMDA (Fig. 1A, 1D, and 1E). All drug treatments had no effect on the number of ERK1/2-labeled neurons. In our culture model, most medium-sized neurons expressed NMDA receptors as evidenced by the fact that > 85% of cells showed detectable levels of NMDA receptor NR1 subunit immunoreactivity (Fig. 1B).
Fig. 1.
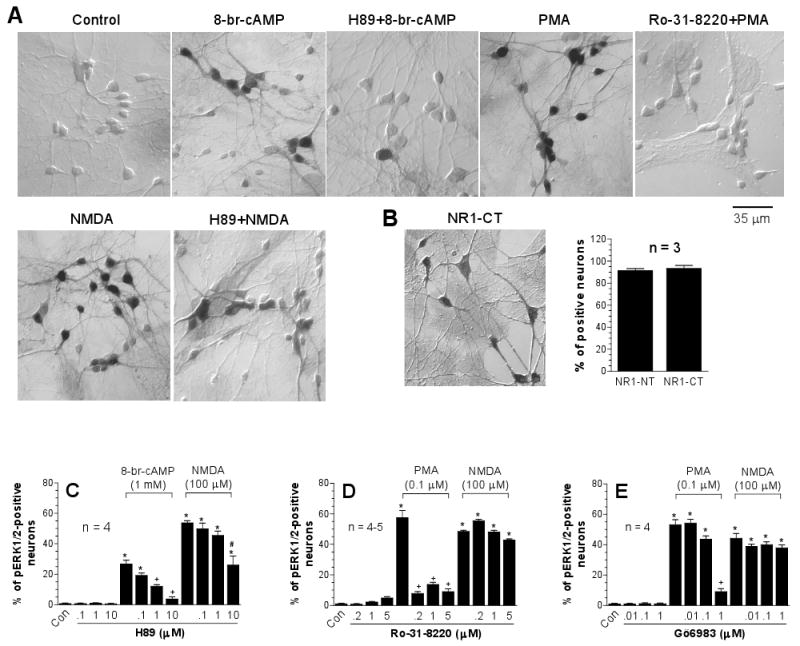
Effects of inhibitors selective for PKA or PKC on basal and NMDA-induced pERK1/2-immunoreactivity in cultured rat striatal neurons. The inhibitors were incubated 20 min prior to and during 15-min NMDA treatment before fixation. (A) Immunocytochemical images illustrating that the PKA inhibitor H89 (10 μM) and the PKC inhibitor Ro-31-8220 (5 μM) blocked 8-br-cAMP (1 mM)- and PMA (0.1 μM)-induced ERK1/2 phosphorylation, respectively. H89 also partially reduced NMDA (100 μM)-stimulated ERK1/2 phosphorylation. (B) Cellular immunostaining of the NMDA receptor NR1 subunit in cultured rat striatal neurons. Note that strong immunoreactivity was seen in cell bodies and enriched processes of medium-sized neurons. (C-E) Data from cell counting are expressed in terms of the mean ± SEM of the percent change in numbers of the pERK1/2-positive neurons. *p < 0.05 vs. control (Con), +p < 0.05 vs. 8-br-cAMP (C) or PMA (D and E) alone, and #p < 0.05 vs. NMDA (C) alone.
3.2. NMDA-induced ERK1/2 phosphorylation is independent on CDK5 and p38 MAPK
CDK5 phosphorylated MEK1 and ERK1/2 in PC12 cells [35]. To detect possible contributions of CDK5 and p38 MAPK, the effect of NMDA on ERK1/2 phosphorylation was tested in the presence of an inhibitor selective for CDK5 (roscovitine) or p38 MAPK (SB203580). Neither roscovitine (0.1-1 μM) nor SB203580 (1.25-20 μM) altered basal levels of pERK1/2 immunoreactivity (data not shown). Similarly, the two inhibitors at all concentrations surveyed had no significant effect on the NMDA-stimulated increases in the number of pERK1/2-labeled neurons (data not shown). These results provide evidence against the involvement of CDK5 and p38 MAPK in the transduction of NMDA receptor signals to ERK1/2.
3.3. NMDA and EGF receptors independently stimulate ERK1/2 phosphorylation
Recent studies reveal the participation of receptor tyrosine kinases, such as the EGF receptor (ErbB1), in transducing the signals from Ca2+ or G-protein-coupled receptors to ERK1/2 [21,29,32]. We then examined the possibility that NMDA receptors transactivate EGF receptors, thereby inducing ERK1/2 phosphorylation. In the first experiment evaluating temporal properties of EGF-mediated ERK1/2 phosphorylation, we found that hEGF (30 ng/ml, 2 to 30 min) induced rapid ERK1/2 phosphorylation, which declined between 20 to 30 min after the commence of incubation (Fig. 2A and 2B). The hEGF-stimulated ERK1/2 phosphorylation was blocked by the EGF selective inhibitor, tyrphostin AG1478 [18], at 0.1 and 1 μM (Fig. 2C). However, AG1478 did not inhibit the increases in pERK1/2 neurons induced by NMDA (Fig. 2D). Neither did AG825, a tyrphostin that selectively inhibits the receptor tyrosine kinase ErbB2 [27] (Fig. 2E). These data suggest an insignificant role of ErbB1/2 in the NMDA-induced phosphorylation of ERK1/2.
Fig. 2.
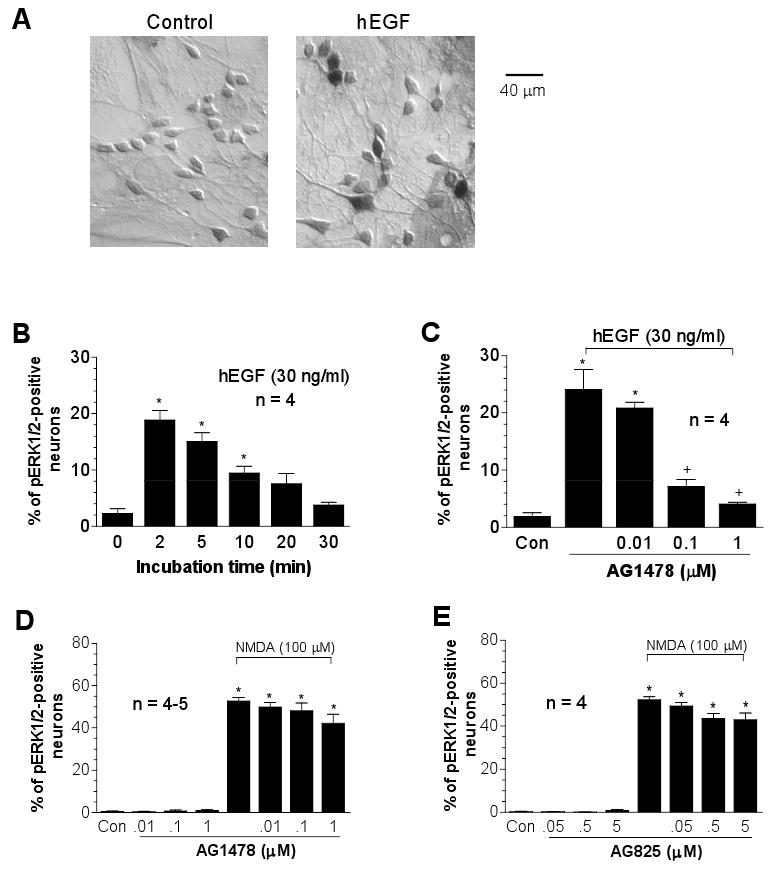
Effects of the receptor tyrosine kinase inhibitors on basal and NMDA-induced pERK1/2-immunoreactivity in cultured rat striatal neurons. (A) Immunocytochemical images illustrating increases in pERK1/2 neurons following hEGF incubation (30 ng/ml, 2 min). (B) Dynamic induction of pERK1/2 neurons following hEGF incubation (30 ng/ml, 2 to 30 min). (C-E) Effects of the EGF/ErbB1 inhibitor AG1478 or the ErbB2 inhibitor AG825 on hEGF- or NMDA-stimulated increases in the number of pERK1/2-positive neurons. The inhibitors were incubated 20 min prior to and during 2-min hEGF treatment or during 15-min NMDA treatment before fixation. Data are expressed in terms of the mean ± SEM of the percent change in numbers of the pERK1/2-positive neurons. *p < 0.05 vs. control (Con), and +p < 0.05 vs. hEGF alone (C).
3.4. NMDA-induced ERK1/2 phosphorylation is independent on non-receptor tyrosine kinases
Non-receptor tyrosine kinases have been demonstrated to be necessary effectors of Ca2+ for ERK activation [7,33,41]. In some models of G-protein-coupled receptors, including metabotropic glutamate receptors, the recruitment of Src non-receptor tyrosine kinases was required for activation of ERK1/2 [21,22,37]. Therefore, the three non-receptor tyrosine kinase inhibitors (genistein, herbimycin A, and PP2) were used to define the importance of tyrosine kinases of this kind. The two general inhibitors genistein [1] at 1-100 μM and herbimycin A at 0.1-10 μM did not inhibit NMDA-induced ERK1/2 phosphorylation (data not shown). A more selective inhibitor for the Src family, PP2 [14], at 0.1-10 μM produced similar results. Thus, non-receptor tyrosine kinases are less likely required for NMDA receptor signaling to ERK1/2.
3.5. Sequential activation of CaMKs and PI3-kinase is required for NMDA phosphorylation of ERK1/2
CaMKs are abundant in the postsynaptic NMDA receptor complex and serve as a major Ca2+-sensitive kinase at excitatory synapses. Inhibition of the kinase prevented glutamate or the group I metabotropic glutamate receptor agonist from inducing detectable ERK1/2 phosphorylation in striatal neurons [9,38]. PI3-kinase is also densely expressed in striatal neurons [36]. Its role as a downstream effector of several surface membrane receptors or channels for ERK activation has been demonstrated in cell lines [13,44]. Perkinton and co-workers [30] identified a mediating role of CaMKs and PI3-kinase in NMDA-stimulated ERK1/2 phosphorylation in mouse striatal neurons. This was confirmed to be the case in this rat culture model. The CaMK selective inhibitor KN93, but not its inactive analog KN92, and the two PI3-kinase inhibitors, LY294002 and wortmannin, blocked NMDA-induced increases in pERK1/2 cells in a concentration-dependent manner in both immunohistochemical (Fig. 3A-C) and immunoblot (Fig. 3D and 3E) analysis.
Fig. 3.
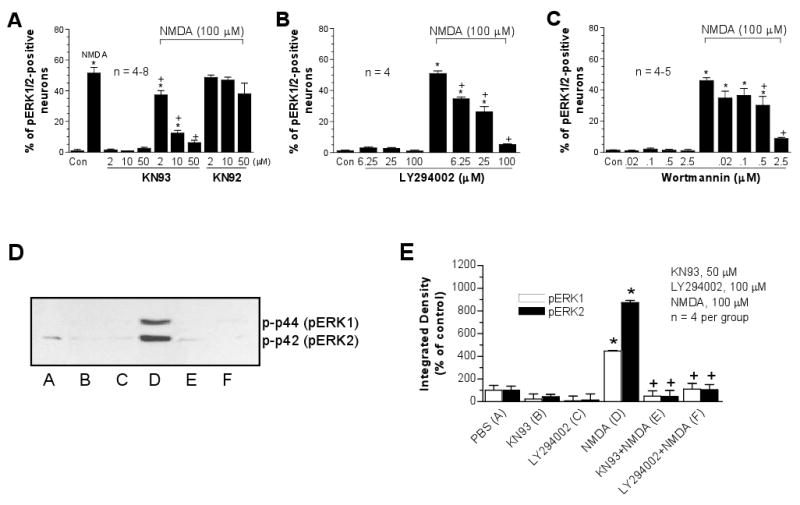
Effects of the inhibitors selective for CaMKs (KN93) or PI3-kinase (LY294002 and wortmannin) on basal and NMDA-induced pERK1/2-immunoreactivity in cultured rat striatal neurons. The inhibitors were incubated 20 min prior to and during 15-min NMDA treatment before fixation. Data from cell counting (A-C) are expressed in terms of the mean ± SEM of the percent change in numbers of the pERK1/2-positive neurons. The quantified data (E) are expressed in terms of the mean ± SEM of the integrated density and are shown below the immunoblots (D). *p < 0.05 vs. control (Con), +p < 0.05 vs. NMDA alone.
We next wanted to examine whether NMDA increases phosphorylation of PI3-kinase as a preceding event upstream to ERK activation. We found that NMDA (100 μM, 5 min) increased the number of cells expressing phosphorylated regulatory subunit (p85α) of PI3-kinase at Tyr508, p-p85α (Fig. 4A). The NMDA effect can be seen at 2 min, peaked at 5-10 min, and declined at 20-30 min after the commence of incubation (Fig. 4B). This time-course seems to kinetically place the PI3-kinase activation as an early event upstream to the ERK1/2 phosphorylation induced by NMDA [23]. Data from western blot also showed a significant increase in p-p85α levels following NMDA stimulation, which was blocked by the PI3-kinase inhibitor LY294002 (Fig. 4C). Interestingly, the NMDA phosphorylation of PI3-kinase was sensitive to the CaMK inhibitor KN93. In the presence of KN93, NMDA failed to induce an increase in p85α phosphorylation (Fig. 4C). This observation appears to support a model in which NMDA increases ERK1/2 phosphorylation by first activating CaMKs followed by PI3-kinase activation. The PI3-kinase immunoreactivity detected in western blot with the antibody raised against unphosphorylated p85α did not show any changes after all drug treatments as compared to a control value (Fig. 4C).
Fig. 4.
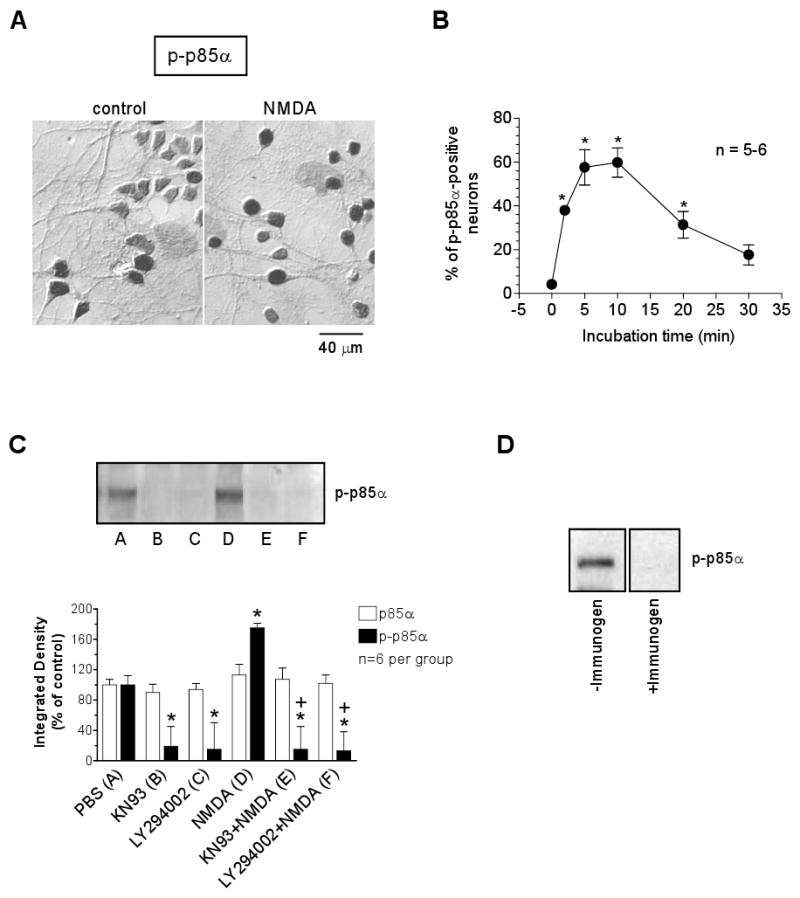
Effects of NMDA on p85α phosphorylation in cultured rat striatal neurons. (A) Immunocytochemical images of p-p85α-labeled cells from cultures treated with control PBS or NMDA (100 μM, 5 min). (B) NMDA (100 μM) induced a time-dependent increase in the number of p-p85α-positive neurons. (C) Effects of KN93 (50 μM) and LY294002 (100 μM) on basal p85α expression and on NMDA-stimulated phosphorylation of p85α as detected with western blot. The quantified data are expressed in terms of the mean ± SEM of the integrated density. (D) The p-p85α band did not shown when the p-p85α antibody was pre-absorbed with a p-p85α immmunogen peptide.
4. Discussion
The present study was conducted to identify protein kinases in a signaling cascade transducing NMDA receptor signals to ERK1/2 in striatal neurons. While a number of kinases surveyed in this study appear to be not important, the two protein kinases, i.e. CaMK and PI3-kinase, were confirmed to be essential links in the cascade. More importantly, we found that NMDA increased PI3-kinase phosphorylation on p85α(Tyr508), which was prevented by the CaMK inhibitor. This indicates a sequential activation of the two responsible kinases to form a pathway coupling NMDA receptors to ERK1/2.
PI3-kinase has been demonstrated to serve as a downstream effector of several surface membrane receptors or channels for ERK activation in cell lines [13,44]. The results from this study confirmed the blocking effect of PI3-kinase inhibitors on the NMDA-induced ERK1/2 phosphorylation [13,30]. In an expanded attempt to explore whether NMDA activates PI3-kinase through increasing its phosphorylation, we found that NMDA increased PI3-kinase (p85α) phosphorylation which is kinetically correlated to concomitant ERK1/2 phosphorylation. Interestingly, the increased PI3-kinase phosphorylation was blocked by the CaMK inhibitor KN93. This finding, together with recent evidence showing a high affinity calmodulin-binding sequence in PI3-kinase [6,12], suggests a signaling model in which NMDA activates ERK1/2 by first activating CaMKs followed by PI3-kinase activation.
PKA mediates ERK activation induced by stimulation of G-protein-coupled receptors in neurons [5,15]. In striatal neurons, PKA induced a Ca2+/ERK-dependent phosphorylation of cAMP response element-binding protein [45]. Our results also support a PKA-dependent mechanism involving NMDA phosphorylation of ERK1/2. Moreover, with immunocytochemical analysis at the cellular level, we were able to visualize that PKA functions in some neurons, but not all, because the PKA inhibitor H89 reduced the number of pERK1/2-positive neurons to about a half of that seen after NMDA treatment alone. Parallel with this observation, the PKA activator 8-br-cAMP increased ERK1/2 phosphorylation in a percentage of neurons equivalent to a half of that induced by NMDA. Thus, PKA-mediated ERK1/2 responses are heterogeneous among striatal neurons. Mechanisms underlying this heterogeneity are unclear. It may reflect differences among different populations of striatal neurons in the composition and efficiency of PKA-related signaling cascades and in the development of NMDA receptors and associative signaling molecules.
Transactivation of the EGF receptor (ErbB1) mediates Ca2+ or G-protein-coupled receptor signals to ERK1/2 in many cell lines or primary rat or mouse astrocytes [21,29,32]. Thus, it is possible that NMDA receptor-mediated Ca2+ influx induces transactivation of EGF, which in turn activates ERK1/2. Our results indeed indicate an early activation of ERK1/2 in response to EGF receptor stimulation with hEGF. However, the EGF inhibitor AG1478 that blocked the hEGF-induced ERK1/2 phosphorylation did not affect the ERK1/2 phosphorylation induced by NMDA. Thus, as a receptor tyrosine kinase, EGF receptors are less likely involved in forming a pathway to ERK1/2 in our cultures of striatal neurons. Like receptor tyrosine kinase, non-receptor tyrosine kinases, such as Src, are shown to act as an essential kinase in Ca2+ signaling to ERK1/2 at least in PC12 cells. For instance, Ca2+ influx through L-type voltage-dependent Ca2+ channels increased Src kinase activity in PC12 cells [33]. A PC12 subline with a stably expressed dominant form of Src did not undergo Ca2+-sensitive MAPK phosphorylation [33]. However, in primary striatal neurons, all effective inhibitors selective for non-receptor tyrosine kinases showed no significant effects on NMDA-induced ERK activation in the present study. This provides evidence against a significant role of non-receptor tyrosine kinases in mediating NMDA receptor signals to ERK1/2 in striatal neurons.
A positive connection between PKC and MAPK cascades exists in cell lines [2,8]. In striatal neurons, the PKC activator induced a robust phosphorylation of ERK1/2 similar to that induced by NMDA (this study). Thus, PKC is among kinases activating ERK1/2 cascades and was hypothesized to be a significant link in the signaling cascade bridging NMDA receptor signals to ERK1/2. However, in contrast to our hypothesis, we found that the inhibitors that inhibited the PKC activator-induced ERK1/2 phosphorylation did not alter the ability of NMDA to phosphorylate ERK1/2. Thus, the positive PKC-MAPK linkage, despite its existence in primary striatal neurons, does not transduce NMDA receptor signals to ERK1/2.
ERK is a superhighway transducing extracellular signals to gene expression. Activation of a variety of surface receptors leads to activation of the ERK pathway [4,28]. Major intracellular signaling pathways are also believed to crosstalk with ERK [10, 40]. Future studies will have to dissect specific signaling pathways that link different extracellular stimuli to ERK and elucidate precise cellular mechanisms underlying the crosstalk between ERK and other signaling pathways.
Acknowledgments
This work was supported by the R01 grants DA10355 (J.Q.W.) and MH61469 (J.Q.W.) from the NIH.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Akiyama T, Ogawara H. Use and specificity of genistein as inhibitor of protein-tyrosine kinases. Methods Enzymol. 1991;201:362–370. doi: 10.1016/0076-6879(91)01032-w. [DOI] [PubMed] [Google Scholar]
- 2.Ambrosini A, Tininini S, Barassi A, Racagni G, Sturani E, Zippel R. cAMP cascade leads to Ras activation in cortical neurons. Mol Brain Res. 2000;75:54–60. doi: 10.1016/s0169-328x(99)00294-6. [DOI] [PubMed] [Google Scholar]
- 3.Arbabi S, Maier RV. Mitogen-activated protein kinases. Crit Care Med. 2002;30:S74–79. [PubMed] [Google Scholar]
- 4.Barbieri R, Alloisio S, Ferroni S, Nobile M. Differential crosstalk between P2X7 and arachidonic acid in activation of mitogen-activated protein kinases. Neurochem Int. 2008 doi: 10.1016/j.neuint.2008.05.001. in press. [DOI] [PubMed] [Google Scholar]
- 5.Bouschet T, Perez V, Fernandez C, Bockaert J, Eychene A, Journot L. Stimulation of the ERK Pathway by GTP-loaded Rap1 Requires the Concomitant Activation of Ras, Protein Kinase C, and Protein Kinase A in Neuronal Cells. J Biol Chem. 2003;278:4778–4785. doi: 10.1074/jbc.M204652200. [DOI] [PubMed] [Google Scholar]
- 6.Chan TO, Rittenhouse SE, Tsichlis PN. AKT/PKB and other D3 phosphoinositide-regulated kinases: kinase activation by phosphoinositide-dependent phosphorylation. Ann Rev Biochem. 1999;68:965–1014. doi: 10.1146/annurev.biochem.68.1.965. [DOI] [PubMed] [Google Scholar]
- 7.Chao TS, Abe M, Hershenson MB, Gomes I, Rosner MR. Src tyrosine kinase mediates stimulation of Raf-1 and mitogen-activated protein kinase by the tumor promoter thapsigargin. Cancer Res. 1997;57:3168–3173. [PubMed] [Google Scholar]
- 8.Cho YJ, Kim JY, Jeong SW, Lee SB, Kim ON. Cyclic AMP induces activation of extracellular signal-regulated kinases in HL-60 cells: role in cAMP-induced differentiation. Leuk Res. 2003;27:51–56. doi: 10.1016/s0145-2126(02)00057-7. [DOI] [PubMed] [Google Scholar]
- 9.Choe ES, Wang JQ. Group I metabotropic glutamate receptors control phosphorylation of CREB, Elk-1 and ERK via a CaMKII-dependent pathway in rat striatum. Neurosci Lett. 2001;313:129–132. doi: 10.1016/s0304-3940(01)02258-3. [DOI] [PubMed] [Google Scholar]
- 10.Crespo-Biel N, Canudas AM, Camins A, Pallas M. Kinate induces AKT, ERK and cdk5/GSK3beta pathway deregulation, phosphorylation tau protein in mouse hippocampus. Neurochem Int. 2007;50:435–442. doi: 10.1016/j.neuint.2006.10.002. [DOI] [PubMed] [Google Scholar]
- 11.Fiore RS, Murphy TH, Sanghera JS, Pelech SL, Baraban JM. Activation of p42 mitogen-activated protein kinase by gluatmate receptor stimulation in rat primary cortical cultures. J Neurochem. 1993;61:1626–1633. doi: 10.1111/j.1471-4159.1993.tb09796.x. [DOI] [PubMed] [Google Scholar]
- 12.Fischer R, Julsgart J, Berchtold MW. High affinity calmodulin target sequence in the signaling molecule PI 3-kinase. FEBS Lett. 1998;425:175–177. doi: 10.1016/s0014-5793(98)00225-7. [DOI] [PubMed] [Google Scholar]
- 13.Fuller G, Veitch K, Ho LK, Cruise L, Morris BJ. Activation of p44/p42 MAP kinase in striatal neurons via kainate receptors and P13 kinase. Mol Brain Res. 2001;89:126–132. doi: 10.1016/s0169-328x(01)00071-7. [DOI] [PubMed] [Google Scholar]
- 14.Hanke JH, Gardner JP, Dow RL, Changelian PS, Brissette WH, Weringer EJ, Pollok BA, Connelly PA. Discovery of a novel, potent, and Src family-selective tyrosine kinase inhibitor. Study of Lck- and FynT-dependent T-cell activation. J Biol Chem. 1996;271:695–701. doi: 10.1074/jbc.271.2.695. [DOI] [PubMed] [Google Scholar]
- 15.Jiang Y, Cypess AM, Muse ED, Wu CR, Unson CG, Merrifield RB, Sakmar TP. Glucagon receptor activates extracellular signal-regulated protein kinase 1/2 via cAMP-dependent protein kinase. Proc Natl Acad Sci USA. 2001;98:10102–10107. doi: 10.1073/pnas.131200398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Kurino M, Fukunaga K, Ushio Y, Miyamoto E. Activation of mitogen-activated protein kinase in cultured rat hippocampal neurons by stimulation of glutamate receptors. J Neurochem. 1995;65:1282–1289. doi: 10.1046/j.1471-4159.1995.65031282.x. [DOI] [PubMed] [Google Scholar]
- 17.Lev S, Moreno H, Martinez R, Canoll P, Peles E, Musacchio JM, Plowman GD, Rudy B, Schlessinger J. Protein tyrosine kinase PYK2 involved in Ca(2+)-induced regulation of ion channel and MAP kinase functions. Nature. 1995;376:737–745. doi: 10.1038/376737a0. [DOI] [PubMed] [Google Scholar]
- 18.Levitzki A, Gazit A. Tyrosine kinase inhibition: an approach to drug development. Science. 1995;267:1782–1788. doi: 10.1126/science.7892601. [DOI] [PubMed] [Google Scholar]
- 19.Liu XY, Chu XP, Mao LM, Wang M, Lan HX, Li MH, Zhang GC, Parelkar NK, Fibuch EE, Haines M, Neve KA, Liu F, Xiong ZG, Wang JQ. Modulation of D2R-NR2B interactions in response to cocaine. Neuron. 2006;52:897–909. doi: 10.1016/j.neuron.2006.10.011. [DOI] [PubMed] [Google Scholar]
- 20.Liu XY, Mao LM, Zhang GC, Papasian CJ, Fibuch EE, LacKamp A, Lan HX, Zhou HF, Xu M, Wang JQ. Activity-dependent modulation of limbic dopamine D3 receptors by CaMKII. Neuron. doi: 10.1016/j.neuron.2008.12.015. in revision. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Luttrell LM, Daaka Y, Lefkowitz RJ. Regulation of tyrosine kinase cascades by G-protein-coupled receptors. Curr Opin Cell Biol. 1999;11:177–183. doi: 10.1016/s0955-0674(99)80023-4. [DOI] [PubMed] [Google Scholar]
- 22.Luttrell LM, Hawes BE, van Biesen T, Luttrell DK, Lansing TJ, Lefkowitz RJ. Role of c-Src tyrosine kinase in G-protein-coupled receptor- and Gβγ subunit-mediated activation of mitogen-activated protein kinases. J Biol Chem. 1996;271:19443–19450. doi: 10.1074/jbc.271.32.19443. [DOI] [PubMed] [Google Scholar]
- 23.Mao L, Tang Q, Samdani S, Liu Z, Wang JQ. Regulation of MAPK/ERK phosphorylation via ionotropic glutamate receptors in cultured rat striatal neurons. Eur J Neurosci. 2004;19:1207–1216. doi: 10.1111/j.1460-9568.2004.03223.x. [DOI] [PubMed] [Google Scholar]
- 24.Mao L, Yang L, Tang Q, Samdani S, Zhang G, Wang JQ. The scaffold protein Homer1b/c links metabotropic glutamate receptor 5 to extracellular signal-regulated protein kinase cascades in neurons. J Neurosci. 2005;25:2741–2752. doi: 10.1523/JNEUROSCI.4360-04.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Mao L, Wang JQ. Glutamate cascade to cAMP response element-binding protein phosphorylation in cultured striatal neurons through calcium-coupled group I mGluRs. Mol Pharmacol. 2002;62:473–484. doi: 10.1124/mol.62.3.473. [DOI] [PubMed] [Google Scholar]
- 26.Mao L, Wang JQ. Group I metabotropic glutamate receptor-mediated calcium signaling and immediate early gene expression in cultured rat striatal neurons. Eur J Neurosci. 2003;17:741–750. doi: 10.1046/j.1460-9568.2003.02495.x. [DOI] [PubMed] [Google Scholar]
- 27.Osherov N, Gazit A, Gilon C, Levitzki A. Selective inhibition of the epidermal growth factor and HER2/neu receptors by tyrphostins. J Biol Chem. 1993;268:11134–11142. [PubMed] [Google Scholar]
- 28.Page G, Khidir FA, Pain S, Barrier L, Fauconneau B, Guillard O, Piriou A, Hugon J. Group I metabotropic glutamate receptors activate the p70S6 kinase via both mammalian target of rapamycin (mTOR) and extracellular signal-regulated kinase (ERK1/2) signaling pathways in rat striatal and hippocampal synaptoneurosomes. Neurochem Int. 2006;49:413–421. doi: 10.1016/j.neuint.2006.01.020. [DOI] [PubMed] [Google Scholar]
- 29.Peavy RD, Chang MSS, Sanders-Bush E, Conn PJ. Metabotropic glutamate receptor 5-induced phosphorylation of extracellular signal-regulated kinase in astrocytes depends on transactivation of the epidermal growth factor receptor. J Neurosci. 2001;21:9619–9628. doi: 10.1523/JNEUROSCI.21-24-09619.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Perkinton MS, Ip JK, Wood GL, Crossthwaite AJ, Williams RJ. Phosphatidylinositol 3-kinase is a central mediator of NMDA receptor signalling to MAP kinase (Erk1/2), Akt/PKB and CREB in striatal neurons. J Neurochem. 2002;80:239–254. doi: 10.1046/j.0022-3042.2001.00699.x. [DOI] [PubMed] [Google Scholar]
- 31.Peyssonnaux C, Eychene AA. The Raf/MEK/ERK pathway: new concepts of activation. Biol Cell. 2001:53–62. doi: 10.1016/s0248-4900(01)01125-x. [DOI] [PubMed] [Google Scholar]
- 32.Rosen LB, Greenberg ME. Stimulation of growth factor receptor signal transduction by activation of voltage-sensitive calcium channels. Proc Natl Acad Sci USA. 1996;93:1113–1118. doi: 10.1073/pnas.93.3.1113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Rusanescu G, Qi H, Thomas SM, Brugge JS, Halegoua S. Calcium influx induces neurite growth through a Src-Ras signaling cassette. Neuron. 1995;15:1415–1425. doi: 10.1016/0896-6273(95)90019-5. [DOI] [PubMed] [Google Scholar]
- 34.Sgambato V, Vanhoutte P, Pages C, Rogard M, Hipskind R, Besson MJ, Caboche J. In vivo expression and regulation of Elk-1, a target of the extracellular-regulated kinase signaling pathway, in the adult rat brain. J Neurosci. 1998;18:214–226. doi: 10.1523/JNEUROSCI.18-01-00214.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Sharma P, Veeranna SM, Amin ND, Sihag RK, Grant P, Ahn N, Kulkarni AB, Pant HC. Phosphorylation of MEK1 by cdk5/p35 down-regulates the mitogen-activated protein kinase pathway. J Biol Chem. 2002;277:528–534. doi: 10.1074/jbc.M109324200. [DOI] [PubMed] [Google Scholar]
- 36.Shin BC, Suzuki M, Inukai K, Anai M, Asano T, Takata K. Multiple isoforms of the regulatory subunit for phosphatidylinositol 3-kinase (PI3-kinase) are expressed in neurons in the rat brain. Biochem Biophys Res Commun. 1998;246:313–319. doi: 10.1006/bbrc.1998.8606. [DOI] [PubMed] [Google Scholar]
- 37.Thandi S, Blank JL, Challiss RA. Group I metabotropic glutamate receptors, mGluR1a and mGluR5 couple to extracellular signal-regulated kinase (ERK) activation via distinct, but overlapping, signaling pathways. J Neurochem. 2002;83:1139–1153. doi: 10.1046/j.1471-4159.2002.01217.x. [DOI] [PubMed] [Google Scholar]
- 38.Vanhoutte P, Barnier JV, Guibert B, Pages C, Besson MJ, Hipskind RA, Caboche J. Glutamate induces phosphorylation of Elk-1 and CREB, along with c-fos activation, via an extracellular signal-regulated kinase-dependent pathway in brain slices. Mol Cell Biol. 1999;19:136–146. doi: 10.1128/mcb.19.1.136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Vincent SR, Sebben M, Dumuis A, Bockaert J. Neurotransmitter regulation of MAP kinase signaling in striatal neurons in primary culture. Synapse. 1998;29:29–36. doi: 10.1002/(SICI)1098-2396(199805)29:1<29::AID-SYN3>3.0.CO;2-D. [DOI] [PubMed] [Google Scholar]
- 40.Wang JQ, Fibuch EE, Mao L. Regulation of mitogen-activated protein kinases by glutamate receptors. J Neurochem. 2007;100:1–11. doi: 10.1111/j.1471-4159.2006.04208.x. [DOI] [PubMed] [Google Scholar]
- 41.Wang H, Reiser G. The role of the Ca2+-sensitive tyrosine kinase Pyk2 and Src in thrombin signalling in rat astrocytes. J Neurochem. 2003;84:1349–1357. doi: 10.1046/j.1471-4159.2003.01637.x. [DOI] [PubMed] [Google Scholar]
- 42.Xia Z, Dudek H, Miranti CK, Greenberg ME. Calcium influx via the NMDA receptor induces immediate early gene transcription by a MAP kinase/ERK-dependent mechanism. J Neurosci. 1996;16:5425–5436. doi: 10.1523/JNEUROSCI.16-17-05425.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Yang L, Mao L, Chen H, Catavsan M, Kozinn J, Arora A, Liu X, Wang JQ. A signaling mechanism from Gαq-protein-coupled metabotropic glutamate receptors to gene expression: role of the c-Jun N-terminal kinase pathway. J Neurosci. 2006;26:971–980. doi: 10.1523/JNEUROSCI.4423-05.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.York RD, Molliver DC, Grewal SS, Stenberg PE, McCleskey EW, Stork PJ. Role of phosphoinositide 3-kinase and endocytosis in nerve growth factor-induced extracellular signal-regulated kinase activation via Ras and Rap1. Mol Cell Biol. 2000;20:8069–8083. doi: 10.1128/mcb.20.21.8069-8083.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Zanassi P, Paolillo M, Feliciello A, Avvedimento EV, Gallo V, Schinelli S. cAMP-dependent protein kinase induces cAMP-response element-binding protein phosphorylation via an intracellular calcium release/ERK-dependent pathway in striatal neurons. J Biol Chem. 2001;276:11487–11495. doi: 10.1074/jbc.M007631200. [DOI] [PubMed] [Google Scholar]