

Abstract
Mortality after influenza is often due to secondary bacterial pneumonia with Streptococcus pneumoniae, particularly in the elderly. The reasons for the high fatality rate seen with this disease are unclear. To further characterize the pathogenesis of pneumonia after influenza in a mouse model, we examined the pathology and immunology that leads to fatal infection. Influenza-infected mice were either euthanized 24 h after secondary infection with S. pneumoniae for determination of pathology, bacterial cultures, and levels of immune effectors or were followed by use of a live imaging system for development of pneumonia. Influenza-infected mice challenged with each of 3 serotypes of pneumococcus developed a severe, necrotic pneumonia and met endpoints for euthanasia in 24 to 60 h. Strikingly elevated levels of both pro- and anti-inflammatory molecules including interleukins 6 and 10, macrophage inflammatory protein 1α, and chemokine KC were present in the blood. High levels of these cytokines and chemokines as well as tumor necrosis factor α, interleukin 1β, and heme oxygenase 1 were present in the lungs, accompanied by a massive influx of neutrophils. Mortality correlated with the development of pneumonia and lung inflammation but not with bacteremia. This model has the potential to help us understand the pathogenesis of severe lung infections.
Streptococcus pneumoniae is a leading cause of community-acquired pneumonia and sepsis in both adults and children.7,18,40 Bacterial pneumonia is a frequent complication of influenza, particularly in the elderly, accounting for approximately 25% of the 36,000 influenza-related deaths that occur in an average season.30 Pneumococcus is the most common bacterial pathogen to complicate influenza in both adults and children. Despite effective antimicrobials and advanced intensive care, the mortality rate from complicated pneumococcal pneumonia is 13%.6,8,22,24 Influenza may predispose patients to more severe manifestations of pneumonia, such as multilobar involvement and bacteremia, thus contributing to the high fatality rate.13,28 The reasons for our inability to effectively treat this manifestation of disease are unclear.
We previously developed mouse models of both secondary bacterial pneumonia and sepsis after influenza.15,17 Mice infected with influenza virus followed 7 d later by infection with S. pneumoniae at low doses gradually meet endpoints for euthanasia due to secondary bacterial pneumonia over 4 to 7 d. Mice typically are not bacteremic at the time the experiment is halted, but effective mortality can be as high as 100%.14,15 When high doses of infectious agents are used in the model, all mice reach euthanasia endpoints within 48 h of the secondary challenge,17 when they have high-grade bacteremia and a severe, necrotic pneumonia. Use of luciferase-expressing pneumococci and sequential imaging of live animals has allowed further characterization of the model.14 Antibiotic treatment of pneumonia is able to clear bacteria from the lungs of mice but does not reduce mortality.13 In many ways this model resembles the clinical and histopathologic picture of acute respiratory distress syndrome, in which the lungs are severely affected, and progression of disease may continue after elimination of the pathogen.38 This model may be a better way to study the role of the immune system in pneumonia than are animal models that use large doses of endotoxin or bacteria to induce lung injury. Therefore, we sought to further characterize the pathology and immunology of influenza-infected mice with severe secondary bacterial pneumonia and sepsis.
Materials and Methods
Infectious agents
The Mount Sinai strain of mouse-adapted influenza virus A/Puerto Rico/8/34 (H1N1), hereafter referred to as PR8, was grown in Madin-Darby canine kidney cells from a stock in the influenza virus repository at St Jude Children's Research Hospital (Memphis, TN). S. pneumoniae strains D39 (type 2), A66.1 (type 3), and Norway T4 (type 4) were kind gifts of Dr Elaine Tuomanen (St Jude Children's Research Hospital). All 3 strains were transformed with the lux operon as described (Kevin Francis and Jun Yu, Xenogen Corporation, Alameda, CA).9 The dose lethal for 50% of mice (MLD50) was identical for each pair of wild-type and luciferase-expressing strains.
Mice
We used 8-week-old female Balb/cJ mice (Mus musculus, Jackson Laboratory, Bar Harbor, ME) in these studies. Mice were housed in high-temperature polycarbonate cages (31.2 cm × 23.5 cm × 15.2 cm) with isolator lids and maintained on a 12:12-h light:dark cycle at 22 ± 2 °C and a humidity of 50% in groups of 4 to 6 mice per cage. Prior to treatment, animals were allowed to acclimate to the animal facility for at least 7 d. Laboratory Autoclavable Rodent Diet (PMI Nutrition International, St Louis, MO) and autoclaved water were available ad libitum. Mice were monitored by serologic testing for specific endo- and ectoparasites including Sendai virus, pneumonia virus of mice, mouse hepatitis virus, Theiler murine encephalomyelitis virus, Mycoplasma pulmonis, mouse parvovirus, lymphocytic choriomeningitis virus, minute virus of mice, reovirus, ectromelia virus, epizootic diarrhea of infant mice virus, and mouse cytomegaly virus, and the results of monitoring were negative during these studies. In addition, mice were monitored by culture for identification of deleterious pathogens. All experimental procedures were done under general anesthesia with 2.5% inhaled isoflurane (Baxter Healthcare Corporation, Deerfield, IL). Animals used in this study were cared for in accordance with the Guide for the Care and Use of Laboratory Animals20 under an approved protocol from the Animal Care and Use Committee of St Jude Children's Research Hospital. All work with animals was carried out in Biosafety Level 2 facilities.
Procedures
Infectious model
Infectious agents were diluted in sterile phosphate-buffered saline (PBS) and administered intranasally in a volume of 100 μl (50 μl per nostril) to anesthetized mice held in an upright position. Groups of 6 to 10 mice were weighed and assessed at least daily for illness and mortality. Mice that were moribund (that is, alive but lacking purposeful movement after stimulation) were euthanized and considered to have died on that day. Influenza virus was given at a dose equivalent to 1000 doses infectious for 50% of tissue culture wells (TCID50) per mouse (equivalent to 0.67 MLD50), followed 7 d later by pneumococcal challenge with 1 × 105 colony-forming units (CFU) per mouse (0.2 MLD50). These high doses led to overwhelming infection and rapid fulfillment of euthanasia endpoints in an earlier study.17 Care was taken to ensure all mice received equal anesthesia during the course of the experiments.
Imaging of live mice
Mice were anesthetized lightly with 2.5% inhaled isoflurane and imaged for 20 s using an IVIS charge-coupled device camera (Xenogen Corporation, Alameda, CA) twice daily, starting 4 h after pneumococcal challenge. Total photon emission from selected and defined areas within the images of each mouse was quantified by use of the LivingImage version 2.20 software package (Xenogen Corporation) as described14 and expressed as the flux of relative light units per min.
Histopathologic examination
At 24 and 48 h after secondary challenge, lungs, brain, heart, liver, and kidneys were removed immediately after euthanasia (2 mice per group per time point). Lungs were not sufflated prior to fixation. Following 24 h fixation, organs were embedded in paraffin, sectioned at 5 μm, and stained with hematoxylin and eosin. At least 6 sections per organ were examined microscopically for histopathologic alterations by an experienced veterinary pathologist (JER), who was blinded regarding the composition of the groups. Lungs were examined to determine the percentage of lung parenchyma with inflammation and whether the airways (bronchioles, alveolar ducts) contained inflammatory cells, epithelial necrosis, or epithelial hyperplasia.
Measurement of cytokines and chemokines
Whole blood collected 24 h after bacterial infection was allowed to separate overnight then centrifuged at 5000 × g for 10 min to recover serum, which was frozen at −20 °C. Lung homogenates collected at the same time point as blood were centrifuged at 10,000 × g for 5 min and the supernatants frozen. Concentrations of interleukin (IL) 1β, IL6, IL10, and chemokines KC, macrophage inflammatory (MIP) 1α, and tumor necrosis factor (TNF) α in serum and lung tissue were measured by using the Mouse 17-Plex cytokine assay (Bio-Rad Laboratories, Hercules, CA), which was read on a Luminex 100 reader (Luminex, Austin, TX) according to the manufacturer's instructions. Samples were diluted 1:4 and run in duplicate in all assays.
Bronchoalveolar lavage (BAL) for cell counts
After euthanasia by CO2 inhalation, the trachea was exposed and cannulated with a 18-gauge plastic catheter (BD Insyte, Becton Dickinson, Sandy, UT). Lungs were lavaged thrice with 1 ml of cold, sterile PBS. Cell counts were done manually by standard methods using a hemocytometer and expressed as number of cells per ml of recovered fluid. BAL fluid was placed on glass slides and stained (Protocol Wright-Giemsa stain, Fisher Diagnostics, Middletown, VA) according to the manufacturer's instructions for manual determination of cell differentials.
Measurement of heme oxygenase 1 (HO1) levels
Whole blood and multiple organs including brain, lung, heart, liver, and kidney were collected 24 h after secondary challenge. Peripheral blood mononuclear cells were separated by centrifugation using Histopaque 1077 (Sigma Diagnostics, St Louis, MO), RNA was isolated from them, and RNA samples were frozen at −80 °C as described.29 RNA was extracted by use of the RNeasy kit (Qiagen, Chatsford, CA) from solid organs, which had been flash-frozen in liquid nitrogen and manually homogenized immediately after necropsy. A real-time polymerase chain reaction assay for HO-1 was used as previously described29 to determine levels of HO-1 expression in mononuclear cells and solid organs, by use of the ABI PRISM 7900 HT Sequence Detection System, the TaqMan One-Step RT-PCR Master Mix Reagent Kit, and the ABI PRISM 7900 HT SDS 2.0 software program (all from Applied Biosystems [Foster City, CA]) . Data are expressed as induction relative to mock-infected, PBS-challenged control mice.
Cultures of lung and blood
Mice were anesthetized lightly with 2.5% inhaled isoflurane, and approximately 100 μl of blood was obtained from each animal via retroorbital puncture with polished, sterile, glass Pasteur pipettes 24 h after infection with pneumococcus and transferred into Isolator 1.5 microbial tubes (Wampole Laboratories, Cranbury, NJ). Anesthetized mice then were euthanized by inhalation of CO2. Preliminary experiments confirmed that compared with cervical dislocation, euthanasia by CO2 does not to interfere with determination of viral titers. Lungs were removed under sterile conditions, washed 3 times in sterile PBS, and placed into 500 μl of sterile PBS. Quantitation of pneumococcal colony counts from blood or from lung homogenates was done by 10-fold serial dilutions on tryptic soy agar plates supplemented with 3% (v/v) sheep erythrocytes.
Statistical analysis
Survival between groups of mice was compared with the log rank test on Kaplan-Meier survival data. Cytokine, chemokine, and HO-1 levels between groups were compared by analysis of variance with Dunn correction. A P value of less than 0.05 was considered to indicate statistical significance in these comparisons. SigmaStat for Windows (SysStat Software, Inc., V3.11) was utilized for all statistical analyses.
Results
Histopathology
To better define the physiology of the fatal infection in this model, we examined multiple organs for pathologic changes. As described before,15,17 the primary pathology was in the lung (Figure 1). The lungs of mice infected with influenza followed by type 2 pneumococcus D39 were characterized by a severe, diffuse pneumonia consisting of bronchiolar and alveolar necrosis, associated with an inflammatory cell infiltrate consisting of mononuclear cells admixed with a preponderance of neutrophils, fibrin, and edema (Figure 1 B, D). The pneumonic process extended to the pleura, resulting in marked pleuritis. The degree of involvement of the parenchyma was greater than 75% at 24 h and approximately 90% at 48 h. Both hearts collected at 24 h had focal epicarditis, likely a result of extension from pericarditis due to the adjacent pleuritis. Other organs were essentially normal.
Figure 1.

Groups of mice were mock-infected with PBS or infected with influenza virus PR8 and then challenged 7 d later with pneumococcus strain D39 or mock-challenged with PBS. Lungs of representative control (A, C) and dual-infected (B, D) mice. The bronchioles (b) and alveoli in the control lung (A, C) do not have any inflammatory cells. In the dual-infected mouse (B, D), a consolidated necrotizing suppurative bronchopneumonia has replaced the normal architecture of the lung. The bronchioles (b with arrowhead) and alveoli are filled with inflammatory cells. In the control mouse (C) the alveolar walls (septa) are intact, and the alveolar air spaces are free of inflammatory cells. In the dual-infected mouse (D), the walls of the alveoli have been destroyed, and the air spaces are filled with neutrophils, mononuclear cells, and cellular debris. Compared with the control bronchiole in (C), the bronchiole in (D) contains neutrophils, mononuclear cells, and cellular debris, and there is hyperplasia and hypertrophy of the bronchiolar epithelium. Magnification, ×4 (A, B); ×20 (C, D)
By comparison, mice mock-infected with PBS had normal findings in all organs (Figure 1 A, C). Mice infected with influenza and then mock-challenged with PBS had multiple small foci of consolidation with bronchiolar epithelial necrosis, alveolar cell hyperplasia, and inflammatory cell infiltrates of mononuclear cells and neutrophils. The degree of involvement was less than 25% at both 24 and 48 h. Other organs were normal. Mice mock-infected with PBS and then challenged with pneumococcus had small foci of mixed inflammatory cells in a perivascular distribution in the lungs, but the degree of involvement was 0% to 10% at both 24 and 48 h. No other noteworthy pathology was seen.
Cytokines and chemokines
To further understand the pathogenesis of the mortality observed during secondary bacterial infection after influenza, we measured a broad array of cytokines and chemokines in the lungs and blood of infected mice. Levels in the lungs and blood of virally infected mice 8 d after infection (24 h after mock-challenge with PBS) were similar to those of controls that received only PBS (Figure 2). As compared with those of mock-infected animals, the lungs and blood of mice infected with the pneumococcal type 2 strain D39 demonstrated significant (P < 0.05) increases in IL6, a cytokine with both pro- and anti-inflammatory properties, and KC, a mouse CXC chemokine that is a homolog of IL-8 and a potent neutrophil chemoattractant. The lungs of mice sequentially infected with influenza virus and then pneumococcus D39 showed striking increases in IL6, the anti-inflammatory cytokine IL10, KC, and the CC macrophage chemotactin MIP1α along with significantly (P < 0.05) elevated values of the proinflammatory cytokines TNFα and IL1β, compared with the other groups (Figure 2 A). In the blood of dually infected mice, similar striking elevations of IL6, IL10, KC, and MIP1α were seen (Figure 2 B). Similar cytokine and chemokine data were seen when the type 3 strain A66.1 was used in the model and when BAL fluid was used instead of lung tissue (data not shown).
Figure 2.

Mean cytokine and chemokine levels in lungs and blood in secondary bacterial infection. Groups of 6 to 8 mice were mock-infected with PBS or infected with influenza virus PR8 and then challenged 7 d later with pneumococcus strain D39 or mock-challenged with PBS. (A) Lung homogenates and (B) serum were collected 24 h after secondary challenge and assayed for immune mediators as indicated. *, P < 0.05 (analysis of variance) versus other groups; **, P < 0.05 (analysis of variance) versus groups mock-infected or infected with virus only. Error bar, 1 standard deviation.
BAL cell counts
Because 2 of the molecules that increased most dramatically were chemokines, we assessed the magnitude of the systemic and local inflammatory response to determine whether leukocyte recruitment contributed to the pathogenesis of the infection. The peripheral white blood cell count was not increased in influenza-infected mice with secondary bacterial infections (4.3 × 103 cells/ml for dually infected mice, compared with 5.7 × 103, 5.6 × 103, and 3.2 × 103 cells/ml for mice mock-infected or given virus or bacteria only, respectively). In BAL fluid, however, the total cell count was significantly (P < 0.05) increased in the dually infected group (Figure 3 A). This increase was mainly due to a large increase in the number of neutrophils in the lung (Figure 3 B); monocyte–macrophage levels were essentially unchanged, and lymphocyte levels were lower than in mock- or singly-infected mice (data not shown).
Figure 3.

Cell counts from bronchoalveolar lavage (BAL) fluid during secondary bacterial infection. Mice were infected with 1000 TCID50 of influenza virus or mock-infected with PBS and then challenged 7 d later with either PBS or 1 × 105 CFU of pneumococcus D39 (serotype 2). The average number of (A) leukocytes and (B) neutrophils in the BAL fluid of 4 mice per group is shown. *, P < 0.05 (analysis of variance with Dunn correction) versus other groups. Error bar, 1 standard deviation
Tissue levels of HO1
We next assayed the anti-inflammatory molecule HO1 because of its association with sepsis and severe lung infections. Concentrations of HO1 were significantly (P < 0.05) greater in the lungs of dually infected mice compared with all other groups (Figure 4). No other sites showed increases in HO1. Serum bilirubin, which is induced through the action of HO1, was 1.9 mg/dl in dually infected mice, as compared with 0.9 mg/dl in mice infected with bacteria only, 0.5 mg/dl in mock-infected animals, and 1.3 mg/dl in virus-infected mice.
Figure 4.

Heme oxygenase 1 (HO1) induction during secondary bacterial infection. Mice were infected with 1000 TCID50 of influenza virus or mock infected with PBS and then challenged 7 d later with either PBS or 1 × 105 CFU of pneumococcus D39 (serotype 2). RNA was extracted from tissues and peripheral blood mononuclear cells (PBMCs) removed from groups of 5 to 7 mice 24 h after secondary challenge and assayed for heme oxygenase 1 by real-time PCR. The relative induction of heme oxygenase 1 is compared with that of mock-infected animals, whose average value was set at 1. *, P < 0.05 (analysis of variance with Dunn correction) versus other groups. Error bar, 1 standard deviation
Strain-dependent differences in secondary infection
Because both bacteremia and pneumonia occur in the model,17 the rapid death seen during secondary infection with pneumococcal strain D39 may have been due to sepsis and vascular collapse or to acute lung injury and inflammation. To distinguish between these possible mechanisms, we used 3 pneumococcal strains representing 3 serotypes that show distinct patterns of infection in mice. The type 3 strain A66.1 is known to cause pneumonia but not bacteremia, and the type 4 strain T4 causes a primary bacteremia, with pneumonia occurring only secondary to seeding of the lungs after systemic spread.21 Similar to previous reports,14,15 all mice met endpoints for euthanasia within 60 h after challenge with the type 2 strain D39 administered at 7 d after influenza infection, compared with only 33% requiring euthanasia when virus infection was followed by mock challenge (Figure 5). In an identical group of 6 mice, blood cultures were positive at 24 h in 5 of 6 mice, with mean titers of 2.8 × 105 CFU/ml. Cultures of lung homogenate were positive in all 6 mice, with mean titers of 3.2 × 107 CFU/ml. The lungs were the primary focus of infection as demonstrated by serial imaging of infected mice (Figure 6).
Figure 5.
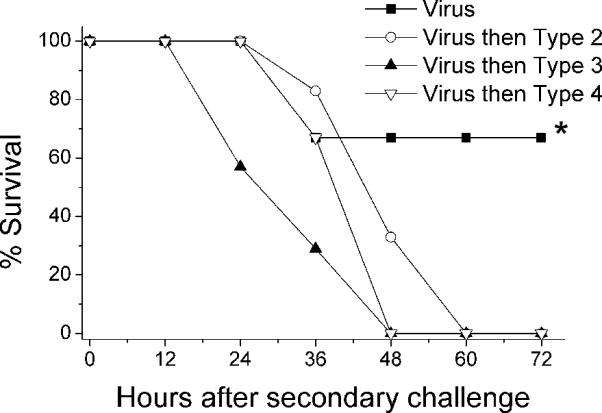
Survival of influenza-infected mice after secondary challenge with Streptococcus pneumoniae. Groups of 6 to 7 mice were infected with 1000 TCID50 of influenza virus and then challenged 7 d later with either PBS (closed squares) or 1 × 105 CFU of pneumococcus D39 (serotype 2, open circles), A66.1 (serotype 3, closed triangles), or T4 (serotype 4, open triangles). *, P < 0.05 (Mantel-Cox χ2) versus other groups.
Figure 6.

Visualization of pneumonia and bacteremia in influenza-infected mice after secondary challenge with Streptococcus pneumoniae. Groups of 6 mice were infected with 1000 TCID50 of influenza virus and then challenged 7 d later with 1 × 105 CFU of pneumococcus D39 (serotype 2), A66.1 (serotype 3), or T4 (serotype 4). Pneumococci had been transformed with luciferase so that they would emit light. Sequential images of live, anesthetized mice were taken every 12 h starting 4 h after secondary challenge. Images shown are from 1 representative mouse per group; the mean relative light units (RLU) detected per min for the group is given below the image. The scale indicates relative light units per pixel.
Infection with the type 3 pneumococcus A66.1 also led to pneumonia (Figure 6) and rapid fulfillment of criteria for euthanasia (Figure 5), but bacteremia was not a prominent feature. All lung cultures were positive (mean titer, 1.7 × 106 CFU/ml), but only 1 of 6 blood cultures was positive (30 CFU/ml). Challenge with the type 4 strain T4 also caused a fatal pneumonia after influenza virus infection (Figures 5, 6). As seen with the type 2 strain, which caused pneumonia and bacteremia, all 6 blood cultures and all 6 lung cultures were positive. However, the mean titers of 1.9 × 104 CFU/ml in blood and 6.8 × 104 CFU/ml in lungs were lower than those in animals infected with other serotypes.
Discussion
In our initial description of this model, mice infected sequentially with influenza and then pneumococcus appeared to develop sepsis, with high-grade bacteremia and rapid demise.17 Further characterization of the model indicates that the pathogenesis of the fatal infections is a massive pneumonia with accompanying local and systemic inflammation. Mice developed severe pneumonia and succumbed rapidly to infection, independent of the presence and degree of bacteremia and regardless of the pneumococcal serotype used. Massive cytokinemia was present, with strikingly increased levels of chemoattractant chemokines and anti-inflammatory cytokines. Concentrations of typical proinflammatory cytokines (TNFα and IL1β) were increased in lung but not blood, consistent with localization of inflammation to this site. Although other organs were essentially unaffected, the lungs of the mice resembled those of patients with acute respiratory distress syndrome, with edema, endothelial and epithelial injury, neutrophil influx, necrosis, and presence of debris.
S. pneumoniae expresses a variety of virulence factors, one of which is the polysaccharide capsule.16 The capsule contributes to invasion and helps bacteria survive in the bloodstream by inhibiting opsonophagocytosis.1,33 We evaluated 3 S. pneumoniae strains with different capsular serotypes and well-characterized differences in the spectrum of disease they cause in mice.21 The type 2 strain that was used in the initial reports, D39,15,17 causes high-grade bacteremia and sepsis when administered to mice, typically without significant involvement of the lung. The type 3 strain A66.1 causes pneumonia without bacteremia.21 The Norway Type 4 strain causes a low-grade bacteremia which typically is not fatal unless it progresses to meningitis; lung infection is an unusual manifestation of disease. In our model of secondary bacterial infection after influenza, all 3 strains caused a severe primary pneumonia of approximately equal intensity (Figure 6), and all mice progressed rapidly to meet endpoints for euthanasia (Figure 5). Mortality did not correlate with bacteremia, because mice secondarily infected with type 2 strain D39 had high-grade bacteremia, type 4 strain T4 had low-grade bacteremia, and type 3 strain A66.1 lacked bacteremia. Therefore, preinfection with influenza virus primes the lung in mice such that the end result of infection is the same, over-riding strain-specific differences among isolates.
The striking synergistic increases of systemic cytokines and chemokines probably contributed to the pathogenesis of these infections. Although the induction of multiple diverse effector molecules, including proinflammatory cytokines and chemokines, is not surprising during such massive infection,2, 23 the magnitudes of increases in IL10, IL6, KC, and MIP1α were unexpected. KC and MIP1α are chemoattractant molecules that enhance recruitment of neutrophils and macrophages to sites of inflammation.4 Both chemokines contribute to the severity of infection by aiding the influx of neutrophils into the lung,10,32 where these activated leukocytes may mediate acute inflammatory tissue damage.39 Histopathologic evaluation revealed accumulation of neutrophils with accompanying tissue destruction and necrosis in the lungs and BALs of dually infected mice in the present study. IL10 is a cytokine that has pleiotropic (but generally anti-inflammatory) effects on the inflammatory response. The effect of IL10 is expressed mainly through decreased or reduced levels of various proinflammatory mediators, including chemokines, TNFα, and IL6.5,12 This negative regulation of the proinflammatory response has deleterious effects on the ability of the host to survive pneumococcal pneumonia.35,36 IL6, in the context of pneumococcal infection, has both pro- and anti-inflammatory effects. Compared with their wild-type littermates, mice deficient in IL-6 succumb more rapidly to pneumococcal pneumonia in association with increased TNFα, IL1β, and IL10.34 However, pneumolysin-dependent influx of neutrophils into the lungs was attenuated in similar studies.26 An emerging concept in the study of severe infections is that a balance of anti- and proinflammatory activity is necessary for resolution of infection and survival.3 The exaggerated responses seen in our model are dysfunctional, and both anti- and proinflammatory effectors are likely responsible for the severity of the disease and the rapid death, with pro-inflammatory molecules contributing to tissue damage and anti-inflammatory mediators interfering with the ability of the host to clear the inciting organisms.
HO1 is an inducible enzyme that is involved in heme metabolism and has anti-inflammatory activity.37 HO1 catalyzes the rate-limiting step in degradation of heme to iron, carbon monoxide, and biliverdin. The anti-inflammatory effects of HO1 may be mediated by the production of the antioxidant bilirubin from biliverdin and of carboxyhemoglobin from the binding of carbon monoxide to hemoglobin.27 We measured HO1 in the present study because the enzyme is increased in severe inflammatory disease states such as acute respiratory distress syndrome and systemic inflammatory response syndrome.19,29 In our mouse model, HO1 was significantly increased in the lungs of mice with secondary bacterial pneumonia, and its downstream product, bilirubin, was elevated in the blood. Because HO1 is induced by a variety of stimuli, including hypoxia, IL6, IL10, and other molecules that contribute to oxidative stress,25 its increase in this model was not surprising. However, these preliminary studies do not indicate whether this increase is a protective response to inflammation11 or contributes to toxicity through the production of heme, as has been suggested.31,37 Further investigation measuring carboxyhemoglobin, biliverdin, ferritin, and free heme is warranted.
Our additional characterization of a mouse model of secondary bacterial infection after influenza suggests that the lung is the primary focus of infection, and poor outcomes are linked to unbridled inflammation at this site. Regardless of the strain of S. pneumoniae tested, the primary manifestation of disease was a severe pneumonia that was rapidly fatal. Exaggerated systemic and local inflammatory responses characterized by elevations of both pro- and anti-inflammatory effector molecules occurred and may provide a basis for further investigation of the innate immune response during severe lung infection and therapeutic approaches.
Acknowledgments
This work was supported by grants AI-49178 and AI-54802 from the National Institutes of Health and by the American Lebanese Syrian Associated Charities (ALSAC). We thank Christine Dietz for technical assistance.
Abbreviations
- IL
interleukin
- MIP
macrophage inflammatory protein
- MLD50
dose lethal for 50% of mice inoculated
- PBS
phosphate-buffered saline
- TCID50
dose infectious for 50% of culture wells treated
- TNF
tumor necrosis factor
References
- 1.Avery OT, Dubos R. The protective action of a specific enzyme against type III pneumococcus infections in mice. J Exp Med. 1931;54:73–89. doi: 10.1084/jem.54.1.73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Bhatia M, Moochhala S. Role of inflammatory mediators in the pathophysiology of acute respiratory distress syndrome. J Pathol. 2004;202:145–156. doi: 10.1002/path.1491. [DOI] [PubMed] [Google Scholar]
- 3.Bone RC. Sir Isaac Newton, sepsis, SIRS, and CARS. Crit Care Med. 1996;24:1125–1128. doi: 10.1097/00003246-199607000-00010. [DOI] [PubMed] [Google Scholar]
- 4.Chensue SW. Molecular machinations: chemokine signals in host-pathogen interactions. Clin Microbiol Rev. 2001;14:821–835. doi: 10.1128/CMR.14.4.821-835.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Donnelly RP, Dickensheets H, Finbloom DS. The interleukin-10 signal transduction pathway and regulation of gene expression in mononuclear phagocytes. J Interferon Cytokine Res. 1999;19:563–573. doi: 10.1089/107999099313695. [DOI] [PubMed] [Google Scholar]
- 6.Dowling HF, Lepper MH. The effect of antibiotics (penicillin, aureomycin, and terramycin) on the fatality rate and incidence of complications in pneumococcic pneumonia—a comparison with other methods of therapy. Am J Med Sci. 1951;222:396–403. doi: 10.1097/00000441-195110000-00005. [DOI] [PubMed] [Google Scholar]
- 7.Fedson DS, Anthony J, Scott G. The burden of pneumococcal disease among adults in developed and developing countries: what is and is not known. Vaccine. 1999;17(Suppl 1):S11–S18. doi: 10.1016/s0264-410x(99)00122-x. [DOI] [PubMed] [Google Scholar]
- 8.Feikin DR, Schuchat A, Kolczak M, Barrett NL, Harrison LH, Lefkowitz L, McGeer A, Farley MM, Vugia DJ, Lexau C, Stefonek KR, Patterson JE, Jorgensen JH. Mortality from invasive pneumococcal pneumonia in the era of antibiotic resistance, 1995−1997. Am J Public Health. 2000;90:223–229. doi: 10.2105/ajph.90.2.223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Francis KP, Yu J, Bellinger-Kawahara C, Joh D, Hawkinson MJ, Xiao G, Purchio TF, Caparon MG, Lipsitch M, Contag PR. Visualizing pneumococcal infections in the lungs of live mice using bioluminescent Streptococcus pneumoniae transformed with a novel gram-positive lux transposon. Infect Immun. 2001;69:3350–3358. doi: 10.1128/IAI.69.5.3350-3358.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Haeberle HA, Kuziel WA, Dieterich HJ, Casola A, Gatalica Z, Garofalo RP. Inducible expression of inflammatory chemokines in respiratory syncytial virus-infected mice: role of MIP-1α in lung pathology. J Virol. 2001;75:878–890. doi: 10.1128/JVI.75.2.878-890.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Kapturczak MH, Wasserfall C, Brusko T, Campbell-Thompson M, Ellis TM, Atkinson MA, Agarwal A. Heme oxygenase-1 modulates early inflammatory responses: evidence from the heme oxygenase-1-deficient mouse. Am J Pathol. 2004;165:1045–1053. doi: 10.1016/S0002-9440(10)63365-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Lang R, Patel D, Morris JJ, Rutschman RL, Murray PJ. Shaping gene expression in activated and resting primary macrophages by IL-10. J Immunol. 2002;169:2253–2263. doi: 10.4049/jimmunol.169.5.2253. [DOI] [PubMed] [Google Scholar]
- 13.McCullers JA. Effect of antiviral treatment on the outcome of secondary bacterial pneumonia after influenza. J Infect Dis. 2004;190:519–526. doi: 10.1086/421525. [DOI] [PubMed] [Google Scholar]
- 14.McCullers JA, Bartmess KC. Role of neuraminidase in lethal synergism between influenza virus and Streptococcus pneumoniae. J Infect Dis. 2003;187:1000–1009. doi: 10.1086/368163. [DOI] [PubMed] [Google Scholar]
- 15.McCullers JA, Rehg JE. Lethal synergism between influenza virus and Streptococcus pneumoniae: characterization of a mouse model and the role of platelet-activating factor receptor. J Infect Dis. 2002;186:341–350. doi: 10.1086/341462. [DOI] [PubMed] [Google Scholar]
- 16.McCullers JA, Tuomanen EI. Molecular pathogenesis of pneumococcal pneumonia. Front Biosci. 2001;6:D877–D889. doi: 10.2741/mccullers. [DOI] [PubMed] [Google Scholar]
- 17.McCullers JA, Webster RG. A mouse model of dual infection with influenza virus and Streptococcus pneumoniae. In: Osterhaus ADME, Cox N, Hampson AW, editors. Options for the control of influenza IV. International Congress Series.; Amsterdam: 2001. pp. 601–607. [Google Scholar]
- 18.Michelow IC, Olsen K, Lozano J, Rollins NK, Duffy LB, Ziegler T, Kauppila J, Leinonen M, McCracken GH., Jr Epidemiology and clinical characteristics of community-acquired pneumonia in hospitalized children. Pediatrics. 2004;113:701–707. doi: 10.1542/peds.113.4.701. [DOI] [PubMed] [Google Scholar]
- 19.Mumby S, Upton RL, Chen Y, Stanford SJ, Quinlan GJ, Nicholson AG, Gutteridge JM, Lamb NJ, Evans TW. Lung heme oxygenase-1 is elevated in acute respiratory distress syndrome. Crit Care Med. 2004;32:1130–1135. doi: 10.1097/01.ccm.0000124869.86399.f2. [DOI] [PubMed] [Google Scholar]
- 20.National Research Council . Guide for the care and use of laboratory animals. National Academy Press; Washington (DC): 1996. [Google Scholar]
- 21.Orihuela CJ, Gao G, McGee M, Yu J, Francis KP, Tuomanen E. Organ-specific models of Streptococcus pneumoniae disease. Scand J Infect Dis. 2003;35:647–652. doi: 10.1080/00365540310015854. [DOI] [PubMed] [Google Scholar]
- 22.Pallares R, Linares J, Vadillo M, Cabellos C, Manresa F, Viladrich PF, Martin R, Gudiol F. Resistance to penicillin and cephalosporin and mortality from severe pneumococcal pneumonia in Barcelona, Spain. N Engl J Med. 1995;333:474–480. doi: 10.1056/NEJM199508243330802. [DOI] [PubMed] [Google Scholar]
- 23.Puneet P, Moochhala S, Bhatia M. Chemokines in acute respiratory distress syndrome. Am J Physiol Lung Cell Mol Physiol. 2005;288:L3–L15. doi: 10.1152/ajplung.00405.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Rahav G, Toledano Y, Engelhard D, Simhon A, Moses AE, Sacks T, Shapiro M. Invasive pneumococcal infections. A comparison between adults and children. Medicine (Baltimore) 1997;76:295–303. doi: 10.1097/00005792-199707000-00007. [DOI] [PubMed] [Google Scholar]
- 25.Ricchetti GA, Williams LM, Foxwell BM. Heme oxygenase 1 expression induced by IL-10 requires STAT-3 and phosphoinositol-3 kinase and is inhibited by lipopolysaccharide. J Leukoc Biol. 2004;76:719–726. doi: 10.1189/jlb.0104046. [DOI] [PubMed] [Google Scholar]
- 26.Rijneveld AW, van den Dobbelsteen GP, Florquin S, Standiford TJ, Speelman van Alphen L, van der PT. Roles of interleukin-6 and macrophage inflammatory protein-2 in pneumolysin-induced lung inflammation in mice. J Infect Dis. 2002;185:123–126. doi: 10.1086/338008. [DOI] [PubMed] [Google Scholar]
- 27.Ryter SW, Otterbein LE, Morse D, Choi AM. Heme oxygenase/carbon monoxide signaling pathways: regulation and functional significance. Mol Cell Biochem. 2002;234−235:249–263. doi: 10.1023/A:1015957026924. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Scadding JG. Lung changes in influenza. Quart J Med. 1937;6:425–465. [Google Scholar]
- 29.Schmidt JE, Morgan JI, Rodriguez-Galindo C, Webb DL, Liang H, Tamburro RF. Heme oxygenase-1 mRNA expression is induced in peripheral blood mononuclear cells of pediatric cancer patients with systemic inflammatory response syndrome. Pediatr Crit Care Med. 2004;5:554–560. doi: 10.1097/01.PCC.0000144709.87365.F0. [DOI] [PubMed] [Google Scholar]
- 30.Simonsen L. The global impact of influenza on morbidity and mortality. Vaccine. 1999;17(Suppl 1):S3–S10. doi: 10.1016/s0264-410x(99)00099-7. [DOI] [PubMed] [Google Scholar]
- 31.Suttner DM, Dennery PA. Reversal of HO-1-related cytoprotection with increased expression is due to reactive iron. FASEB J. 1999;13:1800–1809. doi: 10.1096/fasebj.13.13.1800. [DOI] [PubMed] [Google Scholar]
- 32.Tateda K, Moore TA, Newstead MW, Tsai WC, Zeng X, Deng JC, Chen G, Reddy R, Yamaguchi K, Standiford TJ. Chemokine-dependent neutrophil recruitment in a murine model of Legionella pneumonia: potential role of neutrophils as immunoregulatory cells. Infect Immun. 2001;69:2017–2024. doi: 10.1128/IAI.69.4.2017-2024.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Tuomanen E, Rich R, Zak O. Induction of pulmonary inflammation by components of the pneumococcal cell surface. Am Rev Respir Dis. 1987;135:869–874. doi: 10.1164/arrd.1987.135.4.869. [DOI] [PubMed] [Google Scholar]
- 34.van der Poll T, Keogh CV, Guirao X, Buurman WA, Kopf M, Lowry SF. Interleukin-6 gene-deficient mice show impaired defense against pneumococcal pneumonia. J Infect Dis. 1997;176:439–444. doi: 10.1086/514062. [DOI] [PubMed] [Google Scholar]
- 35.van der Poll T, Marchant A, Keogh CV, Goldman M, Lowry SF. Interleukin-10 impairs host defense in murine pneumococcal pneumonia. J Infect Dis. 1996;174:994–1000. doi: 10.1093/infdis/174.5.994. [DOI] [PubMed] [Google Scholar]
- 36.van der Sluijs KF, van Elden LJ, Nijhuis M, Schuurman R, Pater JM, Florquin S, Goldman M, Jansen HM, Lutter R, van der Poll T. IL-10 is an important mediator of the enhanced susceptibility to pneumococcal pneumonia after influenza infection. J Immunol. 2004;172:7603–7609. doi: 10.4049/jimmunol.172.12.7603. [DOI] [PubMed] [Google Scholar]
- 37.Wagener FA, Volk HD, Willis D, Abraham NG, Soares MP, Adema GJ, Figdor CG. Different faces of the heme–heme oxygenase system in inflammation. Pharmacol Rev. 2003;55:551–571. doi: 10.1124/pr.55.3.5. [DOI] [PubMed] [Google Scholar]
- 38.Ware LB, Matthay MA. The acute respiratory distress syndrome. N Engl J Med. 2000;342:1334–1349. doi: 10.1056/NEJM200005043421806. [DOI] [PubMed] [Google Scholar]
- 39.Weiss SJ. Tissue destruction by neutrophils. N Engl J Med. 1989;320:365–376. doi: 10.1056/NEJM198902093200606. [DOI] [PubMed] [Google Scholar]
- 40.WHO Young Infants Study Group Bacterial etiology of serious infections in young infants in developing countries: results of a multicenter study. Pediatr Infect Dis J. 1999;18:S17–S22. doi: 10.1097/00006454-199910001-00004. [4] [DOI] [PubMed] [Google Scholar]