

Abstract
Mammalian reoviruses infect respiratory and gastrointestinal epithelia and cause disease in neonates. Junctional adhesion molecule-A (JAM-A) is a serotype-independent receptor for reovirus. JAM-A localizes to tight junctions and contributes to paracellular permeability in polarized epithelia. To investigate the mechanisms of reovirus infection of polarized epithelial cells, we assessed reovirus replication, release, and spread after apical and basolateral adsorption to primary human airway epithelial cultures. Reovirus infection of human airway epithelia was more efficient after adsorption to the basolateral surface than after adsorption to the apical surface, and it was dependent on JAM-A. Reovirus binding to carbohydrate coreceptor sialic acid inhibited apical infection, which was partially ameliorated by treatment of the cultures with neuraminidase. Despite the preference for basolateral infection, reovirus was released from the apical surface of respiratory epithelia and did not disrupt tight junctions. These results establish the existence of an infectious circuit for reovirus in polarized human respiratory epithelial cells.
Epithelial surfaces in the respiratory and gastrointestinal tracts serve as the primary site of infection for many viruses [1]. Mucosal surfaces are composed primarily of polarized epithelial cells, but they also contain specialized cells, such as goblet and microfold (M) cells [2- 4] Polarized epithelial cells are knitted together by intercellular junctions that prevent nonspecific passage of solutes. Goblet cells secrete a protective mucus layer, beating cilia sweep mucus-trapped debris in a favorable direction for the host, and M cells immunologically sample the environment. Several pathogenic viruses have found ways to either circumvent or take advantage of epithelial features to achieve productive infection at these sites.
Mammalian reoviruses are nonenveloped, icosahedral viruses with a genome of 10 double-stranded RNA segments [5]. In healthy children and adults, reovirus infects epithelia in the respiratory and intestinal tracts and is shed from both sites. Infected individuals are either asymptomatic or experience mild respiratory or gastrointestinal illnesses. In contrast, reovirus infection in neonates may cause serious disease by means of systemic spread to secondary sites, such as the central nervous system. Differences in receptor-binding specificity among the reovirus serotypes are thought to modulate tropism for distinct types of cells [6, 7].
Reovirus attachment protein σ1 is a filamentous, trimeric molecule that exhibits head-and-tail morphology and extends from each of the 12 vertices of the reovirus capsid [8]. The σ1 tail interacts with carbohydrate moieties that differ among the reovirus serotypes [9]. For type 3 reoviruses, the carbohydrate coreceptor is sialic acid [10, 11]. The σ1 head interacts with the tight junction protein, junctional adhesion molecule-A (JAM-A), which is a receptor for all known reovirus serotypes [12, 13]. JAM-A is a type 1 transmembrane adhesion protein that belongs to the CTX (cortical thymocyte marker of Xenopus protein) family [14] of the immunoglobulin (Ig) superfamily. The extracellular domain of JAM-A is composed of 2 Ig-like domains and forms homodimers [15]. JAM-A is localized to endothelial and epithelial tight junctions and mediates paracellular permeability and tight junction resealing [16-18]. JAM-A also serves as a receptor for feline calicivirus [19].
In the gastrointestinal tract, reovirus gains entry via M cells and replicates in the lymphoid tissue of Peyer patches [20, 21]. Whereas most of the gastrointestinal epithelium is protected by a thick glycoprotein layer, M cells within Peyer patches display unique glycosylation patterns. Binding to cell-surface sialic acid appears to be required for reovirus infection of intestinal M cells [22]. Transcytosis into the basolateral region is hypothesized to result in subsequent infection of Peyer patches and epithelial cells or in lymphoid spread. Similar to its method of access to the gastrointestinal epithelium, reovirus can gain access to the respiratory tract via M cells [23].
In the present study, we investigated infection of primary, polarized, human airway epithelia by reovirus. We evaluated 2 monoreassortant reovirus strains that are isogenic except for the presence of a single polymorphism inσ1 that renders the virus capable (T3SA+) or incapable (T3SA-) of binding to sialic acid [11]; both strains bind to JAM-A [12]. We found that reovirus infects preferentially from the basolateral surface of human airway epithelia, consistent with the localization of JAM-A in polarized epithelial cells [16-18]. Surprisingly, reovirus binding to carbohydrate coreceptor sialic acid inhibits apical infection. Furthermore, reovirus is released apically without disrupting tight junctions. These findings enhance an understanding of how receptor utilization can modulate tropism and suggest that polarized airway epithelial cells are not the primary cell type responsible for spread from the respiratory tract during reovirus infection.
MATERIALS AND METHODS
Cells, viruses, and antibodies
Murine L929 (L) cells were obtained from American Type Culture Collection and were grown in Dulbecco's modified Eagle medium supplemented with 10% fetal calf serum, 100 U/mL penicillin, and 100 μg/mL streptomycin (Invitrogen). Primary human airway epithelia were isolated from the trachea and bronchi of donor lungs; seeded onto collagen-coated, semipermeable membranes (Millipore); and grown at the air-liquid interface as described elsewhere [24]. Approximately 2 weeks after seeding, cultures were well differentiated and contained ciliated, goblet, columnar, and basal cells. Transepithelial electrical resistance was evaluated using an Ohm meter (World Precision Instruments) and converted into conductance. T3/C44-SA- (T3SA-) and T3/C44MA-SA+ (T3SA+) have been previously described and contain the S1 gene segment from the T3 parental virus (T3SA+ binds sialic acid because of a single mutation (Leu204→Pro) within the σ1 tail), with all other gene segments derived from reovirus strain type 1 Lang (T1L) [11]. Purified virion preparations were made using second- or third-passage L-cell lysate stocks of reovirus that had been plaque purified twice [25].
JAM-A extracellular domain-specific monoclonal antibody J10.4 was provided by Charles Parkos. Zonula occludens-1 (ZO-1) antibodies were obtained from Zymed. Alexa-488- and Alexa-568- conjugated goat anti-mouse or anti-rabbit antibodies and TOPRO-3 were obtained from Molecular Probes. RmcB, a monoclonal antibody that was specific for the coxsackievirus and adenovirus receptor (CAR) (i.e., CRL-2379; American Type Cullure Collections) was produced by the University of Iowa Hybridoma Core, and rabbit anti-CAR polyclonal antisera have been described elsewhere [26].
Immunostaining and fluorescent focus assay (FFA)
Immunocytochemical analysis of human airway epithelia cultures or L cells was performed as described elsewhere [11, 26]. In brief, cultures were fixed with methanol/1% paraformaldehyde at -20°C and were blocked with 2% bovine serum albumin in SuperBlock (Pierce). After primary and secondary antibody treatment, nuclei were counterstained with ToPro-3, and coverslips were applied with Vectashield mounting media (Vector Laboratories). Images were acquired using a BioRad MRC-1024 laser scanning confocal microscopy mounted on a Nikon E600 microscope with a 60× oil-immersion lens.
Reovirus infection of human airway epithelia
Primary human airway epithelia were adsorbed apically with reovirus in 50 μL of Eagle minimum essential medium with Earle's salts (EMEM; Invitrogen) at various MOIs at room temperature for 1 h, washed once with PBS, and cultured in fresh media. For basolateral infections, epithelia were inverted and adsorbed with reovirus in 25 μL of EMEM at various MOIs at room temperature for 1 h. Virus was aspirated, and cells were cultured in fresh media. Infection was quantified by FFA performed for 2-3 independent cultures obtained from at least 2 independent experiments, with fluorescent cells counted in 20 random fields of view per epithelia [11]. Epithelia were treated with neuraminidase as described elsewhere [27]. Epithelia were preincubated at 37°C for 4 h with diluent alone or with neuraminidase from Vibrio cholerae (0.02 U/mL [Sigma]; α2,3 sialic acid linkages), Salmonella serotype Typhimurium (10 U/mL [Glyko]; α2,3,6,8,9 sialic acid linkages), or Arthrobactor ureafaciens (1 U/mL [Glyko]; all sialic acid linkages). The apical surfaces of the epithelia were then rinsed and adsorbed apically with T3SA+ or T3SA- at an MOI of 1000 for 1 h. For experiments assessing inhibition of reovirus infection, T3SA- was incubated with 20 μg/mL glutathione S-transferase (GST), soluble GST fused to the extracellular domain of JAM-A (GST-JAM-A), RmcB, or J10.4 at room temperature before basolateral virus adsorption.
Quantitation of reovirus growth
Apical washes using 0.2 mL of PBS and basolateral media from human airway epithelia infected with reovirus were obtained at various intervals and frozen before titration. L cells (105) grown in 24-well plates were adsorbed with reovirus at an MOI of 10-1000 pfu/cell or with serial 10-fold dilutions of epithelial samples. After viral adsorption at room temperature for 1 h, the inoculum was removed, cells were washed once with PBS, 1 mL of fresh medium was added, and cells were incubated at 37°C for 0, 24, or 48 h. After incubation, cells and culture media were frozen (at -70°C) and thawed twice, and viral titers in cell lysates were determined by plaque assay performed using L cell monolayers [25].
RESULTS
Localization of JAM-A to the basolateral surface of human airway epithelia
JAM-A is localized in the tight junction region of several cell types [16, 18, 28]. To define JAM-A localization in human airway epithelia, cultures were stained with monoclonal antibodies raised against JAM-A, tight junction marker ZO-1, or tight junction membrane protein CAR, and then they were counter-stained for nuclei. Confocal microscopic images at the level of the tight junctions (figure 1A-1C) and below the tight junctions at the level of the nuclei (figure 1D-1F) were obtained. ZO-1 staining was limited to the tight junctions in epithelial cultures, as described elsewhere [26]. JAM-A staining was restricted to the basolateral surface juxtaposed to ZO-1 (figure 1G), overlapping with CAR (figure 1H-1J), as well as to the adherens junctions, thereby indicating that polarized airway epithelial cells express JAM-A on basolateral, but not apical, surfaces.
Figure 1.
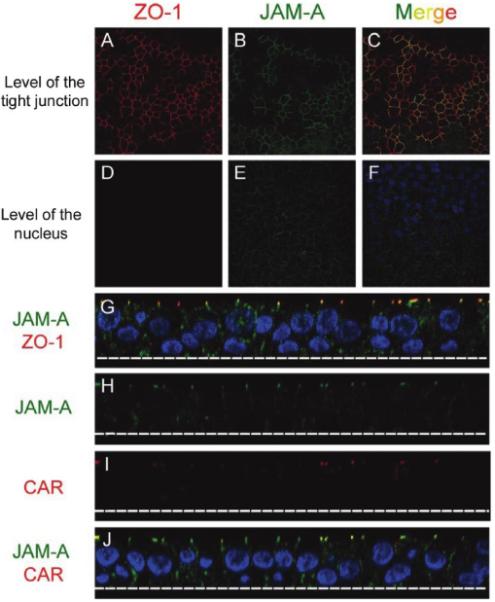
Localization of junctional adhesion molecule-A (JAM-A) in primary human airway epithelia. A-F, Representative x-y confocal microscopic sections of epithelia stained to detect JAM-A (green), zonula occludens-1 (ZO-1) (red), and nuclei (blue). Regions of overlap between JAM-A and ZO-1 are depicted in yellow (C). G-J, Representative x-z confocal microscopic sections of epithelia stained to detect JAM-A (green; G, H, and J), ZO-1 (red; G), the coxsackievirus and adenovirus receptor (CAR) (red; I and J), and nuclei (blue). Regions of overlap between JAM-A and ZO-1 (G) and JAM-A and CAR (J) are depicted in yellow.
Preferential infection of human airway epithelia with reovirus from the basolateral side
Basolateral restriction of viral receptors serves as a form of innate defense against viral infections of air-exposed epithelial surfaces. Considering the basolateral localization of JAM-A and the absence of M cells from cultured airway epithelia, we hypothesized that basolateral infection by reovirus would be more efficient than apical infection. Airway epithelia were adsorbed with sialic acid binding reovirus strain T3SA+ either apically or basolaterally at an MOI of 1, 10, 100, or 1000 pfu/cell. Infected cells were quantified 18 h after infection (figure 2A). As expected, the apical surface was highly resistant to infection. The basolateral surface was also surprisingly resistant; therefore, an MOI of 500 or 50 pfu/cell for apical or basolateral infections, respectively, was chosen to directly compare T3SA+ with T3SA-, which is the non-sialic acid- binding reovirus strain (figure 2B-2K). Despite the addition of 10-fold more virus to the apical surface, basolateral infection was significantly more efficient than apical infection (P < .001). These findings suggest that inefficient apical infection might result from failure of the virus to access JAM-A as a result of either its basolateral localization or the absence of M cells for mediation of initial infection. Furthermore, apical infection by T3SA- was approximately twice as efficient as that by T3SA+ (figure 2J and 2K). This finding suggests that carbohydrate-binding capacity inhibits reovirus infection of airway epithelia.
Figure 2.
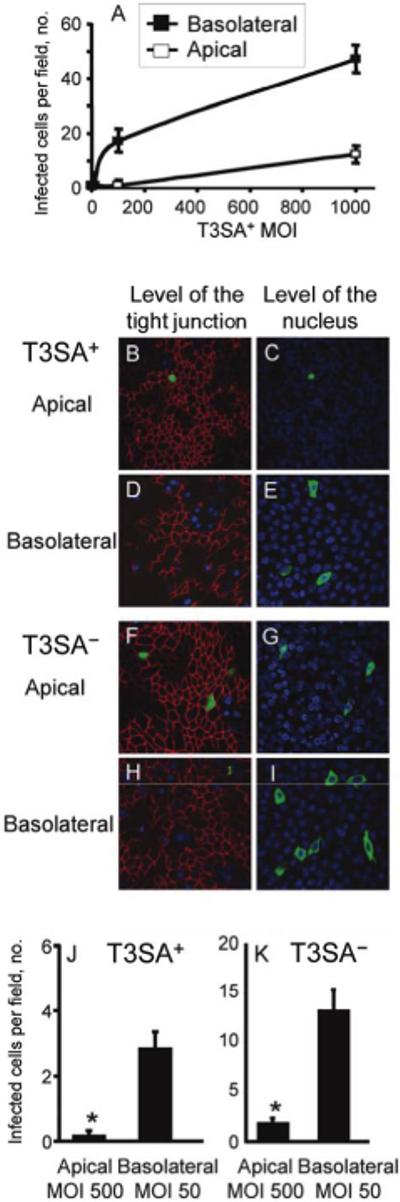
Preferential infection of human airway epithelia by reovirus from the basolateral side. Epithelia were adsorbed either apically or basolaterally with T3SA+ (a monoreassortant reovirus strain that is capable of binding to sialic acid) at an MOI of 1, 10, 100, or 1000 pfu/cell (A), and infected cells were quantified at 18 h after infection. T3SA+ and T3SA- (a monoreassortant reovirus strain that is incapable of binding to sialic acid) were compared from the apical side at an MOI of 500 pfu/cell (T3SA+, B and C; T3SA-, F and G) and from the basolateral side at an MOI of 50 pfu/cell (T3SA+, D and E; T3SA-, H and I) and were immunostained at 18 h after adsorption to detect reovirus proteins (green), zonula occludens-1 (ZO-1) (red), and nuclei (blue). T3SA+- infected cells (J) or T3SA--infected cells (K) were quantified in 20 random fields of view from 4 individual airway cultures. Error bars denote SDs (P < .001).
Basolateral infection of airway epithelia by reovirus via JAM-A
To confirm that infection from the basolateral surface is mediated by JAM-A, GST-JAM-A [13], control GST protein, J10.4, or control antibody directed against CAR (RmcB), a tight junction protein structurally similar to JAM-A [15], were incubated with T3SA- before adsorption. Human airway epithelia were adsorbed via the basolateral surface, and infected cells were quantified after 18 h of incubation. When infected with an MOI of 50 pfu/cell, both GST-JAM-A and J10.4 completely inhibited infection (data not shown); therefore, an MOI of 100 pfu/cell was examined (figure 3A and 3B). In comparison to GST, soluble GST-JAM-A reduced infection by ~3.5-fold, presumably by occupying viral receptor-binding sites. Concordantly, J10.4 inhibited infection by ~10-fold, compared with CAR-specific antibody RmcB. Thus, reovirus infection via the basolateral route is dependent on surface expression of JAM-A.
Figure 3.
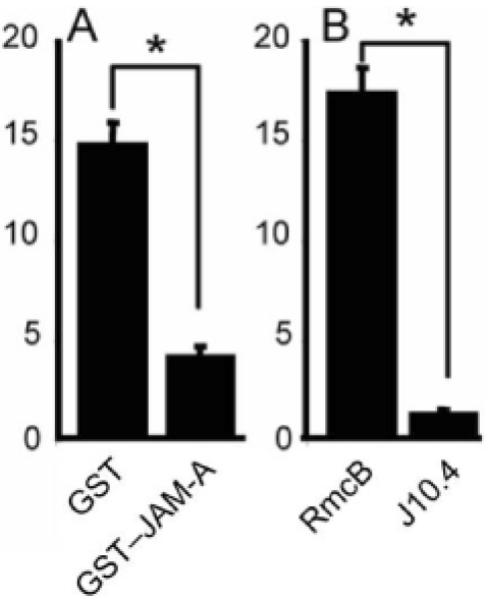
T3SA- (a monoreassortant reovirus strain that is incapable of binding to sialic acid) was preincubated with glutathione S-transferase (GST) or GST-JAM-A (i.e., soluble GST fused to the extracellular domain of junctional adhesion molecule-A [JAM-A]) (A) or with monoclonal antibodies specific for the coxsackievirus and adenovirus receptor (RmcB) or for JAM-A (J10.4) (B). The virus-protein mixtures were adsorbed basolaterally to airway epithelia at an MOI of 100 pfu/cell. Epithelia were immunostained at 18 h after adsorption to detect reovirus proteins, and infected cells were quantified in 20 random fields of view from 3 individual airway cultures. Error bars denote SDs. *, P < .0001.
Augmentation of reovirus T3SA+ apical infection by neuraminidase pretreatment
Mucin and other secreted or cell-surface proteins that are conjugated with sialic acid might inhibit infection of sialic acid-binding reoviruses by capturing virus particles before they reach the apical surface of airway epithelia. To determine whether T3SA+ infection is inhibited by the presence of sialic acid moieties at the apical surface, human airway epithelia were pretreated apically with V. cholerae, S. Typhimurium, or A. ureafaciens neuraminidase or with diluent alone. Epithelia were rinsed and adsorbed apically with T3SA+ at an MOI of 1000 pfu/cell, to observe any reduction of infection. Pretreatment with V. cholerae neuraminidase, which removes α2,3-linked sialic acid [27], but not with neuraminidases with broader specificities, significantly enhanced apical infection (figure 4). Neuraminidase pretreatment of human airway epithelia did not have any effect on infection by the non-sialic acid- binding reovirus T3SA- (24-h FFA after treatment with V. cholerae, +2% [P > .1]; S. Typhimurium, +9% [P > .1]; and A. ureafaciens, +21% [P > .1], compared with vehicle pretreatment). Collectively, these findings suggest that the capacity to engage α2,3-linked sialic acid inhibits apical reovirus infection of respiratory epithelial cells.
Figure 4.
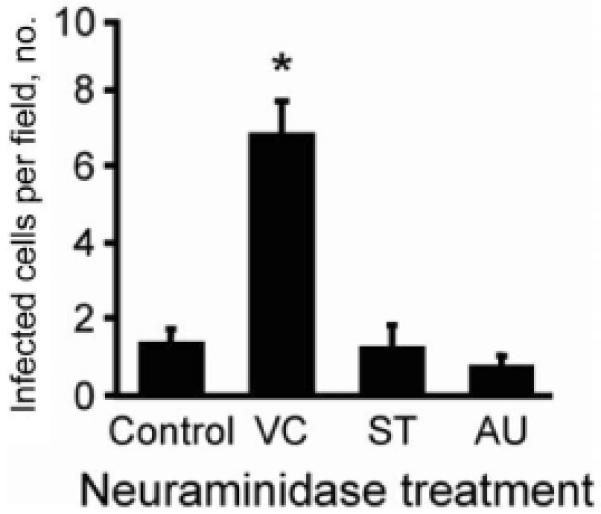
Apical infection of neuraminidase-treated human airway epithelia with T3SA+ (a monoreassortant reovirus strain that is isogenic, except for the presence of a single polymorphism in σ1 that renders the virus capable of binding with sialic acid). The apical surfaces of airway epithelia were pretreated with media alone (Control) or with neuraminidase from V. cholerae (VC), Salmonella serotype Typhimurium (ST), or Arthrobactor ureafaciens (AU) to remove sialic acid. After washing, the epithelia were adsorbed apically with T3SA+ at an MOI of 1000 pfu/cell. Epithelia were immunostained at 18 h after infection to detect reovirus proteins, and infected cells were quantified in 10 random fields of view (n = 4). Error bars denote SDs. *, P < .001.
Lack of effect of reovirus release on junctional integrity
Viral protein production was observed by FFA in individual human airway epithelial cells at 18 h after infection. Therefore we hypothesized that nascent reovirus would be released and would rapidly spread to adjacent cells. Furthermore, we wondered whether reovirus progeny would alter the integrity of the epithelial junctions by binding and disrupting homophilic interactions of JAM-A. To test these hypotheses, human airway epithelia were adsorbed basolaterally with T3SA- at an MOI of 50 pfu/cell or with T3SA+ at an MOI of 200 pfu/cell (to achieve similar numbers of infected cells), or they were mock infected. Transepithelial conductance was determined at 0, 2, and 8 h and at 1, 3, 7, 14, and 21 days after infection. We observed no significant change in conductance at any time point tested (figure 5), indicating that the integrity of the epithelium was maintained. To confirm infection, human airway epithelia from the same experiment were stained for reovirus at 24 h and at 1 week after infection (figure 6). Robust infection was observed at 24 h, with viral protein present throughout the cytoplasm (figure 6A and 6B). At 1 week after infection, the epithelia were stained for both reovirus and ZO-1, to determine the condition of the tight junctions. In spite of the spread of virus through the cultures (figure 6C and 6D), the tight junctions appeared to be completely intact (figure 6E and 6F). In addition, at 1 week after infection, JAM-A staining was not significantly altered (compare figure 6G with figure 1G). Thus, although reovirus replicates and spreads within the epithelium, it does not affect junctional integrity. These findings suggest that (1) infection is nonproductive, (2) basolaterally released reovirus is incapable of disrupting tight junctions, or (3) the majority of nascent virus is released from the apical surface.
Figure 5.

Junctional integrity of polarized human airway epithelia after reovirus infection. Epithelia were adsorbed basolaterally with T3SA+ (a monoreassortant reovirus strain that is capable of binding to sialic acid) (MOI, 200 pfu/cell) or T3SA- (a monoreassortant reovirus strain that is incapable of binding to sialic acid) (MOI, 50 pfu/cell), or they were mock treated. Transepithelial resistance was quantified over time and converted into conductance. The dashed line denotes loss of junctional integrity.
Figure 6.

Junctional integrity and junctional adhesion molecule-A (JAM-A) localization in polarized human airway epithelia. Epithelia were adsorbed basolaterally with T3SA+ (a monoreassortant reovirus strain that is capable of binding to sialic acid) (MOI, 200 pfu/cell) or T3SA- (a monoreassortant reovirus strain that is incapable of binding to sialic acid) (MOI, 50 pfu/cell) and were stained to detect reovirus protein (green, A-F), zonula occludens-1 (ZO-1) (red), JAM-A (green, G), or nuclei (blue). Epithelia were stained at 24 h (A and B) and 1 week (C-G) after adsorption.
Apical shedding of reovirus
To determine whether reovirus infection of human airway epithelia is productive and to define the polarity of viral egress, epithelia were adsorbed basolaterally, and washes of the apical surface and media from the basolateral surface were collected at 24 h and 21 days after infection, for assessment of the viral titer (figure 7). Consistent with basolateral inoculation, the majority of reovirus was found in the basolateral media at 24 h after infection and decreased thereafter (data not shown). By day 21 after infection, reovirus was detected only in the apical washes, not in the basolateral media. These findings indicate that reovirus infection of human airway epithelia results in apical release of nascent viral particles with minimal detectable disruption of intercellular tight junctions.
Figure 7.
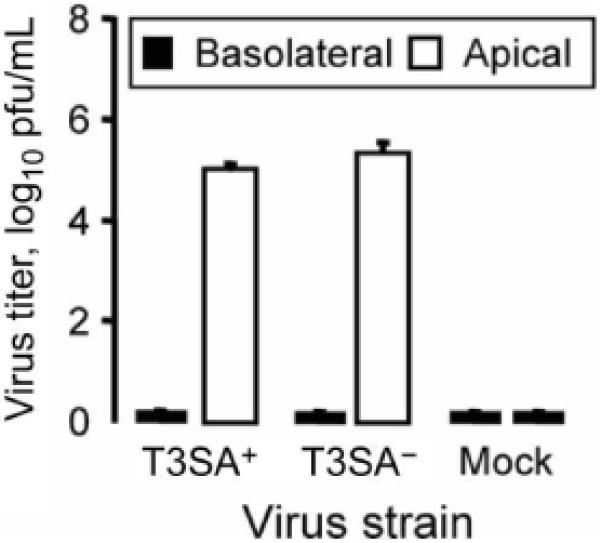
Reovirus shedding from infected airway epithelia. Epithelia were adsorbed basolaterally with T3SA+ (a monoreassortant reovirus strain that is capable of binding to sialic acid) (MOI, 200 pfu/cell) or T3SA- (a monoreassortant reovirus strain that is incapable of binding to sialic acid) (MOI, 50 pfu/cell), or they were mock treated. Washes of the apical surface or basolateral media were collected 21 days after adsorption, and viral titers were determined by plaque assay (n = 3). Error bars denote SDs.
DISCUSSION
In the present study, we defined the route of reovirus infection and spread in primary polarized human airway epithelia. We found that tight junction protein JAM-A localizes to the basolateral surfaces of these epithelial cells. Consistent with JAM-A localization, basolateral reovirus infection of airway epithelia was significantly more efficient than apical infection. In addition, binding to carbohydrate coreceptor sialic acid inhibits apical reovirus infection of airway epithelia. After basolateral infection, reovirus virions are released apically and do not compromise the integrity of epithelial tight junctions.
Our findings regarding reovirus and respiratory epithelial cells are particularly noteworthy when compared with findings for similar viruses. Adenovirus attachment protein fiber and receptor CAR share striking structural similarities with reovirus attachment protein σ1 and receptor JAM-A [15]. Like JAM-A, the CAR extracellular domain is composed of 2 Ig-like domains, forms homodimers, and localizes to epithelial tight junctions [29, 30]. Basolateral reovirus infection was ~20-50 times more efficient than apical infection. This difference in the efficiency of infection is of similar magnitude to the basolateral preference of adenovirus serotypes that utilize CAR as a primary receptor [31]. The preference of both viruses for the basolateral route of infection is likely attributable to the basolateral localization of their receptors (figure 1). However, in contrast to the diminished frequency of infection observed for sialic acid-binding reovirus, apical infection by sialic acid-binding adenovirus serotypes is more robust [32].
For many viruses, the capacity to bind sialic acid is a major determinant of infection efficiency. In the lung, sialic acid may be found within mucus secretions or coupled to cell-surface proteins. Mucins are highly glycosylated, high-molecular-mass macromolecules with surfaces conjugated with sialic acid, primarily in the O-linked configuration. Adeno-associated virus (AAV) serotype 4 binding to O-linked α2,3-linked sialic acid in mucins is inhibitory to apical infection, presumably because of failure of the virus to reach the cell surface [33]. AAV serotype 5 also binds to α2,3-linked sialic acid. However, this AAV serotype binds to N-linked sialic acid associated with the cell surface.
Reovirus strain T3SA+ binds to α2,3- and α2,6-linked sialic acid, which enhances infection [11] and apoptosis induction [34] in cultured cells. Pretreatment of cells expressing JAM-A with A. ureafaciens neuraminidase reduces T3SA+ infection and apoptosis [12]. On the basis of these findings, we anticipated that, compared with T3SA-, T3SA+ would exhibit enhanced apical infectivity of human airway epithelia. However, T3SA+ was almost completely incapable of apical infection of airway epithelia. Moreover, pretreatment with V. cholerae neuraminidase, which removes primarily α2,3-linked sialic acid [27], enhanced apical T3SA+ infection. Our finding that A. ureafaciens neuraminidase, which removes all sialic acid linkages, had no effect on apical T3SA+ infection suggests that sialic acid moieties with α2,6-linkages or other sialic acid moieties on the cell surface can bind T3SA+ and enhance infection. This idea is consistent with the discrepancy between T3SA+ and T3SA- infection from the basolateral surface. Previous studies have demonstrated polarity of reovirus strain type 3 Dearing (T3D) infection and inhibitory interactions with mucin in polarized intestinal epithelial cells [35, 36]. Although reovirus T3D or T1L may behave differently, we speculate that there is a balance between inhibitory α2,3-linked sialic acid in airway surface liquid and carbohydrate coreceptors on the cell surface that enhance infection efficiency.
On the basis of the similar attachment mechanisms used by reovirus and adenovirus [15], we anticipated that reovirus might use an escape mechanism similar to that used by adenovirus. After infection of polarized human airway epithelia, nascent adenovirus spreads basolaterally to infect adjacent cells and breaks down intercellular junctions through interactions with CAR [30]. However, junctional integrity, as assessed by transepithelial conductance and localization of tight junction proteins, was maintained throughout a 21-day course of reovirus infection of airway epithelia, despite productive infection. This result was somewhat unexpected, considering that residues required for reovirus binding are located directly within the JAM-A homodimer interface [37] and that JAM-A is involved in the regulation of colonic epithelial barrier resealing [16]. However, experiments aimed at modulating junctional barriers in airway epithelia through basolateral application of soluble JAM-A or J10.4 antibody also failed to alter conductance (data not shown). This result suggests that JAM-A performs different functions in different types of epithelia. How reovirus gains access to JAM-A in the tight junctions and on the basolateral surface of epithelial cells is unclear, but it is possible that the virus uses M cell transcytosis to access the basolateral surface in vivo [4]
On the basis of experiments performed in this study, it is not clear whether the same cells remained infected throughout the course of study or whether the virus was capable of spread from cell to cell. However, productive infection within airway epithelia persisted for at least 3 weeks after infection, as was evidenced by the continued presence of progeny virions in apical washes obtained over this interval. During the course of infection, JAM-A expression and localization were not altered. We hypothesize that reovirus progeny would bind to JAM-A and infect neighboring cells if released within the basolateral milieu. The presence of new virus in the apical washes, the lack of detectable viral progeny in the basolateral media, the absence of rapid spread of virus to neighboring cells, and the continuous stability of epithelial barriers during a 3-week interval of infection indicate that nascent reovirus is released apically. Taken together, these data suggest that reovirus infection in the lung may be self-limiting, resulting in the mild pathologic findings observed in the respiratory tract in vivo [5]. By comparison, adenovirus infection leads to large regions of cell destruction, immune cell invasion, and high levels of inflammation, consistent with its mode of egress [5, 30].
There are at least 2 potential mechanisms of apical egress that might be used by reovirus. The first is direct apical escape as utilized by rotavirus through interactions between the VP4 spike protein and the apical actin cytoskeleton [38]. Interestingly, rotavirus also displays differential infectivity, replication, and apical viral release in polarized intestinal cell lines, depending on sialic acid-binding capacity [39]. An alternative mechanism is apoptotic extrusion of infected cells [40]. Membrane integrity is maintained throughout the apoptotic process, allowing dying cells to transit through the epithelium into the lumen, where the remnants are sloughed through the action of cilia or removed by macrophages. Reovirus infection leads to NF-κB-mediated apoptosis in a variety of cell types [41-43]. Apoptosis may diminish the initial innate immune response and thus minimize tissue inflammation [40, 44]. Although our histologic analysis did not demonstrate cells with obvious apoptotic morphologic findings, a significant portion of the dead cell debris, whether caught within the mucus or floating in the airway surface liquid, was removed during conductance measurements. Therefore, it is possible that virus-induced apoptosis might contribute to apical reovirus release.
It is remarkable that a wide variety of unrelated viruses have evolved the capacity to use cell adhesion molecules as receptors [1, 45, 46]. In addition to reovirus, viruses that engage cell adhesion molecules include Coxsackie B viruses and most adenovirus serotypes (CAR) [47], rhinoviruses (intercellular adhesion molecule-1) [48, 49], and α-herpesviruses (nectin-1) [50]. Our data suggest that, unlike adenovirus, reovirus does not co-opt the cell adhesion function of its receptor to alter the paracellular path and thereby facilitate spread to other compartments. Instead, reovirus is directly released to the apical surface leaving the tight junction barrier intact. Our ongoing studies of the consequences of virus-receptor interactions will likely enhance an understanding of viral pathogenesis, including tissue invasion and host transmission.
Acknowledgments
We thank Wesley Skelton for providing expert technical assistance, Annukkah Antar for critical review of the manuscript, and Lisa Jorgensen for assistance with manuscript preparation.
Financial support: US Public Health Service (PHS; awards T32 GM08554 [to K.M.G.], T32 CA09385 [to J.A.C.], R37 AI38296 [to T.S.D.], and P01 HL51670 [to J.Z.]) and the Elizabeth B. Lamb Center for Pediatric Research; additional support was provided by PHS awards CA68485 (to the Vanderbilt-Ingram Cancer Center), DK20593 (to the Vanderbilt Diabetes Research and Training Center), and DK54759 (to the University of Iowa In Vitro Cell Models Core).
Footnotes
Potential conflicts of interest: none reported
Presented in part: American Society of Gene Therapy, Seattle, Washington, 2 June 2007 (abstract 809).
References
- 1.Vogelmann R, Amieva MR, Falkow S, Nelson WJ. Breaking into the epithelial apical-junctional complex—news from pathogen hackers. Curr Opin Cell Biol. 2004;16:86–93. doi: 10.1016/j.ceb.2003.12.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Turner JR. Molecular basis of epithelial barrier regulation: from basic mechanisms to clinical application. Am J Pathol. 2006;169:1901–9. doi: 10.2353/ajpath.2006.060681. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Neutra MR, Mantis NJ, Kraehenbuhl JP. Collaboration of epithelial cells with organized mucosal lymphoid tissues. Nat Immunol. 2001;2:1004–9. doi: 10.1038/ni1101-1004. [DOI] [PubMed] [Google Scholar]
- 4.Bienenstock J, McDermott MR. Bronchus- and nasal-associated lymphoid tissues. Immunol Rev. 2005;206:22–31. doi: 10.1111/j.0105-2896.2005.00299.x. [DOI] [PubMed] [Google Scholar]
- 5.Tyler KL. Mammalian reoviruses. In: Knipe DM, Howley PM, editors. Fields virology. 4th ed Vol 2. Lippincott Williams & Wilkins; Philadelphia: 2001. pp. 1729–45. [Google Scholar]
- 6.Weiner HL, Drayna D, Averill DR, Jr, Fields BN. Molecular basis of reovirus virulence: role of the S1 gene. Proc Natl Acad Sci USA. 1977;74:5744–8. doi: 10.1073/pnas.74.12.5744. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Weiner HL, Powers ML, Fields BN. Absolute linkage of virulence and central nervous system cell tropism of reoviruses to viral hemagglutinin. J Infect Dis. 1980;141:609–16. doi: 10.1093/infdis/141.5.609. [DOI] [PubMed] [Google Scholar]
- 8.Chappell JD, Prota AE, Dermody TS, Stehle T. Crystal structure of reovirus attachment protein σ1 reveals evolutionary relationship to adenovirus fiber. EMBO J. 2002;21:1–11. doi: 10.1093/emboj/21.1.1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Chappell JD, Duong JL, Wright BW, Dermody TS. Identification of carbohydrate-binding domains in the attachment proteins of type 1 and type 3 reoviruses. J Virol. 2000;74:8472–9. doi: 10.1128/jvi.74.18.8472-8479.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Gentsch JR, Pacitti AF. Differential interaction of reovirus type 3 with sialylated receptor components on animal cells. Virology. 1987;161:245–8. doi: 10.1016/0042-6822(87)90192-9. [DOI] [PubMed] [Google Scholar]
- 11.Barton ES, Connolly JL, Forrest JC, Chappell JD, Dermody TS. Utilization of sialic acid as a coreceptor enhances reovirus attachment by multistep adhesion strengthening. J Biol Chem. 2001;276:2200–11. doi: 10.1074/jbc.M004680200. [DOI] [PubMed] [Google Scholar]
- 12.Barton ES, Forrest JC, Connolly JL, et al. Junction adhesion molecule is a receptor for reovirus. Cell. 2001;104:441–51. doi: 10.1016/s0092-8674(01)00231-8. [DOI] [PubMed] [Google Scholar]
- 13.Schelling P, Guglielmi KM, Kirchner E, Paetzold B, Dermody TS, Stehle T. The reovirus σ-1 aspartic acid sandwich: a trimerization motif poised for conformational change. J Biol Chem. 2007;282:11582–9. doi: 10.1074/jbc.M610805200. [DOI] [PubMed] [Google Scholar]
- 14.Chretien I, Marcuz A, Courtet M, et al. CTX, a Xenopus thymocyte receptor, defines a molecular family conserved throughout vertebrates. Eur J Immunol. 1998;28:4094–104. doi: 10.1002/(SICI)1521-4141(199812)28:12<4094::AID-IMMU4094>3.0.CO;2-2. [DOI] [PubMed] [Google Scholar]
- 15.Stehle T, Dermody TS. Structural similarities in the cellular receptors used by adenovirus and reovirus. Viral Immunol. 2004;17:129–43. doi: 10.1089/0882824041310621. [DOI] [PubMed] [Google Scholar]
- 16.Mandell KJ, McCall IC, Parkos CA. Involvement of the junctional adhesion molecule-1 (JAM1) homodimer interface in regulation of epithelial barrier function. J Biol Chem. 2004;279:16254–62. doi: 10.1074/jbc.M309483200. [DOI] [PubMed] [Google Scholar]
- 17.Liu Y, Nusrat A, Schnell FJ, et al. Human junction adhesion molecule regulates tight junction resealing in epithelia. J Cell Sci. 2000;113(Pt 13):2363–74. doi: 10.1242/jcs.113.13.2363. [DOI] [PubMed] [Google Scholar]
- 18.Martin-Padura I, Lostaglio S, Schneemann M, et al. Junctional adhesion molecule, a novel member of the immunoglobulin superfamily that distributes at intercellular junctions and modulates monocyte transmigration. J Cell Biol. 1998;142:117–27. doi: 10.1083/jcb.142.1.117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Makino A, Shimojima M, Miyazawa T, Kato K, Tohya Y, Akashi H. Junctional adhesion molecule 1 is a functional receptor for feline calicivirus. J Virol. 2006;80:4482–90. doi: 10.1128/JVI.80.9.4482-4490.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Wolf JL, Rubin DH, Finberg R, et al. Intestinal M cells: a pathway for entry of reovirus into the host. Science. 1981;212:471–2. doi: 10.1126/science.6259737. [DOI] [PubMed] [Google Scholar]
- 21.Wolf JL, Kauffman RS, Finberg R, Dambrauskas R, Fields BN, Trier JS. Determinants of reovirus interaction with the intestinal M cells and absorptive cells of murine intestine. Gastroenterology. 1983;85:291–300. [PubMed] [Google Scholar]
- 22.Helander A, Silvey KJ, Mantis NJ, et al. The viral σ1 protein and glycoconjugates containing α2-3-linked sialic acid are involved in type 1 reovirus adherence to M cell apical surfaces. J Virol. 2003;77:7964–77. doi: 10.1128/JVI.77.14.7964-7977.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Morin MJ, Warner A, Fields BN. A pathway for entry of retroviruses into the host through M cells of the respiratory tract. J Exp Med. 1994;180:1523–7. doi: 10.1084/jem.180.4.1523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Karp PH, Moninger T, Weber SP, et al. An in vitro model of differentiated human airway epithelia: methods and evaluation of primary cultures. In: Wise C, editor. Epithelial cell culture protocols. Vol 188. Humana Press; Totowa, NJ: 2002. pp. 115–37. [DOI] [PubMed] [Google Scholar]
- 25.Virgin HW, Bassel-Duby R, Fields BN, Tyler KL. Antibody protects against lethal infection with the neurally spreading reovirus type 3 (Dearing) J Virol. 1988;62:4594–604. doi: 10.1128/jvi.62.12.4594-4604.1988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Excoffon KJ, Traver GL, Zabner J. The role of the extracellular domain in the biology of the coxsackievirus and adenovirus receptor. Am J Respir Cell Mol Biol. 2005;32:498–503. doi: 10.1165/rcmb.2005-0031OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Walters RW, Yi SM, Keshavjee S, et al. Binding of adeno-associated virus type 5 to 2,3-linked sialic acid is required for gene transfer. J Biol Chem. 2001;276:20610–6. doi: 10.1074/jbc.M101559200. [DOI] [PubMed] [Google Scholar]
- 28.Itoh M, Sasaki H, Furuse M, Ozaki H, Kita T, Tsukita S. Junctional adhesion molecule (JAM) binds to PAR-3: a possible mechanism for the recruitment of PAR-3 to tight junctions. J Cell Biol. 2001;154:491–7. doi: 10.1083/jcb.200103047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Cohen CJ, Shieh JT, Pickles RJ, Okegawa T, Hsieh JT, Bergelson JM. The coxsackievirus and adenovirus receptor is a transmembrane component of the tight junction. Proc Natl Acad Sci USA. 2001;98:15191–6. doi: 10.1073/pnas.261452898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Walters R, Freimuth P, Moninger T, Ganske I, Zabner J, Welsh M. Adenovirus fiber disrupts CAR-mediated intercellular adhesion allowing virus escape. Cell. 2002;110:789–99. doi: 10.1016/s0092-8674(02)00912-1. [DOI] [PubMed] [Google Scholar]
- 31.Walters RW, Grunst T, Bergelson JM, Finberg RW, Welsh MJ, Zabner J. Basolateral localization of fiber receptors limits adenovirus infection from the apical surface of airway epithelia. J Biol Chem. 1999;274:10219–26. doi: 10.1074/jbc.274.15.10219. [DOI] [PubMed] [Google Scholar]
- 32.Zabner J, Chillon M, Grunst T, et al. A chimeric type 2 adenovirus vector with a type 17 fiber enhances gene transfer to human airway epithelia. J Virol. 1999;73:8689–95. doi: 10.1128/jvi.73.10.8689-8695.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Walters RW, Pilewski JM, Chiorini JA, Zabner J. Secreted and transmembrane mucins inhibit gene transfer with AAV4 more efficiently than AAV5. J Biol Chem. 2002;277:23709–13. doi: 10.1074/jbc.M200292200. [DOI] [PubMed] [Google Scholar]
- 34.Connolly JL, Barton ES, Dermody TS. Reovirus binding to cell surface sialic acid potentiates virus-induced apoptosis. J Virol. 2001;75:4029–39. doi: 10.1128/JVI.75.9.4029-4039.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Weiner DB, Girard K, Williams WV, McPhillips T, Rubin DH. Reovirus type 1 and type 3 differ in their binding to isolated intestinal epithelial cells. Microb Pathog. 1988;5:29–40. doi: 10.1016/0882-4010(88)90078-2. [DOI] [PubMed] [Google Scholar]
- 36.Ambler L, Mackay M. Reovirus 1 and 3 bind and internalise at the apical surface of intestinal epithelial cells. Virology. 1991;184:162–9. doi: 10.1016/0042-6822(91)90832-v. [DOI] [PubMed] [Google Scholar]
- 37.Forrest JC, Campbell JA, Schelling P, Stehle T, Dermody TS. Structurefunction analysis of reovirus binding to junctional adhesion molecule 1: implications for the mechanism of reovirus attachment. J Biol Chem. 2003;278:48434–44. doi: 10.1074/jbc.M305649200. [DOI] [PubMed] [Google Scholar]
- 38.Gardet A, Breton M, Trugnan G, Chwetzoff S. Role for actin in the polarized release of rotavirus. J Virol. 2007;81:4892–4. doi: 10.1128/JVI.02698-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Ciarlet M, Crawford SE, Estes MK. Differential infection of polarized epithelial cell lines by sialic acid-dependent and sialic acid-independent rotavirus strains. J Virol. 2001;75:11834–50. doi: 10.1128/JVI.75.23.11834-11850.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Rosenblatt J, Raff MC, Cramer LP. An epithelial cell destined for apoptosis signals its neighbors to extrude it by an actin- and myosindependent mechanism. Curr Biol. 2001;11:1847–57. doi: 10.1016/s0960-9822(01)00587-5. [DOI] [PubMed] [Google Scholar]
- 41.Connolly JL, Rodgers SE, Clarke P, et al. Reovirus-induced apoptosis requires activation of transcription factor NF-κB. J Virol. 2000;74:2981–9. doi: 10.1128/jvi.74.7.2981-2989.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.O'Donnell SM, Hansberger MW, Connolly JL, et al. Organ-specific roles for transcription factor NF-κB in reovirus-induced apoptosis and disease. J Clin Invest. 2005;115:2341–50. doi: 10.1172/JCI22428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Hansberger MW, Campbell JA, Danthi P, et al. IκB kinase subunits α and γ are required for activation of NF-κB and induction of apoptosis by mammalian reovirus. J Virol. 2007;81:1360–71. doi: 10.1128/JVI.01860-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Tesfaigzi Y. Roles of apoptosis in airway epithelia. Am J Respir Cell Mol Biol. 2006;34:537–47. doi: 10.1165/rcmb.2006-0014OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Baranowski E, Ruiz-Jarabo CM, Domingo E. Evolution of cell recognition by viruses. Science. 2001;292:1102–5. doi: 10.1126/science.1058613. [DOI] [PubMed] [Google Scholar]
- 46.White JM, Littman DR. Viral receptors of the immunoglobulin superfamily. Cell. 1989;56:725–8. doi: 10.1016/0092-8674(89)90674-0. [DOI] [PubMed] [Google Scholar]
- 47.Bergelson JM, Cunningham JA, Droguett G, et al. Isolation of a common receptor for Coxsackie B viruses and adenoviruses 2 and 5. Science. 1997;275:1320–3. doi: 10.1126/science.275.5304.1320. [DOI] [PubMed] [Google Scholar]
- 48.Greve JM, Davis G, Meyer AM, et al. The major human rhinovirus receptor is ICAM-1. Cell. 1989;56:839–47. doi: 10.1016/0092-8674(89)90688-0. [DOI] [PubMed] [Google Scholar]
- 49.Staunton DE, Merluzzi VJ, Rothlein R, Barton R, Marlin SD, Springer TA. A cell adhesion molecule, ICAM-1, is the major surface receptor for rhinoviruses. Cell. 1989;56:849–53. doi: 10.1016/0092-8674(89)90689-2. [DOI] [PubMed] [Google Scholar]
- 50.Geraghty RJ, Krummenacher C, Cohen GH, Eisenberg RJ, Spear PG. Entry of αherpesviruses mediated by poliovirus receptor-related protein 1 and poliovirus receptor. Science. 1998;280:1618–20. doi: 10.1126/science.280.5369.1618. [DOI] [PubMed] [Google Scholar]