

Summary
Vascular endothelial growth factor (VEGF)-induced receptor phosphorylation is the crucial step for initiating downstream signaling pathways that lead to angiogenesis or related pathophysiological outcomes. Our previous studies have shown that the neurotransmitter dopamine could inhibit VEGF-induced phosphorylation of VEGF receptor 2 (VEGFR-2), endothelial cell proliferation, migration, microvascular permeability, and thus, angiogenesis. In this study, we address the mechanism by which VEGFR-2 phosphorylation is regulated by dopamine. Here, we demonstrate that D2 dopamine receptor (D2DR) colocalizes with VEGFR-2 at the cell surface. Dopamine pretreatment increases the translocation and colocalization of Src-homology-2-domain-containing protein tyrosine phosphatase (SHP-2) with D2DR at the cell surface. Dopamine administration leads to increased VEGF-induced phosphorylation of SHP-2 and this increased phosphorylation parallels the increased phosphatase activity of SHP-2. Active SHP-2 then dephosphorylates VEGFR-2 at Y951, Y996 and Y1059, but not Y1175. We also observe that SHP-2 knockdown impairs the dopamine-regulated inhibition of VEGF-induced phosphorylation of VEGFR-2 and, subsequently, Src phosphorylation and migration. Our data establish a novel role for SHP-2 phosphatase in the dopamine-mediated regulation of VEGFR-2 phosphorylation.
Keywords: Dopamine, Dopamine D2-receptor (D2DR), SHP-2, Vascular endothelial growth factor receptor 2 (VEGFR-2), Phosphorylation, Migration, Angiogenesis
Introduction
Angiogenesis performs a crucial role in physiological conditions, including embryonic development, wound healing and organ regeneration, as well as in pathological processes such as diabetes retinopathies, atherosclerosis, tumorigenesis and tumor metastasis (Carmeliet, 2003; Ferrara et al., 2003; Folkman, 1971; Folkman, 1995; Sueishi et al., 1997). Vascular endothelial growth factor (VEGF), also known as vascular permeability factor (VPF), is an important mediator of these angiogenic processes in both normal and diseased conditions. As one of the most important cytokines, VEGF is widely expressed by a number of human and animal tumors and has the ability to regulate most of the steps in the angiogenic signal cascade (Dvorak, 1990; Dvorak et al., 1999; Dvorak et al., 1979; Ferrara, 1999; Risau, 1997). The VEGF receptor family consists of three members, namely VEGFR-1, VEGFR-2 and VEGFR-3 (Shibuya, 2008). VEGFR-2, the major positive signal transducer for both physiological and pathological angiogenesis (Mukhopadhyay et al., 2004; Shibuya, 2006), is selectively expressed on vascular endothelial cells. The binding of VEGF to its receptors induces dimerization and subsequent receptor phosphorylation, which then leads to the activation of several intracellular downstream signaling pathways promoting angiogenesis (Olsson et al., 2006; Sakurai et al., 2005; Takahashi et al., 2001; Zachary, 2003).
The role of the neurotransmitter dopamine in modulating VEGF-induced angiogenesis has been well defined (Basu et al., 2001; Sarkar et al., 2004). Dopamine is produced in a wide variety of animals including both vertebrates and invertebrates, and activates the five types of dopamine receptors – D1, D2, D3, D4 and D5 – and their variants (Neve et al., 2004). Dopamine has a role in the management of Parkinson's disease and is also involved in the etiopathogenesis of several neuropsychiatric diseases such as schizophrenia (Egan and Weinberger, 1997; Goldstein and Deutch, 1992; Graybiel et al., 1990; Olanow and Tatton, 1999). Dopamine, when applied to patients with septic shock, can effectively maintain the circulatory stability and promote restoration of renal function (Dellinger et al., 2008). Although it is known that dopamine can inhibit different signaling steps involved in VEGF-mediated angiogenesis, beginning at VEGFR-2 phosphorylation (Basu et al., 2001; Bhattacharya et al., 2008b; Sarkar et al., 2004), the pathophysiological implication of dopamine in the context of VEGF-induced angiogenesis is novel and requires further investigation. Here, we examine how dopamine specifically regulates one of the earliest steps of the angiogenic process, the VEGF-induced phosphorylation of VEGFR-2, and define a novel role of Src-homology-2-domain-containing protein tyrosine phosphatase 2 (SHP-2) in the dopamine-mediated anti-angiogenic pathway.
Results
Dopamine modulates individual tyrosine phosphorylation of VEGFR-2
We previously demonstrated that total tyrosine phosphorylation of VEGFR-2 was inhibited upon dopamine pretreatment (Basu et al., 2001), but the specific tyrosine residues were not identified. Here, we found that phosphorylation of the specific tyrosine residues Y951, Y996, Y1059 and Y1175 on VEGFR-2 were significantly inhibited when HUVEC cells were pretreated with 10 μM dopamine or quinpirole (D2-receptor agonist) before the administration of VEGF (Fig. 1) for 10 minutes. A dose-response experiment showed that a 10 μM concentration of dopamine was more effective than 1 μM (Basu et al., 2001) to inhibit VEGFR-2 phosphorylation (supplementary material Fig. S1A). These results also suggested that the dopamine-mediated effect on VEGFR-2 phosphorylation was through D2DR.
Fig. 1.

Effect of dopamine and quinpirole pretreatment on VEGF-induced specific tyrosine phosphorylation of VEGFR-2. A significant decrease in VEGF-induced phosphorylation of VEGFR-2 at Y1175, Y996, Y951 and Y1059 is seen in HUVECs pretreated with either dopamine (10 μM) or quinpirole (10 μM) for 15 minutes before VEGF (10 ng/ml) treatment. Con, HUVECs without any VEGF or dopamine treatment; +V, HUVECs treated with only VEGF (10 ng/ml) for 10 minutes; D-V, HUVECs pretreated with only 10 μM dopamine for 15 minutes; D+V, HUVECs pretreated with 10 μM dopamine for 15 minutes and then treated with VEGF (10 ng/ml) for 10 minutes; Q-V, HUVECs treated with only 10 μM quinpirole for 15 minutes; Q+V, HUVECs pretreated with 10 μM quinpirole for 15 minutes and then treated with VEGF (10 ng/ml) for 10 minutes. Total VEGFR-2 was used as the loading control. Results from the blots are summarized graphically on the right. Fold change for each treatment in the blots is quantified by NIH image densitometry and is shown in the bar graph. The figures are representative of three separate experiments with similar results.
Whole-cell lysates from serum-starved HUVECs were immunoprecipitated with VEGFR-2 antibody and immunoblotted with antibody against phosphorylated (-P) VEGFR-2(951) to show the specificity of the phospho-antibody against VEGFR-2-P (supplementary material Fig. S1B).
D2DR coprecipitates with VEGFR-2 and SHP-2
As dopamine modulates VEGFR-2 tyrosine phosphorylation, we posited that certain phosphatases might be involved in this modulation. Therefore, we performed coprecipitation experiments to identify specific protein tyrosine phosphatases that are involved in the dopamine-mediated VEGFR-2 dephosphorylation. Whole-cell lysates from serum-starved HUVECs were immunoprecipitated with D2DR antibody and immunoblotted with antibodies against VEGFR-2 and SHP-2. We found constitutive association between D2DR, VEGFR-2 and SHP-2. The association between D2DR and VEGFR-2 decreased upon VEGF (10 ng/ml) induction but increased significantly with dopamine (10 μM) pretreatment, followed by VEGF induction (P=0.05) (Fig. 2A). Interestingly, the association between D2DR and SHP-2 markedly increased upon VEGF treatment and remained unchanged even with dopamine pretreatment (Fig. 2A). We also found that VEGFR-2 associated with SHP-2 and that this association increased upon treatment with VEGF for 10 minutes; however, dopamine pretreatment had no additive effect on the VEGF-induced association of VEGFR-2 and SHP-2. (supplementary material Fig. S2). These results were further corroborated using confocal microscopy. Cells were stained without permeabilization and we found that VEGFR-2 and D2DR remained associated with each other at the cell surface when cells were untreated. This localization and association at the cell surface markedly decreased (P=0.021) upon stimulation with VEGF and again increased significantly (P=0.004) with dopamine pretreatment followed by VEGF induction (Fig. 2B). In untreated cells, little SHP-2 staining and colocalization with D2DR were observed at the cell surface. However, after 10 minutes of VEGF treatment, SHP-2 appeared at the surface, indicating that SHP-2 translocated from the cytoplasm to the cell surface and became associated with D2DR at the surface (P=0.036). Dopamine pretreatment caused a significant induction in the VEGF-stimulated colocalization of SHP-2 with D2DR (P=0.009) at the cell surface (Fig. 2C).
Fig. 2.

(A) D2DR coprecipitates with VEGFR-2 and SHP-2. Serum-starved (0.1% serum, for 24 hours) HUVECs were pretreated with dopamine (10 μM) for 15 minutes before VEGF (10 ng/ml) stimulation and then the lysates were immunoprecipitated with D2DR antibody and immunoblotted with antibodies against VEGFR-2 and SHP-2. Con, HUVECs without VEGF or dopamine treatment; +V5, HUVECs treated with only VEGF (10 ng/ml) for 5 minutes; +V10, HUVECs treated with only VEGF (10 ng/ml) for 10 minutes; D+V5, HUVEC pretreated with 10 μM dopamine for 15 minutes and then treated with VEGF (10 ng/ml) for 5 minutes; D+V10, HUVECs pretreated with 10 μM dopamine for 15 minutes and then treated with VEGF (10 ng/ml) for 10 minutes. The figures are representative of three separate experiments with similar results. Results from the blots are summarized graphically on the right. (B) D2DR colocalizes with VEGFR-2. Serum-starved HUVECs were pretreated with dopamine for 15 minutes and then stimulated with VEGF (10 ng/ml) for 10 minutes and stained with D2DR (green) and VEGFR-2 (red) antibodies. The D2DR localizes with VEGFR-2 at the cell surface. The intensity of complex formation is shown in the lower chamber (arrow). (a) VEGFR-2 and D2DR colocalize at the cell surface without any VEGF or dopamine treatment. (b) Colocalization of VEGFR-2 and D2DR decreases with VEGF stimulation. (c) Pretreatment with dopamine followed by VEGF induction results in an increase in the colocalization of D2DR and VEGFR-2 at the cell surface. The figures are representative of three separate experiments with similar results. Quantification of surface colocalization is shown in bar graph on the right (mean ± s.d.). (C) D2DR colocalizes with SHP-2. Serum-starved HUVECs were pretreated with dopamine for 15 minutes and then stimulated with VEGF (10 ng/ml) for 10 minutes and stained with D2DR (green) and SHP-2 (red) antibodies. The intensity of complex formation is shown in the lower chamber (arrowhead). (a) Intense D2DR and minor SHP-2 staining were observed at the cell surface without any VEGF or dopamine treatment. (b) Upon VEGF induction, SHP-2 translocated more from the cytosol to the cell surface and localized with D2DR. (c) Pretreatment with dopamine, followed by VEGF induction, leads to a marked increase in colocalization of D2DR with SHP-2 at the cell surface. The figures are representative of three separate experiments with similar results. Quantification of surface colocalization is shown in bar graph on the right (mean ± s.d.). (D) Cofractionation of VEGFR-2 and SHP-2 in the light density membrane fraction of HUVECs. Cell light density membrane fractions were purified using the hyperosmotic carbonate method. Equal volumes of each fraction were separated by SDS-PAGE electrophoresis, immunoblotted, and tested for VEGFR-2 and SHP-2. Presence of D2DR was monitored by immunoprecipitation with an anti-D2DR antibody. Dopamine pretreatment causes increased colocalization of VEGFR-2 and SHP-2 to the low-density domain. (E) Effect of dopamine treatment on biotinylated VEGFR-2 and D2DR. Serum-starved HUVECs were pretreated with dopamine for 15 minutes, then stimulated with VEGF (10 ng/ml) for 10 minutes, and then subjected to cell surface biotinylation following the manufacturer's instructions. After VEGF induction, less biotinylated VEGFR-2 is detected on the cell surface. However, with dopamine pretreatment, increased levels of VEGFR-2 and decreased levels of D2DR are found. From the biotinylation experiment, cell-surface-bound proteins were also recorded. VEGF induction increases surface recruitment of SHP-2 protein and dopamine pretreatment significantly enhances this localization.
VEGFR-2, SHP-2 and D2DR in membrane fractions
To further support our immunoprecipitation and confocal data, sucrose density gradient and cell surface biotinylation experiments were performed. To isolate endothelial cell low-density membrane fractions, HUVEC homogenates were subjected to a sucrose density gradient and 12 fractions were recovered. Eight fractions were subjected to western blot analysis. Under these conditions (see the Materials and Methods), low-density membranes were sedimented at the 5% and 35% sucrose interface (fractions 3-6; Fig. 2D). In the absence of any treatment, SHP-2 was not detected in the low-density membrane fraction. With VEGF induction, colocalization of SHP-2 with VEGFR-2 was observed in the low-density membrane fraction (fraction 4; Fig. 2D). Dopamine pretreatment followed by VEGF induction resulted in further enrichment of both VEGFR-2 and SHP-2 in the low-density fraction (fraction 4). For the detection of dopamine receptor D2, the immunoprecipitation experiment was done with D2DR antibody and seven fractions were analyzed (fractions 2-8; Fig. 2D). We observed expression of D2DR in all fractions, with the moderate enrichment of D2DR in fractions 3 and 4 with dopamine treatment.
Cell-surface-bound VEGFR-2 and D2DR were also measured with and without dopamine or VEGF treatment via biotinylation. As shown in Fig. 2E, after the addition of VEGF, the concentration of biotinylated VEGFR-2 decreased. However, upon dopamine pretreatment, increased VEGFR-2 and decreased D2DR levels were observed at the cell surface (Fig. 2E). Fig. 2E also indicated an increased surface recruitment of SHP-2 upon dopamine pretreatment followed by VEGF induction for 10 minutes.
Knockdown of SHP-2 interferes with the inhibitory role of dopamine towards VEGFR-2 phosphorylation
Previous reports (Gallicchio et al., 2005; Mitola et al., 2006) showed that the tyrosine phosphatase SHP-2 limited VEGF-induced VEGFR-2 activation, and we observed an increased association of SHP-2 with both VEGFR-2 and D2DR in HUVECs after dopamine treatment. We used SHP-2 siRNA to knock down SHP-2 and observed the effect of dopamine on VEGF-induced phosphorylation of VEGFR-2. We found that dopamine could not block the VEGF-induced phosphorylation of the tyrosine residues Y951, Y996 and Y1059 in SHP-2 siRNA-treated HUVECs (Fig. 3), but dopamine-mediated dephosphorylation of Y1175 was not affected by SHP-2 knockdown (Fig. 3). Our data suggest that dopamine selectively regulates the dephosphorylation of VEGFR-2 Y951, Y996 and Y1059, but not Y1175, by engaging the protein tyrosine phosphatase SHP-2.
Fig. 3.
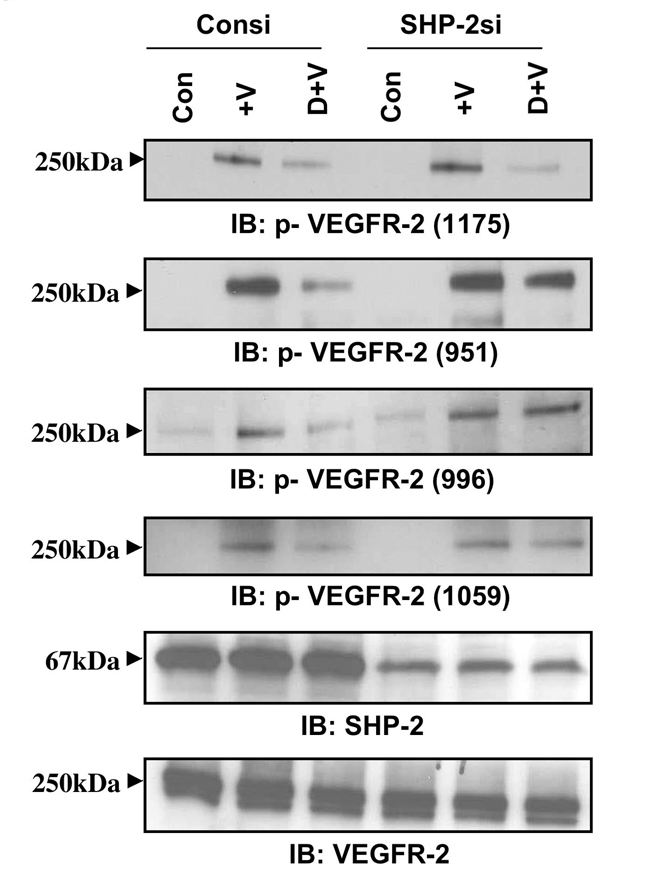
Effect of dopamine on VEGFR-2 phosphorylation in SHP-2-knockdown cells. HUVECs were transfected with a scrambled control (Consi) or SHP-2 siRNA (SHP-2si) using Oligofectamine. After 48 hours, cells were serum-starved and pretreated with dopamine for 15 minutes and then stimulated with VEGF for 10 minutes. In SHP-2-knockdown cells, dopamine cannot inhibit the VEGF-induced phosphorylation of VEGFR-2 at Y951, Y996 and Y1059, but successfully blocks Y1175 phosphorylation. Total VEGFR-2 was used as the loading control. The figures are representative of three separate experiments with similar results.
SHP-2 phosphorylation and phosphatase activity upon dopamine pretreatment in HUVECs
Catalytic activity of SHP-2 is crucial for its ability to modulate different signaling cascades. Previous reports have shown that the phosphatase activity of SHP-2 increases upon tyrosine phosphorylation (Tang et al., 1999; Vogel et al., 1993). There are two important phosphorylation sites in SHP-2, Y542 and Y580 (Bennett et al., 1994), and the phosphorylation at Y542 (the major phosphorylation site) (Bennett et al., 1994) has been proposed to relieve the basal inhibition of SHP-2 phosphatase activity (Lu et al., 2001). Compared with untreated cells, the total phosphorylation of SHP-2 significantly increased in cells treated with VEGF for both 5 and 10 minutes (Fig. 4A). When pretreated with dopamine, the phosphorylation of SHP-2 was even higher than that observed in cells treated with VEGF alone (Fig. 4A), translating into a 1.5-fold upregulation in the VEGF-induced phosphorylation of Y542 (data not shown). Therefore, dopamine treatment promotes the VEGF-induced total and Y542 phosphorylation of SHP-2. We did not observe any effect of dopamine on the phosphorylation of SHP-2 at Y580. Total phosphorylation of VEGFR-2 at these time points is also indicated (Fig. 4A, upper panel; supplementary material Fig. S3).
Fig. 4.

Effect of dopamine on SHP-2 phosphorylation and phosphatase activity. (A) Dopamine leads to an increase in VEGF-induced tyrosine phosphorylation of SHP-2. Serum-starved HUVEC were pretreated with dopamine (10 μM) for 15 minutes before VEGF (10 ng/ml) treatment for 5 or 10 minutes. Cell lysates were then immunoprecipitated with the Tyr-P antibody and immunoblotted with antibodies against VEGFR-2 and SHP-2. The figures are representative of three separate experiments with similar results. (B) SHP-2 phosphatase assay using pNPP. Serum-starved HUVECs were pretreated with 10 μM dopamine for 15 minutes followed by treatment with VEGF (10 ng/ml) for 10 minutes. Lysates were then immunoprecipitated with antibody against SHP-2. One half was run on a gel and the other half used for the phosphatase assay. SHP-2 phosphatase activity is markedly increased (P=0.01) after dopamine pretreatment, when compared with VEGF treatment alone. Data represent an average of three independent determinations (± s.d.) normalized against the negative control.
We also measured the SHP-2 phosphatase activity in response to dopamine. As shown in Fig. 4B, SHP-2 phosphatase activity was markedly increased (P=0.01) after a 15 minute dopamine pretreatment when compared with VEGF or dopamine treatment alone. In summary, dopamine treatment in endothelial cells resulted in an upregulation of VEGF-induced phosphorylation of SHP-2, increased phosphatase activity and decreased phosphorylation of VEGFR-2 at Y951, Y996 and Y1059.
Role of Fyn in dopamine-mediated regulation of SHP-2 and VEGFR-2 phosphorylation
Next, we looked at whether Fyn kinase or Src tyrosine kinase was involved in phosphorylation of SHP-2 or VEGFR-2. There have been reports describing a role for Fyn kinase upstream of SHP-2 and in the regulation of SHP-2 phosphorylation and phosphatase activity (Samayawardhena et al., 2006; Tang et al., 1999). We first examined the status of Fyn phosphorylation in HUVECs in the presence or absence of dopamine and VEGF treatment. Dopamine pretreatment in HUVECs did not cause any change in VEGF-induced Fyn phosphorylation, and thus, its kinase activity (supplementary material Fig. S5A). To rule out the role of Fyn in dopamine-mediated regulation of VEGFR-2 dephosphorylation, we then transfected HUVECs with Fyn siRNA. This knockdown of Fyn did not affect the phosphorylation of SHP-2 at Y542 nor on the phosphorylation of VEGFR-2 at Y1175 and Y951 after dopamine pretreatment and VEGF stimulation (supplementary material Fig. S5B).
Src regulates the phosphorylation SHP-2 at Y542
HUVECs were pretreated with either 5 μM PP2 (a Src tyrosine kinase inhibitor) (Bhattacharya et al., 2008a) or PP3 (an inactive analog of PP2) for 1 hour in the presence or absence of dopamine and VEGF. Cells were also pretreated with 10 μM SU6656 (a Src tyrosine kinase inhibitor) for 1 hour before treatment with either dopamine or VEGF. Without PP2, PP3 or SU6656 treatment, we observed significant VEGF-induced phosphorylation of SHP-2 at Y542 in the presence or absence of dopamine (Fig. 5A). Fig. 5A clearly shows that VEGF-induced phosphorylation of SHP-2 at Y542 was inhibited with PP2 treatment but not with PP3 even in dopamine-pretreated cells. VEGF stimulated phosphorylation of SHP-2 at Y542 was also blocked by the Src inhibitor SU6656 in the presence or absence of dopamine (supplementary material Fig. S4A). To further support our data we also checked the phosphorylation of SHP-2 at Y542 after Src knockdown with siRNA. We observed decreased phosphorylation of SHP-2 at Y542 after 24 hours of siRNA treatment with or without VEGF or dopamine treatment (supplementary material Fig. S4B). These findings suggest that although Src kinase is downstream of SHP-2, it regulates the phosphorylation of SHP-2 in a feedback mechanism that is independent of dopamine pretreatment.
Fig. 5.

(A) Effect of PP2 and PP3 on SHP-2 Y542 phosphorylation. Serum-starved HUVECs were pretreated with PP2 or PP3 (5 μM) for 1 hour before treatment with either 10 μM dopamine or VEGF (10 ng/ml). PP2, a Src kinase inhibitor, significantly inhibits phosphorylation of SHP-2 at Y542. Total SHP-2 was used as a loading control. The phosphorylation of Src at Y418 is significantly inhibited by treatment with 5 μM PP2. The figures are representative of three separate experiments with similar results. (B) Effect of dopamine on Src phosphorylation at Y418 after SHP-2 knockdown. 48 hours after siRNA transfection, HUVECs were serum starved and pretreated with dopamine for 15 minutes, followed by VEGF stimulation for 10 minutes. Dopamine pretreatment does not inhibit Src phosphorylation at Y418 in the absence of SHP-2. β-actin was used as a loading control. The figures are representative of three separate experiments with similar results.
Effect of dopamine on Src phosphorylation at Y418
We have already shown (Bhattacharya et al., 2008b) that dopamine inhibits the association of VEGFR-2 and Src by decreasing VEGFR-2 phosphorylation, and that this resulted in impaired Src phosphorylation at Y418 (the activation domain). We found that after SHP-2 knockdown, the basal level and the VEGF-induced phosphorylation of Src at Y418 increased compared with levels in control siRNA-treated cells (Fig. 5B). Dopamine pretreatment led to a significant upregulation of phosphorylation of Src at Y418 in SHP-2-knockout cells (Fig. 5B). Therefore, we concluded that SHP-2 has a prominent role in dopamine-mediated phosphorylation of VEGFR-2, which ultimately affects Src phosphorylation at Y418 and its kinase activity.
Effect of dopamine on migration in SHP-2-knockdown HUVECs
Phosphorylation of different tyrosine residues on VEGFR-2 results in the activation of different signaling pathways. VEGF-dependent phosphorylation at Y1175 leads to DNA synthesis and endothelial cell proliferation (Sakurai et al., 2005). Phosphorylation of Y951 and Y1059 are essential for VEGF-induced migration and proliferation in HUVECs (Zeng et al., 2001). Here, we show that dopamine was unable to block the VEGF-induced Y951 phosphorylation in SHP-2-knockdown cells (Fig. 3). Because VEGF-induced Y951 phosphorylation is responsible for HUVEC migration (Zeng et al., 2001), we performed a Boyden chamber migration assay with SHP-2 siRNA-transfected HUVEC cells in the presence or absence of dopamine and VEGF treatments. SHP-2 knockdown in HUVECs led to an increase in the basal level and the VEGF-induced level of cell migration compared with levels in control cells (Fig. 6). Dopamine significantly inhibited the VEGF-induced HUVEC migration (P<0.001) (Fig. 6) in control siRNA treated cells. However, dopamine could not affect the VEGF-induced migration (P=0.304) in SHP-2-knockdown cells (Fig. 6).
Fig. 6.
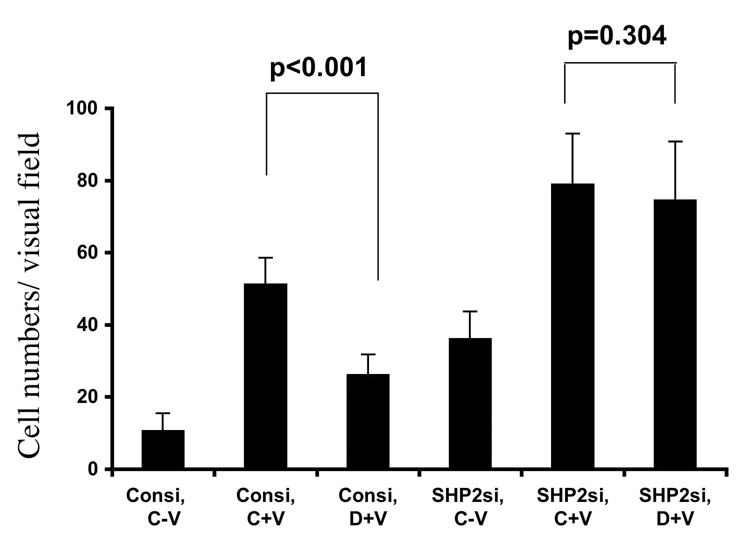
Effect of dopamine on migration in SHP-2-knockdown HUVECs. Cells were transfected with the scrambled control or SHP-2 siRNA using Oligofectamine. After 48 hours, cells were serum starved and a migration experiment was carried out as described in the Materials and Methods. After SHP-2 knockdown, dopamine does not inhibit the VEGF-induced HUVEC migration. Data represent an average of three independent determinations (± s.d.), each in triplicate.
Collectively, these data suggest that through SHP-2, dopamine modulates the phosphorylation of VEGFR-2 at Y951, Y996 and Y1059, but not at Y1175. The phosphorylation of VEGFR-2 at Y951 regulates Src kinase activity and thereby influences VEGF-mediated migration in endothelial cells (Fig. 7).
Fig. 7.

Model for dopamine-mediated regulation of VEGFR-2 phosphorylation by recruiting SHP-2. (A) In untreated HUVECs, D2DR and VEGFR-2 remain associated with each other. (B) Upon VEGF induction, SHP-2 translocates from the cytosol to the cell surface and becomes associated with both VEGFR-2 and D2DR. However, VEGF treatment promotes the dissociation of VEGFR-2 from D2DR to subsequently induce VEGFR-2 phosphorylation and downstream signaling. (C) Dopamine pretreatment, followed by VEGF stimulation for 10 minutes, leads to an increase in the association of D2DR with VEGFR-2. Dopamine treatment also induces an increased association between SHP-2 and D2DR at the cell surface and stimulates the phosphorylation of SHP-2 and its phosphatase activity. Active SHP-2 then inhibits the phosphorylation of VEGFR-2 at Y951, Y996 and Y1059, but not at Y1175. Decreased phosphorylation of VEGFR-2 at Y951 leads to a subsequent decrease in Src phosphorylation at Y418 and its kinase activity, effectively blocking VEGF-induced migration.
Discussion
Dopamine and its related molecules have been reported to inhibit different types of tumor growth in mouse models (Basu and Dasgupta, 2000; Sarkar et al., 2008; Wick, 1978; Wick, 1979). This probably occurs through the inhibition of angiogenesis, because we have previously shown that dopamine can inhibit VEGF-regulated angiogenesis in vitro and in vivo (Basu et al., 2001). Furthermore, a pharmacological dose of dopamine can inhibit VEGF-mediated microvascular permeability, proliferation and migration of endothelial cells (Basu et al., 2001; Bhattacharya et al., 2008b). It has also been shown that the neurotransmitter dopamine, when acting through the D2 receptor, blocks VEGF-induced focal adhesion kinase (FAK) and mitogen-activated protein kinase (MAPK) phosphorylation in endothelial cells (Sarkar et al., 2004). Recently, it has been reported that dopamine, by acting through D2DR, can regulate the mobilization of bone-marrow-derived endothelial progenitor cells and tumor growth (Chakroborty et al., 2008). However, the basic molecular mechanism involved in this regulation is still unclear. Moreover, phosphorylation of different VEGFR-2 tyrosine residues lead to activation of different downstream signaling pathways (Sakurai et al., 2005; Zeng et al., 2001). It is therefore important to understand the molecular mechanism by which dopamine regulates the phosphorylation of each residue. Our aim was to define the crucial molecular signaling partners engaged by dopamine to bring about the inhibition of VEGF-induced VEGFR-2 phosphorylation and the subsequent blockade of angiogenesis.
The involvement of different phosphatases, including DEP-1, SHP-1 and SHP-2 have been described in the regulation of VEGF-induced VEGFR-2 phosphorylation and subsequent downstream signaling (Bhattacharya et al., 2008a; Gallicchio et al., 2005; Grazia Lampugnani et al., 2003; Mitola et al., 2006; Seo et al., 2003). In mammalian cells, there are two tyrosine phosphatases containing SH2 domains, SHP-1 and SHP-2. However, unlike SHP-1, SHP-2 is ubiquitously expressed. The SH2 domains of SHP-2 target this enzyme to the phosphotyrosine residues of a variety of growth factor receptors and other signaling molecules (Neel et al., 2003). Here, we have delineated the unique role of SHP-2 phosphatase, recruited by D2DR in the dopamine-regulated dephosphorylation of VEGFR-2 and this regulation is a time-dependent phenomenon. With immunoprecipitation experiments, we showed increased association of VEGFR-2 with D2DR upon dopamine pretreatment, but we could not detect any change in the association of SHP-2 with D2DR and VEGFR-2 in the presence or absence of dopamine by this method. With confocal microscopy, it was evident that dopamine pretreatment followed by VEGF induction significantly enhanced the translocation of SHP-2 from the cytoplasm to the cell surface, and its association with D2DR on the surface. However, because immunoprecipitation was performed with whole-cell lysates and because confocal microscopy only highlights changes at the cell surface, it is not possible to draw a direct correlation between the immunoprecipitation and confocal data.
Cell-surface biotinylation also confirms increased recruitment of SHP-2 to the cell surface, and decreased VEGFR-2 internalization upon dopamine pretreatment. From sucrose density gradient experiments, we showed that VEGFR-2 and SHP-2 are in the same lipid-enriched membrane fraction and we found that both VEGFR-2 and SHP-2 were enriched in this fraction upon dopamine pretreatment. Further studies will define the recruitment mechanism of SHP-2 into the D2DR complex and the proper compartmentalization that leads to its phosphatase function.
We propose that after being engaged by D2DR, SHP-2 becomes phosphorylated and activated. The active SHP-2 then targets the specific tyrosine residues on VEGFR-2, which renders the dissociation of VEGFR-2 from D2DR; however, SHP-2 is not responsible for the dopamine-mediated dephosphorylation of VEGFR-2 at Y1175. Therefore, we speculate that other phosphatases are involved in this dopamine-mediated differential regulation of VEGFR-2 phosphorylation. We have previously reported that dopamine regulates VEGFR-2 phosphorylation, which in turn controls Src kinase activity (Bhattacharya et al., 2008b). It has also been reported that phosphorylation of VEGFR-2 at Y951 allows for the binding and tyrosine phosphorylation of T-cell-specific adapter (TSAd) (Matsumoto et al., 2005). TSAd then binds to the cytoplasmic tyrosine kinase Src and regulates the migration of endothelial cells in response to VEGF (Matsumoto et al., 2005). After knockdown of SHP-2, dopamine could not inhibit the VEGFR-2 phosphorylation at Y951, downstream Src phosphorylation or Src kinase activity: thus HUVEC migration is permitted. From this data, we infer that SHP-2 has a crucial role in the dopamine-modulated inhibition of VEGF-induced endothelial cell migration.
We were also interested in determining how SHP-2 activity is regulated after dopamine challenge. Although previous reports suggest that Fyn kinase is upstream of SHP-2 (Samayawardhena et al., 2006; Tang et al., 1999), dopamine pretreatment did not alter Fyn phosphorylation, and the knockdown of Fyn by siRNA in HUVECs did not show any effect on the phosphorylation of SHP-2 at Y542. However, inhibition of Src kinase activity by PP2 or SU6656 caused a significant reduction in the phosphorylation of SHP-2 at Y542, with or without dopamine pretreatment. We also found inhibition of SHP-2 phosphorylation at Y542 with or without VEGF or dopamine treatment using siRNA against Src. Therefore, Src kinase is probably responsible for the control of SHP-2 phosphorylation in a feedback mechanism after both VEGF and dopamine treatment. Additional in-depth studies are ongoing to elucidate the feedback loop between SHP-2 and Src in this context.
Our current hypothesis derived from our data is presented in Fig. 7. We propose that D2DR and VEGFR-2 remain associated with each other at the cell surface. Upon VEGF induction, SHP-2 translocates from the cytosol to the cell surface and becomes associated with both VEGFR-2 and D2DR. VEGF treatment also promotes the dissociation of VEGFR-2 from D2DR and induces VEGFR-2 phosphorylation and downstream signaling. Dopamine pretreatment prevents VEGFR-2 internalization, resulting in an increase association between D2DR with VEGFR-2. Dopamine treatment also induces an increased association between SHP-2 and D2DR at the cell surface and stimulates the phosphorylation of SHP-2 and its phosphatase activity. Active SHP-2 then inhibits the phosphorylation of VEGFR-2 at Y951, Y996 and Y1059, but not at Y1175. Decreased phosphorylation of VEGFR-2 at Y951 leads to subsequent decrease in the phosphorylation of Src at Y418 and its kinase activity, and that in turn, blocks VEGF-induced migration.
Overall, our results provide a novel mechanistic insight into the regulatory role of SHP-2 as a key mediator in the dopamine-regulated D2DR and VEGFR-2 crosstalk and ultimately, angiogenesis.
Materials and Methods
Reagents
VEGF-A was obtained from R&D Systems, Minneapolis, MN. The antibodies to VEGFR-2 (sc-504), SHP-2 (sc-280), D2DR (sc-7522), Src (sc-5266), VEGFR-2(996)-P (sc-16629) and VEGFR-2(951)-P (sc-16628) were purchased from Santa Cruz Biotechnology (Santa Cruz, CA); VEGFR-2(1059)-P, Tyr-P 4G10 and Src antibodies were purchased from Upstate. Antibodies against VEGFR-2(1175)-P (cat. no. 2478), SHP-2(542)-P (3751), Fyn (4023) and Src(527)-P (2105) were purchased from Cell Signaling. The Src(Tyr418-P) antibody was from BioSource International.Antibody SHP-2 and β-actin were from BD Biosciences. siRNAs against SHP-2 (sc-36488) and Fyn (sc-29321) were from Santa Cruz Biotechnology. siRNA against Src was from Dharmacon (Lafayette, CO). PP2, PP3 and SU6656 were from EMD Biosciences (San Diego, CA). Dopamine hydrochloride, quinpirole and all other chemicals not listed were from Sigma.
Immunoprecipitation and western blot analysis
Serum-starved (0.1% serum, for 24 hours) HUVECs were pretreated with 5 μM PP2 or PP3 or 10 μM SU6656 for 1 hour and then treated either with 10 μM dopamine or quinpirole for 15 minutes before treatment with VEGF (10 ng/ml) for 5 or 10 minutes. Whole-cell lysates in RIPA buffer supplemented with protease inhibitor cocktail and with or without phosphatase inhibitor were prepared from HUVECs. The lysates were collected after centrifugation at 14,000 g for 10 minutes at 4°C. 250 μg protein was incubated with 2 μg respective antibody for 1 hour and 50 μl of proteinA/G-conjugated agarose beads overnight at 4°C. Beads were washed with RIPA buffer three times and immunoprecipitates were resuspended in SDS sample buffer and electrophoresis was performed. All signals were detected using the chemiluminescent detection method according to the manufacturer's protocol (Amersham, UK) and quantified using the NIH Image densitometry software. Experiments were repeated at least three times.
Cell fractionation
The separation of light and heavy membrane fractions from HUVECs was performed as described (Song et al., 1996). Briefly, the serum-starved HUVECs were pretreated with 10 μM dopamine for 15 minutes before treatment with VEGF (10 ng/ml) for 10 minutes, and then cells were suspended in 2 ml of 0.5 M sodium carbonate buffer (pH, 11.0) containing phosphatase and protease inhibitor. The cell suspension was homogenized, adjusted to 45% sucrose by the addition of 2 ml of 90% sucrose prepared in MBS buffer, and placed into ultracentrifuge tubes. A 5-45% discontinuous sucrose gradient was then formed on top by the addition of 35% and 5% sucrose in MBS buffer (4 ml each). The samples were centrifuged at 40,000 r.p.m. for 18 hours in a SW41-Ti rotor (Beckman Instruments; Palo Alto, CA). The fractions were collected from the top in 1 ml amounts. Equal volumes of each fraction from each sample were then separated by SDS-PAGE, and transferred to nitrocellulose membranes. The membranes were immunoblotted with the desired antibodies. All signals were detected by the chemiluminescent detection method according to the manufacturer's protocol (Amersham).
Cell-surface biotinylation
Serum-starved HUVECs were pretreated with 10 μM dopamine for 15 minutes before treatment with VEGF (10 ng/ml) for 10 minutes. The biotinylation experiment was performed without permeabilization at 4°C, following the manufacturer's protocol (Pierce, Rockford, IL). After VEGF or dopamine treatment, the cells were lysed with a mild detergent and then cell surface biotinylated proteins were isolated with immobilized neutravidin gel. The bound proteins released by incubating the resin with SDS-PAGE sample buffer containing 50 mM DTT.
siRNA transfection
1×105 HUVECs were seeded in 60 mm plates and cultured for 24 hours in EGM. The next day, cells were washed with OPTI-MEM reduced-serum medium and transfected with 50 nM SHP-2 or Fyn siRNA and 100 nM Src siRNA using Oligofectamine (Invitrogen). After 4 hours, antibiotic-free EGM was added and cell lysates were prepared 48 or 24 hours after transfection.
Immunofluorescence
The anti-VEGFR-2 monoclonal antibody used for immunofluorescence was from Sigma and the anti-SHP-2 antibody was from BD Biosciences (San Jose, CA). Alexa Fluor 488 anti-goat (green) or Alexa Fluor 647 anti-mouse (red) secondary antibodies were purchased from Molecular Probes (Eugene, OR). 2×104 HUVECs were seeded on collagen-coated Lab-Tek chamber slides. After 24 hours, cells were serum starved. The next day, cells were pretreated with 10 μM of dopamine before induction with VEGF (10 ng/ml). Further steps were carried out at 4°C. Slides were washed in PBS and fixed in 4% paraformaldehyde (PFA) without permeabilization. Slides were washed in PBS, blocked in 10% goat serum, and stained with the respective primary antibody in 1% goat serum for 2 hours. Slides were then washed in PBS and incubated for 1 hour with the respective secondary antibody at a dilution of 1:200, followed by post-fixing in 4% PFA and mounting in Vectashield (Vector Labs, CA). Confocal microscopy was performed using a Zeiss LSM 510 confocal laser-scanning microscope with a C-Apochromat 100× NA 1.4 oil-immersion lens. Absence of signal crossover was established using single-labeled samples and quantification of colocalization was done using the Axio KS400 software (≥15 cells/group).
SHP-2 phosphatase assay
The phosphatase assay was performed according to the manufacturer's protocol (Stratagene, Signal Scout Phosphatase Profiling System). Briefly, starved HUVECs were pretreated for 15 minutes with or without dopamine and then with or without 10 ng/ml VEGF for 10 minutes. Cells were lysed (100 mM NaCl, 10 mM Tris-HCl, pH 8.0, 1 mM EDTA, 1% Triton X-100, 5 mM DTT) and the resulting lysates were precleared; protein concentration was determined using the Bradford assay. An equal amount of protein from each sample was then immunoprecipitated with an SHP-2 antibody. One part was then subjected to western blot and another part was treated as follows: 80 μl complete assay buffer (14 mM HEPES, pH 7.4, 30 mM NaCl, 1.5 mM EDTA, 5 mM DTT) was added and eluted from the column. 120 μl pNPP substrate (20 mM) was then added to each sample and absorbance at 405 nm was recorded on a Tecan SpectraFluor Plus after a 30 minute incubation at 30°C. Data presented were normalized with respect to the negative control. Three independent experiments were performed.
Migration assay
5×104 SHP-2 siRNA-treated serum-starved HUVEC cells were seeded into collagen-coated Transwell chambers with a diameter of 6.5 mm and a pore size of 8 μm (Corning CoStar Corporation, Cambridge, MA), which were then inserted into 24-well plates containing serum-starved EGM. After incubation at 37°C for 1 hour, 10 μM dopamine was added to the cells in the Transwells, which were then incubated for a further 15 minutes. VEGF was then added to a final concentration of 10 ng/ml in the lower chamber. Following incubation for 4 hours at 37°C in a CO2 incubator, cells that remained in the upper chamber were gently removed with a cotton swab. Cells that had invaded through the filter were fixed in 4% PFA and stained with 0.2% crystal violet dissolved in 2% ethanol. Migration was quantified by counting the number of cells on the filter using bright-field optics with a Nikon Diaphot microscope equipped with a 16-square reticule (1 mm2). Four separate fields were counted for each filter. An average of three separate experiments was recorded.
Statistical analysis
All values are expressed as means ± s.d. Statistical significance was determined using the two-sided Student's t-test and a value of P≤0.05 was considered significant. Error bars are given on the basis of calculated s.d. values.
Supplementary Material
Supplementary material available online at http://jcs.biologists.org/cgi/content/full/122/18/3385/DC1
We would like to thank Jim Tarara for help with the confocal microscopy experiments and Julie Lau for discussions. This work is partly supported by NIH grants HL70567, HL72178 and CA78383, and also a grant from American Cancer Society to D.M. Deposited in PMC for release after 12 months.
References
- Basu, S. and Dasgupta, P. S. (2000). Role of dopamine in malignant tumor growth. Endocrine 12, 237-241. [DOI] [PubMed] [Google Scholar]
- Basu, S., Nagy, J. A., Pal, S., Vasile, E., Eckelhoefer, I. A., Bliss, V. S., Manseau, E. J., Dasgupta, P. S., Dvorak, H. F. and Mukhopadhyay, D. (2001). The neurotransmitter dopamine inhibits angiogenesis induced by vascular permeability factor/vascular endothelial growth factor. Nat. Med. 7, 569-574. [DOI] [PubMed] [Google Scholar]
- Bennett, A. M., Tang, T. L., Sugimoto, S., Walsh, C. T. and Neel, B. G. (1994). Protein-tyrosine-phosphatase SHPTP2 couples platelet-derived growth factor receptor beta to Ras. Proc. Natl. Acad. Sci. USA 91, 7335-7339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bhattacharya, R., Kwon, J., Wang, E., Mukherjee, P. and Mukhopadhyay, D. (2008a). Src homology 2 (SH2) domain containing protein tyrosine phosphatase-1 (SHP-1) dephosphorylates VEGF Receptor-2 and attenuates endothelial DNA synthesis, but not migration. J. Mol. Signal. 3, 8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bhattacharya, R., Sinha, S., Yang, S. P., Patra, C., Dutta, S., Wang, E. and Mukhopadhyay, D. (2008b). The neurotransmitter dopamine modulates vascular permeability in the endothelium. J. Mol. Signal. 3, 14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carmeliet, P. (2003). Angiogenesis in health and disease. Nat. Med. 9, 653-660. [DOI] [PubMed] [Google Scholar]
- Chakroborty, D., Chowdhury, U. R., Sarkar, C., Baral, R., Dasgupta, P. S. and Basu, S. (2008). Dopamine regulates endothelial progenitor cell mobilization from mouse bone marrow in tumor vascularization. J. Clin. Invest. 118, 1380-1389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dellinger, R. P., Levy, M. M., Carlet, J. M., Bion, J., Parker, M. M., Jaeschke, R., Reinhart, K., Angus, D. C., Brun-Buisson, C., Beale, R. et al. (2008). Surviving Sepsis Campaign: international guidelines for management of severe sepsis and septic shock: 2008. Crit. Care Med. 36, 296-327. [DOI] [PubMed] [Google Scholar]
- Dvorak, H. F. (1990). Leaky tumor vessels: consequences for tumor stroma generation and for solid tumor therapy. Prog. Clin. Biol. Res. 354A, 317-330. [PubMed] [Google Scholar]
- Dvorak, H. F., Orenstein, N. S., Carvalho, A. C., Churchill, W. H., Dvorak, A. M., Galli, S. J., Feder, J., Bitzer, A. M., Rypysc, J. and Giovinco, P. (1979). Induction of a fibrin-gel investment: an early event in line 10 hepatocarcinoma growth mediated by tumor-secreted products. J. Immunol. 122, 166-174. [PubMed] [Google Scholar]
- Dvorak, H. F., Nagy, J. A., Feng, D., Brown, L. F. and Dvorak, A. M. (1999). Vascular permeability factor/vascular endothelial growth factor and the significance of microvascular hyperpermeability in angiogenesis. Curr. Top. Microbiol. Immunol. 237, 97-132. [DOI] [PubMed] [Google Scholar]
- Egan, M. F. and Weinberger, D. R. (1997). Neurobiology of schizophrenia. Curr. Opin. Neurobiol. 7, 701-707. [DOI] [PubMed] [Google Scholar]
- Ferrara, N. (1999). Vascular endothelial growth factor: molecular and biological aspects. Curr. Top. Microbiol. Immunol. 237, 1-30. [DOI] [PubMed] [Google Scholar]
- Ferrara, N., Gerber, H. P. and LeCouter, J. (2003). The biology of VEGF and its receptors. Nat. Med. 9, 669-676. [DOI] [PubMed] [Google Scholar]
- Folkman, J. (1971). Tumor angiogenesis: therapeutic implications. N. Engl. J. Med. 285, 1182-1186. [DOI] [PubMed] [Google Scholar]
- Folkman, J. (1995). Angiogenesis in cancer, vascular, rheumatoid and other disease. Nat. Med. 1, 27-31. [DOI] [PubMed] [Google Scholar]
- Gallicchio, M., Mitola, S., Valdembri, D., Fantozzi, R., Varnum, B., Avanzi, G. C. and Bussolino, F. (2005). Inhibition of vascular endothelial growth factor receptor 2-mediated endothelial cell activation by Axl tyrosine kinase receptor. Blood 105, 1970-1976. [DOI] [PubMed] [Google Scholar]
- Goldstein, M. and Deutch, A. Y. (1992). Dopaminergic mechanisms in the pathogenesis of schizophrenia. FASEB J. 6, 2413-2421. [PubMed] [Google Scholar]
- Graybiel, A. M., Hirsch, E. C. and Agid, Y. (1990). The nigrostriatal system in Parkinson's disease. Adv. Neurol. 53, 17-29. [PubMed] [Google Scholar]
- Grazia Lampugnani, M., Zanetti, A., Corada, M., Takahashi, T., Balconi, G., Breviario, F., Orsenigo, F., Cattelino, A., Kemler, R., Daniel, T. O. et al. (2003). Contact inhibition of VEGF-induced proliferation requires vascular endothelial cadherin, beta-catenin, and the phosphatase DEP-1/CD148. J. Cell Biol. 161, 793-804. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu, W., Gong, D., Bar-Sagi, D. and Cole, P. A. (2001). Site-specific incorporation of a phosphotyrosine mimetic reveals a role for tyrosine phosphorylation of SHP-2 in cell signaling. Mol. Cell 8, 759-769. [DOI] [PubMed] [Google Scholar]
- Matsumoto, T., Bohman, S., Dixelius, J., Berge, T., Dimberg, A., Magnusson, P., Wang, L., Wikner, C., Qi, J. H., Wernstedt, C. et al. (2005). VEGF receptor-2 Y951 signaling and a role for the adapter molecule TSAd in tumor angiogenesis. EMBO J. 24, 2342-2353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mitola, S., Brenchio, B., Piccinini, M., Tertoolen, L., Zammataro, L., Breier, G., Rinaudo, M. T., den Hertog, J., Arese, M. and Bussolino, F. (2006). Type I collagen limits VEGFR-2 signaling by a SHP2 protein-tyrosine phosphatase-dependent mechanism 1. Circ. Res. 98, 45-54. [DOI] [PubMed] [Google Scholar]
- Mukhopadhyay, D., Zeng, H. and Bhattacharya, R. (2004). Complexity in the vascular permeability factor/vascular endothelial growth factor (VPF/VEGF)-receptors signaling. Mol. Cell Biochem. 264, 51-61. [DOI] [PubMed] [Google Scholar]
- Neel, B. G., Gu, H. and Pao, L. (2003). The `Shp'ing news: SH2 domain-containing tyrosine phosphatases in cell signaling. Trends Biochem. Sci. 28, 284-293. [DOI] [PubMed] [Google Scholar]
- Neve, K. A., Seamans, J. K. and Trantham-Davidson, H. (2004). Dopamine receptor signaling. J. Recept. Signal Transduct. Res. 24, 165-205. [DOI] [PubMed] [Google Scholar]
- Olanow, C. W. and Tatton, W. G. (1999). Etiology and pathogenesis of Parkinson's disease. Annu. Rev. Neurosci. 22, 123-144. [DOI] [PubMed] [Google Scholar]
- Olsson, A. K., Dimberg, A., Kreuger, J. and Claesson-Welsh, L. (2006). VEGF receptor signalling-in control of vascular function. Nat. Rev. Mol. Cell Biol. 7, 359-371. [DOI] [PubMed] [Google Scholar]
- Risau, W. (1997). Mechanisms of angiogenesis. Nature 386, 671-674. [DOI] [PubMed] [Google Scholar]
- Sakurai, Y., Ohgimoto, K., Kataoka, Y., Yoshida, N. and Shibuya, M. (2005). Essential role of Flk-1 (VEGF receptor 2) tyrosine residue 1173 in vasculogenesis in mice. Proc. Natl. Acad. Sci. USA 102, 1076-1081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Samayawardhena, L. A., Hu, J., Stein, P. L. and Craig, A. W. (2006). Fyn kinase acts upstream of Shp2 and p38 mitogen-activated protein kinase to promote chemotaxis of mast cells towards stem cell factor. Cell. Signal. 18, 1447-1454. [DOI] [PubMed] [Google Scholar]
- Sarkar, C., Chakroborty, D., Mitra, R. B., Banerjee, S., Dasgupta, P. S. and Basu, S. (2004). Dopamine in vivo inhibits VEGF-induced phosphorylation of VEGFR-2, MAPK, and focal adhesion kinase in endothelial cells. Am. J. Physiol. Heart Circ. Physiol. 287, H1554-H1560. [DOI] [PubMed] [Google Scholar]
- Sarkar, C., Chakroborty, D., Chowdhury, U. R., Dasgupta, P. S. and Basu, S. (2008). Dopamine increases the efficacy of anticancer drugs in breast and colon cancer preclinical models. Clin. Cancer Res. 14, 2502-2510. [DOI] [PubMed] [Google Scholar]
- Seo, D. W., Li, H., Guedez, L., Wingfield, P. T., Diaz, T., Salloum, R., Wei, B. Y. and Stetler-Stevenson, W. G. (2003). TIMP-2 mediated inhibition of angiogenesis: an MMP-independent mechanism. Cell 114, 171-180. [DOI] [PubMed] [Google Scholar]
- Shibuya, M. (2006). Differential roles of vascular endothelial growth factor receptor-1 and receptor-2 in angiogenesis. J. Biochem. Mol. Biol. 39, 469-478. [DOI] [PubMed] [Google Scholar]
- Shibuya, M. (2008). Vascular endothelial growth factor-dependent and -independent regulation of angiogenesis. BMB Rep. 41, 278-286. [DOI] [PubMed] [Google Scholar]
- Song, K. S., Scherer, P. E., Tang, Z., Okamoto, T., Li, S., Chafel, M., Chu, C., Kohtz, D. S. and Lisanti, M. P. (1996). Expression of caveolin-3 in skeletal, cardiac, and smooth muscle cells. Caveolin-3 is a component of the sarcolemma and co-fractionates with dystrophin and dystrophin-associated glycoproteins. J. Biol. Chem. 271, 15160-15165. [DOI] [PubMed] [Google Scholar]
- Sueishi, K., Yonemitsu, Y., Nakagawa, K., Kaneda, Y., Kumamoto, M. and Nakashima, Y. (1997). Atherosclerosis and angiogenesis: its pathophysiological significance in humans as well as in an animal model induced by the gene transfer of vascular endothelial growth factor. Ann. NY Acad. Sci. 811, 311-322; 322-324. [DOI] [PubMed] [Google Scholar]
- Takahashi, T., Yamaguchi, S., Chida, K. and Shibuya, M. (2001). A single autophosphorylation site on KDR/Flk-1 is essential for VEGF-A-dependent activation of PLC-gamma and DNA synthesis in vascular endothelial cells. EMBO J. 20, 2768-2778. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tang, H., Zhao, Z. J., Huang, X. Y., Landon, E. J. and Inagami, T. (1999). Fyn kinase-directed activation of SH2 domain-containing protein-tyrosine phosphatase SHP-2 by Gi protein-coupled receptors in Madin-Darby canine kidney cells. J. Biol. Chem. 274, 12401-12407. [DOI] [PubMed] [Google Scholar]
- Vogel, W., Lammers, R., Huang, J. and Ullrich, A. (1993). Activation of a phosphotyrosine phosphatase by tyrosine phosphorylation. Science 259, 1611-1614. [DOI] [PubMed] [Google Scholar]
- Wick, M. M. (1978). Dopamine: a novel antitumor agent active against B-16 melanoma in vivo. J. Invest. Dermatol. 71, 163-164. [DOI] [PubMed] [Google Scholar]
- Wick, M. M. (1979). Levodopa and dopamine analogs: melanin precursors as antitumor agents in experimental human and murine leukemia. Cancer Treat. Rep. 63, 991-997. [PubMed] [Google Scholar]
- Zachary, I. (2003). VEGF signalling: integration and multi-tasking in endothelial cell biology. Biochem. Soc. Trans. 31, 1171-1177. [DOI] [PubMed] [Google Scholar]
- Zeng, H., Sanyal, S. and Mukhopadhyay, D. (2001). Tyrosine residues 951 and 1059 of vascular endothelial growth factor receptor-2 (KDR) are essential for vascular permeability factor/vascular endothelial growth factor-induced endothelium migration and proliferation, respectively. J. Biol. Chem. 276, 32714-32719. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}