

Abstract
Purpose
To identify children with acute lymphoblastic leukemia (ALL) at initial diagnosis who are at risk for inferior response to therapy by using molecular signatures.
Patients and Methods
Gene expression profiles were generated from bone marrow blasts at initial diagnosis from a cohort of 99 children with National Cancer Institute–defined high-risk ALL who were treated uniformly on the Children's Oncology Group (COG) 1961 study. For prediction of early response, genes that correlated to marrow status on day 7 were identified on a training set and were validated on a test set. An additional signature was correlated with long-term outcome, and the predictive models were validated on three large, independent patient cohorts.
Results
We identified a 24–probe set signature that was highly predictive of day 7 marrow status on the test set (P = .0061). Pathways were identified that may play a role in early blast regression. We have also identified a 47–probe set signature (which represents 41 unique genes) that was predictive of long-term outcome in our data set as well as three large independent data sets of patients with childhood ALL who were treated on different protocols. However, we did not find sufficient evidence for the added significance of these genes and the derived predictive models when other known prognostic features, such as age, WBC, and karyotype, were included in a multivariate analysis.
Conclusion
Genes and pathways that play a role in early blast regression may identify patients who may be at risk for inferior responses to treatment. A fully validated predictive gene expression signature was defined for high-risk ALL that provided insight into the biologic mechanisms of treatment failure.
INTRODUCTION
The current management of children with acute lymphoblastic leukemia (ALL) modulates treatment intensity according to the risk of relapse, which thereby maximizes opportunities for cure and minimizes adverse effects.1
A number of variables have been shown to be predictive of outcome in childhood ALL, including clinical and laboratory features, cytogenetic characteristics of the blast, and early response to chemotherapy.2 These variables are routinely used for treatment assignment, but approximately 20% of children unpredictably suffer a relapse.3
Global gene expression profiling has facilitated the discovery of biologic subgroups in a variety of cancers.4,5 This technique has been shown to accurately classify ALL into cohorts that correspond to known biologic subgroups.6,7 However, it has proved more difficult to identify signatures that are globally predictive of outcome. In the present study, we performed gene expression profiles on leukemic blasts from children who were treated on a single, contemporary Children's Oncology Group (COG) protocol for high-risk (HR) ALL to discover gene expression signatures that are predictive of early response and outcome.
PATIENTS AND METHODS
Diagnostic marrow samples from 99 children (age 1 to 18 years) with National Cancer Institute–defined HR B-precursor ALL (age ≥10 years and/or presenting WBC ≥ 50,000/μL) who were treated on the COG 1961 protocol were analyzed.8 We focused on this particular group of patients, because many lack known genetic subtypes predictive of outcome. All patients received a standard four-drug induction and were further classified as slow early responders (SER)—day 7 marrow was M3 (> 25% blasts)—or rapid early responders (RER)—day 7 marrow was M1 (< 5% blasts) or M2 (5% to 25% blasts).
To determine genetic profiles associated with early response to therapy, we analyzed 82 of 99 patients: 42 patients who had M1 marrow on day 7 were compared with 40 patients who had M3 marrow on day 7. Patients with M2 marrow (n = 17) were excluded to maximize the distinction between responders. To study the genes associated with long-term outcome, we analyzed expression profiles of 59 patients who fulfilled the following criteria: 28 patients who remained in complete continuous remission (CCR) for at least 4 years and 31 patients with marrow relapse within the first 3 years of initial diagnosis. Forty-two samples were common to both the early response and outcome analyses. Patient characteristics are listed in Appendix Table A1 (online only).
RNA Extraction and Amplification and DNA Arrays
Total RNA was extracted from cryopreserved blasts from the COG cell bank by using RNeasy Midi kits (Qiagen, Valencia, CA) followed by the MinEluate kit (Qiagen). Fifty nanograms of total RNA were used as template in a double-amplification protocol by using the RiboAmp OA kit (Arcturus, Mountain View, CA) according to the manufacturer's recommendations. In vitro transcription was completed with biotinylated UTP and CTP for labeling by using the Enzo BioArray HighYield RNA Transcript Labeling kit (Enzo Diagnostics, Farmingdale, NJ). Twenty micrograms of labeled cRNA were fragmented and hybridized to Affymetrix U133Plus2.0 microarrays (Affymetrix, Santa Clara, CA). These arrays contain 54,675 probe sets, which represented approximately 38,500 genes.
Screening Analysis for Cytogenetic Risk Group
Patients were tested by reverse transcriptase polymerase chain reaction (RT-PCR) for the presence of each of four common prognostic translocations: t(1;19), t(4;11), t(9;22), and t(12;21). The t(1;19), t(4;11), and t(12;21) fusion products were assayed by qualitative RT-PCR, whereas the t(9;22) analysis was done quantitatively by using TaqMan technology (Applied Biosystems, Foster City, CA). Primers are listed in Appendix Table A2 and methods for the assays detailed in the Appendix (online only).
Data Analysis
Data generated from the COG 1961 samples discussed in this publication have been deposited in the National Center for Biotechnology Information Gene Expression Omnibus (http://www.ncbi.nlm.nih.gov/geo) and are accessible through series accession number GSE7440.
Gene expression values were generated by using Affymetrix MAS 5.0 Software. Expression levels were scaled to an average value of 1,000 per gene chip9 and were log transformed. In each analysis, the probe sets of 53 nonhuman genes and those that did not receive present calls in at least 30% of the samples were removed from the study.
For prediction of early response, the samples (n = 82) were randomly divided into a training set (28 RER, 26 SER) and a test set (14 RER, 14 SER). A nearest shrunken centroids prediction model with a subset of genes that were best associated with early response (RER v SER) was determined by utilizing Prediction Analysis of Microarrays (PAM)10 packaged in R (Stanford University Labs, Palo Alto, CA; www.r-project.org/) with a 200 × 10-fold cross validation procedure on the training data set. This model was used to make predictions on the test set. Logistic regression was utilized to test the significances of the subset of genes and the class predictor when analysis was adjusted for clinical covariates, such as age and presenting WBC.
For long-term outcome prediction, t test and adjusted P value (or false discovery rate [FDR]), as proposed by Benjamini and Hochberg,11 were utilized to select a subset of probe sets that were statistically associated with outcome. A logistic regression12 model was used to test whether each of the genes added prognostic value beyond that of known clinical covariates. Logistic regression with various variable selection options11 was utilized to build the best models for predicting outcome on the basis of clinical covariates and the genes identified by the t test with the adjusted P value (or FDR). Prediction accuracies of these models were estimated by using an unbiased, leave-one-out cross validation (LOOCV).
Three independent microarray data sets of childhood B-precursor ALL were used for validation of the outcome signature: a set of 220 patients treated on Pediatric Oncology Group (POG) trials,13 145 patients treated on German Cooperative Study Group for Childhood ALL (COALL) protocols,14 and 92 patients from Dutch Childhood Oncology Group (DCOG) protocols.15 The samples of the POG, COALL, and DCOG data sets were hybridized to Affymetrix U95Av2, U133A, and U133Plus2.0 arrays, respectively. Logistic regression was used to determine the association of the significant probe sets in the POG data set, and Cox regression was used in the COALL and DCOG data sets. We next built models for outcome prediction. Of the 47 probe sets identified in the COG 1961 data set, 18 could be matched by 20 probe sets of the U95Av2 microarrays. We constructed logistic regression models with these 20 probe sets and with three clinical covariates (sex, age, and WBC) as predictors of outcome. Briefly, model I (LP1) was based on three genes, model II (LP2) on five genes, and model III (LP3) on four genes. Receiver operating characteristic (ROC) accuracy, t test, Mann-Whitney U test, Cox proportional hazards regression, and logistic regression were used to validate these predictive models on the independent data sets. In addition to the three statistical models mentioned above, we considered a simple linear combination of the expression values of the probe sets that match the 47 probe sets in each of the three validation cohorts (LPV).
RESULTS
Prediction of Early Response
Analysis with PAM on the training set (n = 54) led to a model comprised of 24 probe sets with a minimal average cross validated error rate of 0.38 that best characterized early response (FDR, 3.6%). The Affymetrix probe set identifications and gene descriptions in rank order can be found in Table 1.
Table 1.
Significant Probe Sets Predictive of Early Response
| Rank by Response Type | Training Set P Adjusted for Clinical Covariates | Test Set
|
Affymetrix Identification | Gene Symbol | Gene Description | |
|---|---|---|---|---|---|---|
| P of t Test | P Adjusted for Clinical Covariates | |||||
| RER | ||||||
| 1 | .001 | .066 | .252 | 219489_s_at | RHBDL2 | Rhomboid, veinlet-like 2 (Drosophila) |
| 2 | < .001 | .232 | .208 | 228346_at | Transcribed sequence with strong similarity to protein sp:P00722 (Escherichia coli) BGAL_ECOLI β-galactosidase | |
| 3 | .002 | .008* | .035* | 225606_at | BCL2L11 | BCL2-like 11 (apoptosis facilitator) |
| 4 | < .001 | .300 | .885 | 203588_s_at | TFDP2 | Transcription factor Dp-2 (E2F dimerization partner 2) |
| 5 | < .001 | .391 | .889 | 203505_at | ABCA1 | ATP-binding cassette, sub-family A (ABC1), member 1 |
| 6 | .001 | .001* | .004* | 1555372_at | BCL2L11 | BCL2-like 11 (apoptosis facilitator) |
| 7 | .007 | .019* | .018* | 1569110_x_at | PDCD6 | Programmed cell death 6 |
| SER | ||||||
| 1 | < .001 | .064 | .083 | 227353_at | EVER2 | Epidermodysplasia verruciformis 2 |
| 2 | .001 | .158 | .670 | 214255_at | ATP10A | ATPase, Class V, type 10A |
| 3 | .004 | .173 | .740 | 219667_s_at | BANK1 | B-cell scaffold protein with ankyrin repeats 1 |
| 4 | .001 | .023* | .028* | 206940_s_at | POU4F1 | POU domain, class 4, transcription factor 1 |
| 5 | .002 | .346 | .863 | 223562_at | PARVG | Parvin, γ |
| 6 | .001 | .059 | .378 | 203373_at | SOCS2 | Suppressor of cytokine signaling 2 |
| 7 | < .001 | .324 | .707 | 226869_at | Full-length insert cDNA clone ZD77F06 | |
| 8 | .015 | .013* | .110 | 211675_s_at | HIC | I-mfa domain-containing protein |
| 9 | < .001 | .432 | .799 | 232614_at | MRNA; cDNA DKFZp686K02231 (from clone DKFZp686K02231) | |
| 10 | .005 | .121 | .208 | 242644_at | EVER2 | Epidermodysplasia verruciformis 2 |
| 11 | .017 | .292 | .590 | 205290_s_at | BMP2 | Bone morphogenetic protein 2 |
| 12 | .001 | .505 | .306 | 223451_s_at | CKLF | Chemokine-like factor |
| 13 | .030 | .139 | .418 | 207339_s_at | LTB | Lymphotoxin beta (tumor necrosis factor superfamily, member 3) |
| 14 | .023 | .015* | .043* | 229390_at | Full length insert cDNA clone ZA84A12 | |
| 15 | .014 | .017* | .087 | 212070_at | GPR56 | G protein–coupled receptor 56 |
| 16 | .016 | .030* | .044* | 204198_s_at | RUNX3 | Runt-related transcription factor 3 |
| 17 | .008 | .129 | .184 | 227013_at | LATS2 | LATS, large tumor suppressor, homolog 2 (Drosophila) |
Abbreviations: RER, rapid early responder; SER, slow early responder.
Statistically significant in the test set.
To validate the significance of the 24 probe sets, we performed t test and logistic regression analyses on the expression values in the test data set (n = 28). Although there was a positive trend of association between all probes in the test and training sets, eight reached statistical significance. The estimated ROC accuracy of the predicted score on the test set was 0.7755 (P = .0061; Fig 1). The overall misclassification rate was 0.25 (sensitivity = .7143 and specificity = .7857). The observed and predicted early responses significantly correlated with each other (odds ratio, 8.33; P (one-sided Fisher's exact test) = .011).
Fig 1.

Receiver operating characteristic (ROC) curve of the predicted score of early response on the test set. The model that comprised 24 probe sets that were derived from the training set was used for the prediction of early response on the test set. The ROC accuracy (ie, the area under the curve) is A = 0.77, which is significantly larger than that of noninformative prediction (P = .006). By using a threshold of C = 0.4 (determined in training set), the model correctly predicted 21 of 28 patient cases (success rate, 0.75). RER, rapid early responder; SER, slow early responder.
Functional Analysis of Genes Related to Early Response
A list of 188 differentially expressed probe sets (RER v SER) that were selected by PAM on the entire data set (N = 82; FDR ≤ 10%) was used for the detection of the relative enrichment of genes according to GeneOntology15a terms with the help of the L2L tool.16 Genes significantly over-represented in RER patients included those involved with induction of apoptosis and hematopoeitic development, whereas genes involved with cell growth and metabolism were over-represented in SER patients (Table 2).
Table 2.
Enrichment Analysis of Genes Associated With Early Response
| GeneOntology Term | GeneOntology Number | P |
|---|---|---|
| Hemocyte development | GO:0007516 | .00007 |
| Vesicle targeting | GO:0006903 | < .001 |
| Intercellular junction assembly | GO:0007043 | .001 |
| Induction of apoptosis | GO:0006917 | .003 |
| Cytoplasm organization and biogenesis | GO:0007028 | .004 |
| Hemopoiesis | GO:0030097 | .005 |
| Membrane fusion | GO:0006944 | .005 |
| Hemopoietic or lymphoid organ development | GO:0048534 | .005 |
| Nucleotide catabolism | GO:0009166 | .00004 |
| Growth | GO:0040007 | < .001 |
| Hormone-mediated signaling | GO:0009755 | < .001 |
| Cellular morphogenesis | GO:0000902 | .002 |
| Leukotriene biosynthesis | GO:0019370 | .002 |
| Regulation of neuron differentiation | GO:0045664 | .003 |
| Alkene biosynthesis | GO:0043450 | .004 |
| Nucleosome assembly | GO:0006334 | .006 |
| Cell growth | GO:0016049 | .007 |
| Alkene metabolism | GO:0043449 | .007 |
| Chromatin assembly | GO:0031497 | .009 |
| Phospholipase C activation | GO:0007202 | .009 |
NOTE. The pathways upregulated in rapid early responders are hemocyte development through hematopoietic or lymphoid organ development; those upregulated in slow early responders are nucleotide catabolism through phospholipase C activation.
Prediction of Long-Term Outcome
Gene expression profiles from 59 patients (28 CCR; 31 relapse) were analyzed to identify genes related to long-term outcome. By using a threshold FDR of 5%, we identified 47 probe sets (which represented 41 unique genes) that were significantly associated with outcome. The Affymetrix identifications, which are descriptions for the genes, are listed in rank order in Table 3 with t test P values that compare CCR and relapse on the selected genes. Figure 2 represents the heatmap of the expression values.
Table 3.
Probe Sets Differentially Expressed Between Patients in CCR and Those Who Experienced Relapse
| Rank |
t Test
|
Likelihood Ratio Test (P)
|
Affymextrix Probe Set Identification | Gene Name | Gene Description | |||
|---|---|---|---|---|---|---|---|---|
| P | FDR | Response Associated With High Expression | Significance of Gene | Significance of Clinical Variables | ||||
| 1 | < .001 | .013 | CCR | < .001 | < .001 | 35666_at | SEMA3F | SEMA domain, immunoglobulin domain (Ig), short basic domain, secreted, (semaphoring) 3F |
| 2 | < .001 | .015 | Fail | < .001 | < .001 | 227877_at | Similar to annexin II receptor (LOC389289), mRNA | |
| 3 | < .001 | .015 | CCR | < .001 | < .001 | 227131_at | MAP3K3 | Mitogen-activated protein kinase kinase kinase 3 |
| 4 | < .001 | .015 | Fail | .004 | .001 | 205401_at | AGPS | Alkylglycerone phosphate synthase |
| 5 | < .001 | .028 | Fail | .001 | < .001 | 208687_x_at | HSPA8 | Heat shock 70 kDa protein 8 |
| 6 | < .001 | .028 | CCR | .002 | < .001 | 212229_s_at | FBXO21 | F-box only protein 21 |
| 7 | < .001 | .028 | CCR | .001 | < .001 | 212576_at | MGRN1 | Mahogunin, ring finger 1 |
| 8 | < .001 | .028 | CCR | .004 | < .001 | 225446_at | C21orf107 | Chromosome 21 open reading frame 107 |
| 9 | < .001 | .031 | CCR | .004 | < .001 | 224793_s_at | TGFBR1 | Transforming growth factor, β receptor I (activin A receptor type II–like kinase, 53 kDa) |
| 10 | < .001 | .033 | CCR | .001 | < .001 | 221840_at | PTPRE | Protein tyrosine phosphatase, receptor type, E |
| 11 | < .001 | .033 | CCR | < .001 | < .001 | 203514_at | MAP3K3 | Mitogen-activated protein kinase kinase kinase 3 |
| 12 | < .001 | .034 | CCR | .003 | < .001 | 1559018_at | PTPRE | Protein tyrosine phosphatase, receptor type, E |
| 13 | < .001 | .034 | Fail | < .001 | < .001 | 217499_x_at | OR7E47P | Olfactory receptor, family 7, subfamily E, member 47 pseudogene |
| 14 | < .001 | .034 | Fail | .007 | < .001 | 224187_x_at | HSPA8 | Heat shock 70 kDa protein 8 |
| 15 | < .001 | .034 | Fail | .007 | < .001 | 221891_x_at | HSPA8 | Heat shock 70 kDa protein 8 |
| 16 | < .001 | .034 | CCR | < .001 | < .001 | 201642_at | IFNGR2 | Interferon γ receptor 2 (interferon gamma transducer 1) |
| 17 | < .001 | .034 | CCR | .002 | < .001 | 218418_s_at | ANKRD25 | Ankyrin repeat domain 25 |
| 18 | < .001 | .034 | Fail | < .001 | < .001 | 242305_at | cDNA FLJ42757 fis, clone BRAWH3001712 | |
| 19 | < .001 | .034 | CCR | .002 | < .001 | 216035_x_at | TCF7L2 | Transcription factor 7-like 2 (T-cell specific, high mobility group box) |
| 20 | < .001 | .034 | CCR | .001 | < .001 | 1556321_a_at | MRNA full length insert cDNA clone EUROIMAGE 283668 | |
| 21 | < .001 | .034 | Fail | < .001 | < .001 | 235014_at | LOC147727 | Hypothetical protein LOC147727 |
| 22 | < .001 | .034 | CCR | .002 | < .001 | 208820_at | PTK2 protein tyrosine kinase 2 | |
| 23 | < .001 | .034 | CCR | .004 | < .001 | 212231_at | FBXO21 | F-box only protein 21 |
| 24 | < .001 | .034 | CCR | .004 | < .001 | 229618_at | SNX16 | Sorting nexin 16 |
| 25 | < .001 | .034 | CCR | .005 | < .001 | 209033_s_at | DYRK1A | Dual-specificity tyrosine-(Y)-phosphorylation regulated kinase 1A |
| 26 | < .001 | .034 | CCR | < .001 | < .001 | 200641_s_at | YWHAZ | Tyrosine 3-monooxygenase/tryptophan 5-monooxygenase activation protein, zeta polypeptide |
| 27 | < .001 | .034 | Fail | .015 | < .001 | 202657_s_at | SERTAD2 | SERTA domain containing 2 |
| 28 | < .001 | .034 | CCR | .015 | < .001 | 201099_at | USP9X | Ubiquitin specific protease 9, X-linked (fat facets-like, Drosophila) |
| 29 | < .001 | .034 | CCR | .004 | < .001 | 201542_at | SARA1 | SAR1a gene homolog 1 (S. cerevisiae) |
| 30 | < .001 | .034 | CCR | .005 | < .001 | 227068_at | PGK1 | Phosphoglycerate kinase 1 |
| 31 | < .001 | .037 | CCR | < .001 | < .001 | 213944_x_at | GNA11 | Guanine nucleotide binding protein (G protein), α-11 (Gq class) |
| 32 | < .001 | .039 | CCR | .015 | < .001 | 201472_at | VBP1 | von Hippel-Lindau binding protein 1 |
| 33 | < .001 | .039 | CCR | .018 | < .001 | 202806_at | DBN1 | Drebrin 1 |
| 34 | < .001 | .039 | CCR | .022 | < .001 | 221918_at | PCTK2 | PCTAIRE protein kinase 2 |
| 35 | < .001 | .039 | Fail | < .001 | < .001 | 214585_s_at | VPS52 | Vacuolar protein sorting 52 (yeast) |
| 36 | < .001 | .039 | Fail | < .001 | < .001 | 219078_at | GPATC2 | G patch domain containing 2 |
| 37 | < .001 | .039 | Fail | < .001 | < .001 | 219133_at | FLJ20604 | Hypothetical protein FLJ20604 |
| 38 | < .001 | .040 | Fail | < .001 | < .001 | 1558111_at | MBNL1 | Muscleblind–like (Drosophila) |
| 39 | < .001 | .040 | CCR | .001 | < .001 | 221773_at | ELK3, ETS-domain protein (SRF accessory protein 2) | |
| 40 | < .001 | .040 | CCR | .003 | < .001 | 1558732_at | MAP4K2 | mitogen-activated protein kinase kinase kinase kinase 4 |
| 41 | < .001 | .043 | CCR | .032 | < .001 | 212441_at | KIAA0232 | KIAA0232 gene product |
| 42 | < .001 | .045 | CCR | .015 | < .001 | 226775_at | e(y)2 | e(y)2 protein |
| 43 | < .001 | .045 | Fail | .001 | < .001 | 208498_s_at | AMY2B | Amylase, α 2B; pancreatic |
| 44 | < .001 | .049 | CCR | .016 | < .001 | 201121_s_at | PGRMC1 | Progesterone receptor membrane component 1 |
| 45 | < .001 | .049 | CCR | .004 | < .001 | 202984_s_at | BAG5 | BCL2-associated athanogene 5 |
| 46 | < .001 | .049 | Fail | .032 | < .001 | 210338_s_at | HSPA8 | Heat shock 70 kDa protein 8 |
| 47 | < .001 | .049 | CCR | .002 | < .001 | 206548_at | FLJ23556 | Hypothetical protein FLJ23556 |
Abbreviations: CCR, complete continuous remission; fail, relapse.
Fig 2.

Genes differentially expressed in patients that remained in complete continuous remission v in those that relapsed. Heatmap of the 47–probe set signature that was predictive of outcome (which represented 41 unique genes).
To get an unbiased estimate for the prediction accuracy of each of the three models (LP1 through 3), we performed LOOCV. The misclassification rates for the three models were 0.2542, 0.3051, and 0.2881, respectively (sensitivity = 0.643, 0.642, and 0.643, respectively; specificity = 0.839, 0.742, and 0.774, respectively). The ROC accuracies were 0.8065, 0.7154, and 0.7316 (P < .0001, < .002, and < .001, respectively). These LOOCV results indicated that the three models were significantly predictive of outcome.
Validation of Outcome Prediction Models on Independent Patient Cohorts
Three large patient cohorts—POG, COALL, and DCOG—were used as independent sets for validation of the 47–probe set signature. Notably, the trend in the DCOG and COALL data sets of the association of the matched probe sets all agree with that observed in the 1961 data set, and this was also true of the POG data set with three exceptions (Appendix Table A2, online only).
The POG data set consisted of 220 patient cases (4-year CCR, n = 95; relapse, n = 125) of childhood B-precursor ALL. The estimated ROC accuracies for the three prediction models were 0.6119, 0.5820, and 0.5674, respectively. By using the one-sided Mann-Whitney U test, the P values were .00226, .0187, and .0436, respectively, which indicated that each of the predicted LP values of the three models were significantly predictive of outcome in the independent POG set. To further validate the predictive value of the three models, we fit the univariate and multivariate logistic regression models (Table 4). The LPs of all the three models were significantly associated with outcome (P < .05 for all). However, we did not find statistical evidence for the prognostic significance of the majority of the models when analysis was adjusted for age and WBC or for karyotype. Only model I retained prognostic significance when age, WBC, and karyotype were considered. Models II and III were significant after analysis was adjusted for karyotype but not for age and WBC. Similar results were obtained when only the HR subset of patients was analyzed (data not shown). Logistic regression with LPV (ie, the weighted sum of expression values of 20 probe sets common in the COG 1961 and POG datasets) as the explanatory variable indicated that LPV also was associated with a good outcome; P (one-tailed Wald test) = .007.
Table 4.
Validation of the Outcome Signature on POG Data Set
| Model | Analyses
|
|||||
|---|---|---|---|---|---|---|
| Univariate
|
Multivariate Adjusted for Age and WBC
|
Multivariate Adjusted for ALL Subtype*
|
||||
| Odds Ratio | P | Odds Ratio | P | Odds Ratio | P | |
| I (LP1) | 1.468 | .004 | 1.311 | .035 | 1.314 | .033 |
| II (LP2) | 1.363 | .016 | 1.242 | .070 | 1.346 | .026 |
| III (LP3) | 1.312 | .029 | 1.202 | .105 | 1.281 | .048 |
NOTE. Total number of patients in data set = 220. Data were analyzed with the U95Av2 microarray.
Abbreviations: POG, Pediatric Oncology Group; ALL, acute lymphoblastic leukemia.
Analysis was adjusted for TEL/AML1.
We next validated the three prediction models by using COALL data with Cox proportional hazards regression (Table 5). We again noted that the predicted LP values of all three models were significantly associated with outcome (P < .05), and they remained significant after analysis was adjusted for age and WBC but not for karyotype. Cox regression with LPV was significantly associated with outcome (P = .0002). The DCOG data set comprised of 92 (4-year CCR, n = 67; relapse, n = 25) B-lineage diagnostic samples. By using the one-sided Wilcoxon rank sum test, the P values were .030, .020, and .0635, respectively, which provided a significant or marginal association with outcome. Cox PH regression was performed to additionally validate the association of the predicted values with outcome (Table 6). We noted again that the hazard ratios were all less than 1, which indicated a consistent trend that the high predicted values were associated with good outcome. In the DCOG data set, the three models were statistically significant when considered on their own (univariate analysis) but were not after analysis was adjusted for WBC, age, and karyotype. Logistic regression yielded similar results (Appendix Table A3, online only). LPV with all 47 probe sets was significant (P = .02, Appendix Table A5, online only).
Table 5.
Validation of the Outcome Signature on COALL Data Set
| Model | Analyses
|
|||||
|---|---|---|---|---|---|---|
| Univariate
|
Multivariate Adjusted for Age and WBC
|
Multivariate Adjusted for ALL Subtype*
|
||||
| Hazard Ratio | P | Hazard Ratio | P | Hazard Ratio | P | |
| I (LP1) | 0.898 | .015 | 0.917 | .041 | 0.934 | .135 |
| II (LP2) | 0.993 | .009 | 0.994 | .017 | 0.997 | .17 |
| III (LP3) | 0.961 | .020 | 0.967 | .036 | 0.983 | .23 |
NOTE. Total number of patients in data set = 145. Data were analyzed with the U133A microarray.
Abbreviations: COALL, German Cooperative Study Group for Childhood Acute Lymphoblastic Leukemia; ALL, acute lymphoblastic leukemia; MLL, mixed lineage leukemia; BCR, break point cluster region; ABL, Abelson murine leukemia viral oncogene; TEL, translocation ETS leukemia, AML, acute myeloid leukemia.
Analysis was adjusted for MLL, BCRABL, TEL/AML1, hyperdiploid, and E2A subtypes.
Table 6.
Validation of the Outcome Signature on DCOG Data Set
| Model | Analyses
|
|||||
|---|---|---|---|---|---|---|
| Univariate
|
Multivariate Adjusted for Age and WBC
|
Multivariate Adjusted for ALL Subtype*
|
||||
| Hazard Ratio | P | Hazard Ratio | P | Hazard Ratio | P | |
| I (LP1) | 0.836 | .010 | 0.919 | .155 | 0.981 | .415 |
| II (LP2) | 0.987 | .016 | 0.991 | .090 | 0.991 | .100 |
| III (LP3) | 0.938 | .047 | 0.968 | .215 | 0.981 | .350 |
NOTE. Total number of patients in data set = 92. Data were analyzed with the U133Plus2.0 microarray.
Abbreviations: COALL, German Cooperative Study Group for childhood acute lymphoblastic leukemia; ALL, acute lymphoblastic leukemia; MLL, mixed lineage leukemia; BCR, break point cluster region; ABL, Abelson murine leukemia viral oncogene; TEL, translocation ETS leukemia; AML, acute myeloid leukemia.
Analysis was adjusted for MLL, BCRABL, TEL/AML1, hyperdiploid, and E2A subtypes.
DISCUSSION
The goal of our study was to identify gene expression signatures in diagnostic samples that are predictive of early response to therapy and overall outcome in children with National Cancer Institute–defined HR ALL. All samples studied in these experiments were from patients who were treated on a single, contemporary protocol and who received intensified therapy according to a COG-modified Berlin-Frankfurt-Munster backbone, which thus minimized the effects of treatment variables.
Early response to therapy has proven to be one of the strongest predictors of outcome and now is routinely used to stratify patients according to the risk of relapse.17 We were able to identify and validate a gene expression signature that correlated with the kinetics of regression of tumor burden, as assessed by the bone marrow blast content on day 7. Apoptosis-facilitating genes, such as BIM and PDCD6, were upregulated in RER patients, whereas multiple genes involved in cell adhesion (eg, GPR56, PARVG), cell proliferation (eg, CKLF, BMP2), and antiapoptosis (eg, BCL2, SOCS2) were upregulated in SER patients. If this signature is validated with additional research, more rapid approaches to assessment of gene expression could be used so that augmented therapy might be deployed early—within the first few days of diagnosis—to overcome slow response and possibly the emergence of drug-resistant clones and, ultimately, to improve outcome.
Other investigators also have sought to identify gene expression profiles associated with early response to therapy. Two recent publications from Flotho et al18,19 have portrayed signatures that correlated with minimal residual disease at day 1918 and at day 4619 of induction. Though only five of 44 probe sets from the day 19 signature reached statistical significance in our data set of day 7 response, the trend of association for all the probe sets was remarkably strong. Not surprisingly, this trend was not observed with the day 46 signature (data not shown). Previous studies show that the kinetics of blast reduction is quite steep in the first 2 weeks of induction and is much slower thereafter.20 Thus, although day 7 bone marrow morphology and end induction minimal residual disease may correlate,21 it is likely that fundamental differences exist in the mechanisms of leukemia cell death that occurs in early compared with late induction.
Though various groups have performed microarray experiments on childhood ALL samples, it has proved difficult to identify a prognostic signature at diagnosis. For example, Yeoh et al7 were able to detect distinct expression profiles that predicted relapse in T-cell acute lymphoblastic leukemia and hyperdiploid ALL but not in other subtypes.7 Although expression of OPAL1 predicts ALL in some studies, it has not been validated in others, which suggests that differences in treatment may influence the prognostic impact of expression profiles.22 Other investigators have correlated gene expression signatures with in vitro drug response.14,23 However, this drug resistance profile was not selected for its prognostic value and, hence, may not represent the best selection of outcome-predictive genes. Despite these challenges, we have identified a gene expression signature that was predictive of long-term outcome and was validated in three independent cohorts of diagnostic samples from children who were treated on different protocols, which thus yielded an accurate perspective on the validity and reproducibility of the results.
Almost all of the genes that comprised our predictive signature were not identified in the studies mentioned above that looked at drug resistance and/or outcome. However, studies that have used microarray methodology to discover predictive signatures in other cancers also have shown little overlap in gene lists. Although these gene lists may not always be concordant between data sets, each signature still may be significantly predictive across the data sets. For example, five recently published predictive gene sets for outcome in breast cancer showed little overlap between sets.24 However, four of five were predictive of outcome in a single data set of 295 women, which emphasizes that, despite the lack of overlap, the signatures are reflective of common biologic subsets. This is consistent with our findings that demonstrated the ability of individual gene expression signatures and the derived models by using the COG samples to predict outcome on three different cohorts of patients.
The utilization of predictive signatures in clinical cancer trials is eagerly awaited. The application of array technology to define additional patients with ALL who have a poor outcome may be more difficult given the high cure rate of ALL and the elucidation of many well-established risk factors to date. One of the most crucial findings of our study was that, although gene expression signatures correlated with outcome in univariate analyses in multiple data sets, they lost much significance when well-known outcome predictors, like age, initial WBC, and genotype, were taken into account. A logical interpretation of these findings is that the most important variables associated with treatment failure in ALL have been identified already. However, the inability to accurately predict outcome uniformly by using these conventional variables may be related in many instances to host factors. In addition, measurements of gene expression do not take into account important events, such as post-translational modifications. Another explanation is that prognostic signatures may exist within biologic subtypes of ALL only. It has been established that gene expression profiles correlate with ALL cohorts defined by molecular changes, such as translocations and ploidy. We specifically focused our efforts on National Cancer Institute–defined HR ALL, because known genetic subtypes account for only a minority of patients in this cohort, and we sought to identify novel biologic subtypes associated with outcome by using gene expression profiling. Our inability to define such a group might reflect the existence of smaller biologic subsets within this population that may not be possible to detect with the number of patient cases studied here. However, our study and similar ones by others, even if not predictive in multivariate analysis, are likely to lead to a biologic understanding of why certain clinical and laboratory variables are associated with clinical outcome. Such information is essential to derive more effective, tumor-specific therapies.
In summary, we have identified a gene expression signature that is significantly predictive of outcome in childhood ALL, but it does not seem to provide additional information beyond that contained in already established prognostic variables. The analysis of a larger number of samples may allow investigators to discover gene signatures that provide additional prognostic information. Strict adherence to uniform protocols for sample acquisition, processing, and array experimentation may facilitate comparison between data sets.25 In addition, analysis of gene expression profiles may lead to a biologic understanding of why clinical and laboratory variables are associated with outcome, and this information potentially may be exploited therapeutically.
AUTHORS' DISCLOSURES OF POTENTIAL CONFLICTS OF INTEREST
The authors indicated no potential conflicts of interest.
AUTHOR CONTRIBUTIONS
Conception and design: William L. Carroll, Stephen P. Hunger
Financial support: William L. Carroll, Cheryl L. Willman
Administrative support: Harland Sather, Stephen P. Hunger, William L. Carroll
Provision of study materials or patients: Stephen P. Hunger, Nita Seibel, Rob Pieters, Monique L. den Boer, Martin A. Horstmann, Cheryl L. Willman
Collection and assembly of data: Deepa Bhojwani, Huining Kang, Harland Sather, Wenjian Yang, Monique L. den Boer, Renee X. Menezes, Jeffrey W. Potter
Data analysis and interpretation: Deepa Bhojwani, Huining Kang, Monique L. den Boer, Renee X. Menezes, Wenjian Yang, Naomi P. Moskowitz, Dong-Joon Min, Richard Harvey
Manuscript writing: Deepa Bhojwani, Huining Kang, Monique L. den Boer, Elizabeth A. Raetz, Mary V. Relling, Stephen P. Hunger, William L. Carroll
Final approval of manuscript: Deepa Bhojwani, Huining Kang, Stephen P. Hunger, Monique L. den Boer, Rob Pieters, Mary V. Relling, Cheryl L. Willman, William L. Carroll
Supplementary Material
Appendix
Methods for polymerase chain reaction.
Five hundred nanograms of patient RNA was converted into cDNA using Maloney murine leukemia virus–reverse transcriptase in a 20-μL reaction volume (Invitrogen Corp, Carlsbad, CA). This cDNA was diluted to a final volume of 50 μL by the addition of 30 μL 1× TE. For the qualitative polymerase chain reaction (PCR) analysis, 5 μL of this diluted cDNA (equivalent to 50 ng of starting RNA) was subjected to 40 cycles of amplification with the appropriate primers for each particular translocation in a model 9700 thermocycler (Applied Biosystems, Foster City, CA).
After amplification, the products of the t(1;19), t(12;21), and t(4;11) reactions were analyzed using capillary electrophoresis with the DNA 1,000 chips and a model 2100 Bioanalyzer (Agilent Technologies, Santa Clara, CA). Those samples showing products consistent with the predicted translocation sizes were verified by Southern blot analysis and hybridization with fluorescein-labeled oligonucleotide probes. Detection was done using a chemiluminescent detection kit (DAKO Corp, Carpinteria, CA). Quantitative PCR for the t(9;22) translocations was performed on the ABI model 7900 (Applied Biosystems) using primers to detect both the e1a2 and b2a2/b3a2 forms as well as an endogenous control gene, EEF2. The equivalent of 50 ng of starting RNA (5 μL of the diluted cDNA) was used for each of the three reactions. A fusion probe for the e1a2 product was used to quantify its levels. The b2a2 and b3a2 products were quantified together with a consensus probe in the b2 exon that is common to both forms. The EEF2 gene was used to show the quality and quantity of the cDNA as well as to normalize the samples. Although the detection of the t(9;22) products was performed quantitatively, the results for the purposes of assignment are scored as either positive or negative.
Statistical models for outcome prediction.
The logistic regression model can be written as
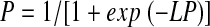 |
(1) |
where P is the probability of complete continuous remission in the selected population and LP is a linear combination of the predictors. An optimal subset of variables was selected to build the model using three variable selection methods (backward, forward, and stepwise) with a significance level of .05 for predictor to enter or to stay in the model. Three different best models, which included several genes and no clinical variables as predictors of outcome, were identified (Appendix Table A3).
In addition, LPV was the simple linear combination of the expression values of the probe sets that match the 47 probe sets in each of the three validation cohorts.
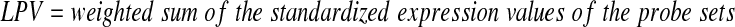 |
(2) |
where the weights were the t test statistics calculated in the Children's Oncology Group 1961 data set. Included in each of the three LPV models were 20 probe sets in the Pediatric Oncology Group data set, 31 probe sets in German Cooperative Study Group for Childhood ALL (acute lymphoblastic leukemia) or 47 probe sets in the Dutch Childhood Oncology Group data sets separately.
Table A1.
Patient Characteristics
| Patient | Sex | Age (months) | WBC (×109/L) | Translocation | Description |
|---|---|---|---|---|---|
| O 1 | Male | 46 | 59,300 | RER | |
| O 2 | Female | 134 | 8,700 | t(1;19) | RER, CCR |
| O 3 | Male | 25 | 70,000 | SER | |
| O 4 | Male | 170 | 732,000 | t(4;11) | RER |
| O 5 | Female | 208 | 314,600 | SER | |
| O 6 | Female | 161 | 2,100 | t(12;21) | RER |
| O 7 | Female | 132 | 99,500 | t(12;21) | SER |
| O 8 | Male | 118 | 66,700 | RER | |
| O 9 | Female | 19 | 71,500 | SER | |
| O 10 | Female | 27 | 65,200 | SER | |
| O 11 | Female | 198 | 44,580 | RER | |
| O 12 | Female | 16 | 161,700 | SER | |
| O 13 | Male | 61 | 65,800 | RER | |
| O 14 | Male | 187 | 2,950 | SER | |
| O 15 | Male | 202 | 68,000 | t(9;22) | RER |
| O 16 | Male | 80 | 144,000 | RER | |
| O 17 | Female | 18 | 64,100 | SER | |
| O 18 | Female | 31 | 92,800 | t(1;19) | RER |
| O 19 | Female | 171 | 9,800 | t(1;19) | RER |
| O 20 | Male | 177 | 61,800 | SER, relapse | |
| O 21 | Female | 129 | 2,500 | RER | |
| O 22 | Female | 209 | 4,000 | RER | |
| O 23 | Male | 133 | 8,000 | t(1;19) | RER |
| O 24 | Female | 134 | 30,700 | t(1;19) | RER |
| O 25 | Male | 144 | 15,600 | SER | |
| O 26 | Female | 191 | 10,500 | RER | |
| O 27 | Male | 34 | 84,700 | t(1;19) | RER |
| O 28 | Female | 39 | 97,000 | SER | |
| O 29 | Female | 16 | 191,000 | RER | |
| O 30 | Male | 158 | 50,000 | t(9;22) | SER |
| O 31 | Female | 117 | 300,000 | SER | |
| O 32 | Female | 14 | 279,000 | RER | |
| O 33 | Male | 213 | 68,300 | SER | |
| O 34 | Female | 39 | 64,900 | SER | |
| O 35 | Male | 66 | 88,100 | SER | |
| O 36 | Female | 150 | 34,400 | RER | |
| O 37 | Male | 154 | 54,000 | t(9;22) | SER, relapse |
| O 38 | Female | 50 | 76,400 | t(12;21) | RER, CCR |
| O 39 | Female | 136 | 50,300 | SER, CCR | |
| O 40 | Female | 125 | 6,900 | t(12;21) | RER, CCR |
| O 41 | Male | 126 | 10,700 | SER, relapse | |
| O 42 | Female | 129 | 87,600 | SER, relapse | |
| O 43 | Male | 177 | 45,900 | SER, CCR | |
| O 44 | Female | 41 | 90,800 | SER, CCR | |
| O 45 | Male | 167 | 4,400 | t(12;21) | SER, CCR |
| O 46 | Male | 176 | 93,500 | RER, relapse | |
| O 47 | Male | 52 | 165,000 | t(12;21) | SER, relapse |
| O 48 | Male | 70 | 121,500 | t(9;22) | SER, relapse |
| O 49 | Male | 39 | 86,700 | RER | |
| O 50 | Male | 109 | 253,100 | t(4;11) | RER, relapse |
| O 51 | Male | 128 | 68,100 | t(1;19) | RER, relapse |
| O 52 | Male | 158 | 164,000 | SER, relapse | |
| O 53 | Male | 193 | 28,000 | RER, relapse | |
| O 54 | Female | 185 | 1,800 | RER, relapse | |
| O 55 | Female | 41 | 64,500 | RER, CCR | |
| O 56 | Male | 83 | 65,000 | SER, CCR | |
| O 57 | Female | 29 | 178,000 | t(12;21) | SER, CCR |
| O 58 | Male | 139 | 9,440 | RER, CCR | |
| O 59 | Male | 101 | 58,700 | RER, relapse | |
| O 60 | Male | 40 | 106,000 | SER, CCR | |
| O 61 | Male | 225 | 262,800 | SER, relapse | |
| O 62 | Female | 125 | 12,600 | t(12;21) | RER, CCR |
| O 63 | Male | 12 | 189,300 | SER, relapse | |
| O 64 | Male | 135 | 71,670 | SER, relapse | |
| O 65 | Female | 109 | 672,000 | t(9;22) | SER, CCR |
| O 66 | Male | 189 | 82,400 | RER, relapse | |
| O 67 | Male | 199 | 6,000 | RER, CCR | |
| O 68 | Female | 116 | 15,8000 | t(4;11) | SER, relapse |
| O 69 | Male | 191 | 91,800 | RER, relapse | |
| O 70 | Male | 183 | 36,000 | RER, CCR | |
| O 71 | Male | 188 | 303,900 | t(9;22) | SER, relapse |
| O 72 | Female | 126 | 165,900 | RER | |
| O 73 | Male | 169 | 4,600 | RER, CCR | |
| O 74 | Male | 106 | 271,700 | SER, relapse | |
| O 75 | Male | 138 | 9,900 | RER | |
| O 76 | Male | 80 | 55,000 | SER | |
| O 77 | Male | 214 | 17,900 | RER | |
| O 78 | Female | 158 | 6,700 | SER | |
| O 79 | Male | 153 | 62,800 | SER, relapse | |
| O 80 | Male | 75 | 51,100 | SER, CCR | |
| O 81 | Male | 191 | 31,100 | RER, CCR | |
| O 82 | Female | 19 | 138,550 | CCR | |
| O 83 | Male | 179 | 4,250 | Relapse | |
| O 84 | Male | 79 | 325,900 | Relapse | |
| O 85 | Male | 27 | 209,000 | t(1;19) | Relapse |
| O 86 | Male | 36 | 113,000 | CCR | |
| O 87 | Female | 146 | 315,200 | Relapse | |
| O 88 | Male | 141 | 19,900 | t(1;19) | CCR |
| O 89 | Male | 146 | 158,000 | Relapse | |
| O 90 | Female | 148 | 28,400 | CCR | |
| O 91 | Male | 173 | 44,400 | CCR | |
| O 92 | Male | 213 | 98,600 | Relapse | |
| O 93 | Male | 138 | 1,800 | CCR | |
| O 94 | Male | 125 | 12,900 | RER, CCR | |
| O 95 | Male | 126 | 2,200 | CCR | |
| O 96 | Male | 208 | 108,000 | Relapse | |
| O 97 | Male | 152 | 170,400 | Relapse | |
| O 98 | Female | 189 | 60,200 | CCR | |
| O 99 | Male | 178 | 260,500 | t(4;11) | Relapse |
NOTE. Bold text indicates patients were common in both analyses (early response and outcome).
Abbreviations: RER, rapid early responder; SER, slow early responder; CCR, complete continuous remission.
Table A2.
Primer Sequences for RT-PCR
| Assay | Primer Sequence |
|---|---|
| EEF2 (TaqMan) | |
| EEF2 10(+)a | GAAGCGGCTGGCCAAGTCC |
| EEF2 12(−)a | CGACTCTTCACTGACCGTCTCG |
| EEF2 Probe (BHQ) | CCATGGTGCAGTGCATCATCGAGGAGTCGG |
| TEL-AML | |
| AML4 | CAGAGTGCCATCTGGAACAT |
| TEL5 | AACCTCTCTCATCGGGAAGA |
| TEL-AML2 FLU | GCAGAATGCATACTTGGAATG |
| TEL-AML3 FLU | ATAGCAGATGCCAGCACGAGC |
| TEL-AML4 FLU | ATAGCAGGTGGTGGCCCTAGG |
| BCR-ABL (TaqMan for both B2/3 and E1 forms) | |
| E1(+)A | CTGCCCGGTTGTCGTGTC |
| BCR2(+)a | CTGACCAACTCGTGTGTGAAAC |
| ABL2(−) | CTCAGACCCTGAGGCTCAAAG |
| E1 Probe | CAAGACCGGGCAGATCTGGCCCAAC |
| B2 Probe | CTGTCCACAGCATTCCGCTGACCATCA |
| E2A-PBX | |
| E2A | CTCCACGGCCTGCAGAGTAAG |
| PBX | GCCACGCCTTCCGCTAACA |
| E2A-PBX FLU | ACAGTGTTTTGAGTATCCGAGG |
| MLL-AF4 (nested) | |
| MLL-I | GGTCTCCCAGCCAGCACTGG |
| AF4-I | GCATGGATGACGTTCCTTGCTG |
| MLL-II | GCCTCAGCCACCTACTACAG |
| AF4-II | TTTTGGTTTTGGGTTACAGA |
| MLL FLU | TCCCAAAACCACTCCTAGTGAGC |
| AF4 FLU | GACTCTCAGCATGTCAGTTCTG |
Abbreviations: RT-PCR, reverse transcriptase polymerase chain reaction; EEF2, eukaryotic translation elongation factor 2; TEL, translocation ETS leukemia; AML, acute myeloid leukemia; BCR, break point cluster region; ABL, Abelson murine leukemia viral oncogene; PBX, pre B-cell leukemia transcription factor; MLL, mixed linage leukemia; AF4, ALL1 fused gene from chromosome 1.
Table A3.
Logistic Regression Models
| Model I (backward) | LP1 = −66.128 + 4.0681 × (VBP1) − 2.1351 × (HSPA8) + 4.6574 × (MGRN1) |
|---|---|
| Model II (forward) | LP2 = −1,170.3 + 47.9138 × (YWHAZ) + 32.0034 × (VBP1) − 16.0348 × (AGPS) + 4.8997 × (PTK2) + 45.9633 × (MGRN1) |
| Model III (stepwise) | LP3 = −238.6 + 6.3478 × (YWHAZ) + 7.2844 × (VBP1) + 0.8561 × (PTK2) + 8.5699 × (MGRN1) |
Table A4.
Validation for 47 Probe Sets (1)
| U133 ID | High → | U95 ID | Validation With POG Data
|
Validation With COALL
|
Validation With DCOG
|
Symbol | Gene Description | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Odds Ratio of CCR | High → | P | P Adjusting for Subtype | Hazard Ratio | High → | P | P Adjusting for Subtype | Hazard Ratio | High → | P | P Adjusting for Subtype | |||||
| 35666_at | CCR | 0.8270 | CCR | .1652 | .2244 | 0.5347 | CCR | .0083 | .0150 | SEMA3F | Sema domain, immunoglobulin domain (Ig), short basic domain, secreted, (semaphorin) 3F | |||||
| 227877_at | Fail | No match | 3.4569 | Fail | .0003 | .0079 | Similar to annexin II receptor (LOC389289), mRNA | |||||||||
| 227131_at | CCR | No match | 0.3778 | CCR | .0073 | .1170 | MAP3K3 | Mitogen-activated protein kinase kinase kinase 3 | ||||||||
| 205401_at | Fail | 39225_at | 0.7709 | Fail | .0308 | .0444 | 1.4191 | Fail | .0227 | .2417 | 1.7024 | Fail | .0114 | .0302 | AGPS | Alkylglycerone phosphate synthase |
| 208687_x_at | Fail | 1179_at | 0.8418 | Fail | .1050 | .3281 | 1.5373 | Fail | .0246 | .2826 | 1.7072 | Fail | .1138 | .1809 | HSPA8 | Heat shock 70 kDa protein 8 |
| 212229_s_at | CCR | No match | 0.1971 | CCR | .0005 | .0023 | FBXO21 | F-box only protein 21 | ||||||||
| 212576_at | CCR | 32235_at | 1.4168 | CCR | .0073 | .0526 | 0.5066 | CCR | .0355 | .0157 | 0.1713 | CCR | .0061 | .1709 | MGRN1 | Mahogunin, ring finger 1 |
| 225446_at | CCR | No match | 0.7464 | CCR | .2590 | .3139 | C21orf107 | Chromosome 21 open reading frame 107 | ||||||||
| 224793_s_at | CCR | No match | 0.5579 | CCR | .0332 | .0333 | TGFBR1 | Transforming growth factor, β receptor I (activin A receptor type II-like kinase, 53 kDa) | ||||||||
| 221840_at | CCR | No match | 0.5599 | CCR | .0001 | .1409 | 0.4861 | CCR | .0073 | .0796 | PTPRE | Protein tyrosine phosphatase, receptor type, E | ||||
| 203514_at | CCR | No match | 0.2618 | CCR | < .0001 | .0103 | 0.5114 | CCR | .0498 | .4390 | MAP3K3 | Mitogen-activated protein kinase kinase kinase 3 | ||||
| 1559018_at | CCR | No match | 0.7534 | CCR | .1851 | .2120 | PTPRE | Protein tyrosine phosphatase, receptor type, E | ||||||||
| 217499_x_at | Fail | No match | 1.6161 | Fail | .0358 | .3588 | 2.9133 | Fail | .0661 | .4768 | OR7E47P | Olfactory receptor, family 7, subfamily E, member 47 pseudogene | ||||
| 224187_x_at | Fail | 1180_g_at | 0.7093 | Fail | .0078 | .0846 | 2.0577 | Fail | .0639 | .0990 | HSPA8 | Heat shock 70 kDa protein 8 | ||||
| 40637_at | 0.8986 | Fail | .2164 | .5032 | ||||||||||||
| 221891_x_at | Fail | 33820_g_at | 0.8539 | Fail | .1247 | .4082 | 1.6161 | Fail | .0399 | .0533 | 1.6902 | Fail | .1898 | .2258 | HSPA8 | Heat shock 70 kDa protein 8 |
| 201642_at | CCR | 41140_at | 0.8839 | Fail | .8166 | .8323 | 0.8781 | CCR | .2963 | .4960 | 1.0325 | Fail | .5284 | .6506 | IFNGR2 | Interferon γ receptor 2 (interferon γ transducer 1 |
| 218418_s_at | CCR | No match | 0.6570 | CCR | .0065 | .1875 | 0.4928 | CCR | .0008 | .0135 | ANKRD25 | Ankyrin repeat domain 25 | ||||
| 242305_at | Fail | No match | 4.5128 | Fail | .0103 | .0057 | CDNA FLJ42757 fis, clone BRAWH3001712 | |||||||||
| 216035_x_at | CCR | No match | 0.5543 | CCR | .0000 | .0222 | 0.5389 | CCR | .0099 | .0514 | TCF7L2 | Transcription factor 7-like 2 (T-cell specific, HMG-box) | ||||
| 1556321_a_at | CCR | No match | 0.2974 | CCR | .0037 | .0127 | mRNA full-length insert cDNA clone EUROIMAGE 283668 | |||||||||
| 235014_at | Fail | No match | 2.6793 | Fail | .0476 | .0921 | LOC147727 | Hypothetical protein LOC147727 | ||||||||
| 208820_at | CCR | 36117_at | 1.1822 | CCR | .1137 | .2828 | 0.8187 | CCR | .0481 | .1307 | 0.6916 | CCR | .0725 | .4788 | PTK2 | PTK2 protein tyrosine kinase 2 |
| 212231_at | CCR | 32169_at | 1.3503 | CCR | .0194 | .1161 | 0.6376 | CCR | .0545 | .3078 | 0.3293 | CCR | .0068 | .0121 | FBXO21 | F-box only protein 21 |
| 229618_at | CCR | No match | 0.8772 | CCR | .3791 | .2522 | SNX16 | Sorting nexin 16 | ||||||||
| 209033_s_at | CCR | 1512_at | 1.0524 | CCR | .3541 | .2722 | 0.7866 | CCR | .2308 | .4579 | 0.3686 | CCR | .0303 | .0066 | DYRK1A | Dual-specificity tyrosine-(Y)-phosphorylation regulated kinase 1A |
| 36946_at | 1.0186 | CCR | .4464 | .4112 | ||||||||||||
| 200641_s_at | CCR | 34642_at | 1.0255 | CCR | .4269 | .3071 | 0.9139 | CCR | .3670 | .3979 | 1.1687 | Fail | .6451 | .6822 | YWHAZ | Tyrosine 3-monooxygenase/tryptophan 5-monooxygenase activation protein, zeta polypeptide |
| 202657_s_at | Fail | 37312_at | 1.0611 | CCR | .6674 | .6212 | 1.3910 | Fail | .1106 | .2672 | 3.4313 | Fail | .0050 | .1299 | SERTAD2 | SERTA domain containing 2 |
| 201099_at | CCR | No match | 0.9324 | CCR | .3890 | .3773 | 0.3949 | CCR | .0241 | .0022 | USP9X | Ubiquitin-specific protease 9, X-linked (fat facets-like, Drosophila) | ||||
| 201542_at | CCR | No match | 0.4317 | CCR | .0086 | .1713 | 0.2702 | CCR | .0050 | .0048 | SARA1 | SAR1a gene homolog 1 (S. cerevisiae) | ||||
| 227068_at | CCR | No match | 0.5209 | CCR | .0565 | .0508 | PGK1 | Phosphoglycerate kinase 1 | ||||||||
| 213944_x_at | CCR | 41476_at | 0.8669 | Fail | .8518 | .7282 | 0.9231 | CCR | .4079 | .3152 | 0.4148 | CCR | .0360 | .4760 | GNA11 | Guanine nucleotide binding protein (G protein), alpha 11 (Gq class) |
| 201472_at | CCR | 171_at | 1.1376 | CCR | .1773 | .1634 | 0.7118 | CCR | .0734 | .4987 | 0.6076 | CCR | .1698 | .1764 | VBP1 | von Hippel-Lindau binding protein |
| 202806_at | CCR | 37981_at | 1.8523 | CCR | .0001 | .0007 | 0.7634 | CCR | .0785 | .3280 | 0.4670 | CCR | .0244 | .2112 | DBN1 | drebrin 1 |
| 221918_at | CCR | No match | 0.5827 | CCR | .0124 | .1428 | 0.4768 | CCR | .0492 | .0195 | PCTK2 | PCTAIRE protein kinase 2 | ||||
| 214585_s_at | Fail | 32658_at | 0.9215 | Fail | .2743 | .2793 | 1.3231 | Fail | .0597 | .0929 | 4.3420 | Fail | .0448 | .0391 | VPS52 | Vacuolar protein sorting 52 (yeast) |
| 219078_at | Fail | No match | 1.0618 | Fail | .3451 | .3739 | 2.9254 | Fail | .0090 | .2201 | GPATC2 | G patch domain containing 2 | ||||
| 219133_at | Fail | No match | 2.3405 | Fail | .0612 | .0998 | FLJ20604 | Hypothetical protein FLJ20604 | ||||||||
| 1558111_at | Fail | No match | 2.0333 | Fail | .0287 | .1407 | MBNL1 | Muscleblind-like (Drosophila) | ||||||||
| 221773_at | CCR | No match | 0.7483 | CCR | .0001 | .4428 | 0.5803 | CCR | .0046 | .3046 | ELK3 | ELK3, ETS-domain protein (SRF accessory protein 2) | ||||
| 1558732_at | CCR | No match | 0.3631 | CCR | .0015 | .0369 | gb:AK074900.1/DB_XREF = gi:22760646/TID = Hs2.382077.1/CNT = 11/FEA = mRNA/... | |||||||||
| 212441_at | CCR | 37748_at | 1.4004 | CCR | .0118 | .0312 | 0.5488 | CCR | .0010 | .1296 | 0.3673 | CCR | .0171 | .0250 | KIAA0232 | KIAA0232 gene product |
| 226775_at | CCR | No match | 0.3359 | CCR | .0221 | .0963 | e(y)2 | e(y)2 protein | ||||||||
| 208498_s_at | Fail | 36680_at | 0.9858 | Fail | .4582 | .7287 | 1.0305 | Fail | .4201 | .0957 | 1.6438 | Fail | .0508 | .3211 | AMY2B | Amylase, α 2B; pancreatic |
| 201121_s_at | CCR | 38802_at | 1.1054 | CCR | .2349 | .1222 | 0.3499 | CCR | .0007 | .0377 | 0.3753 | CCR | .0256 | .0257 | PGRMC1 | Progesterone receptor membrane component 1 |
| 202984_s_at | CCR | No match | 0.8270 | CCR | .1357 | .3122 | 0.3916 | CCR | .0152 | .0197 | BAG5 | BCL2-associated athanogene 5 | ||||
| 210338_s_at | Fail | No match | 1.4333 | Fail | .0590 | .1634 | 1.6945 | Fail | .1165 | .1925 | HSPA8 | Heat shock 70 kDa protein 8 | ||||
| 206548_at | CCR | No match | 0.7866 | CCR | .0156 | .3473 | 0.4007 | CCR | .0003 | .0025 | FLJ23556 | Hypothetical protein FLJ23556 | ||||
NOTE. P values are one sided and uncorrected for multiple testing.
Abbreviations: ID, identification; POG, Pediatric Oncology Group; COALL, German Cooperative Study Group for Childhood ALL; ALL, acute lymphoblastic leukemia; DCOG, Dutch Childhood Oncology Group; CCR, complete continuous remission; Fail, relapse.
Table A5.
Validation of the Outcome Signature on DCOG Data Set Using Logistic Regression
| Model | Univariate
|
Multivariate Adjusting for Age and WBC
|
Multivariate Adjusting for ALL Subtype
|
|||
|---|---|---|---|---|---|---|
| Odds Ratio | P | Odds Ratio | P | Odds ratio | P | |
| I (LP1) | 1.233 | .011 | 1.175 | .059 | 0.653 | .874 |
| II (LP2) | 1.016 | .015 | 1.011 | .079 | 0.744 | .898 |
| III (LP3) | 1.078 | .046 | 1.054 | .139 | 0.781 | .771 |
Abbreviations: DCOG, Dutch Childhood Oncology Group; ALL, acute lymphoblastic leukemia.
Supported by Grants No. U01 CA114762, CA21765 (W.Y. and M.V.R.), and CA51001 (W.Y. and M.V.R.) from the National Cancer Institute; Director's Challenge Grant No. U01 CA88361 (C.L.W., W.L.C.); by the Penelope London Foundation; the Friedman Fund for Childhood Leukemia; the Walter Family Pediatric Leukemia Fund; the Garrett B. Smith Foundation (N.P.M.); the Pediatric Cancer Foundation; the Dutch Cancer Society and the Pediatric Oncology Foundation of Rotterdam (M.L.D., R.X.M., and R.P.); the Center of Medical Systems Biology, established by the Netherlands Genomics Initiative/Netherlands Organization for Scientific Research (R.X.M.); Grants No. U01 GM61393 and U01 GM61374 from the National Institutes of Health National Institute of General Medical Sciences Pharmacogenetics Research Network and Database (W.Y. and M.V.R.); and the American-Lebanese-Syrian Associated Charities (W.Y. and M.V.R.).
R.P. reports on behalf of the Dutch Childhood Oncology Group, The Hague, the Netherlands; M.A.H. reports on behalf of the German Cooperative Study Group for Childhood ALL, Hamburg, Germany.
Authors' disclosures of potential conflicts of interest and author contributions are found at the end of this article.
REFERENCES
- 1.Pui CH, Evans WE: Treatment of acute lymphoblastic leukemia. N Engl J Med 354:166-178, 2006 [DOI] [PubMed] [Google Scholar]
- 2.Schultz KR, Pullen DJ, Sather HN, et al: Risk and response-based classification of childhood B-precursor acute lymphoblastic leukemia: A combined analysis of prognostic markers from the Pediatric Oncology Group (POG) and Children's Cancer Group (CCG). Blood 109:926-935, 2007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Gaynon PS: Childhood acute lymphoblastic leukaemia and relapse. Br J Haematol 131:579-587, 2005 [DOI] [PubMed] [Google Scholar]
- 4.Alizadeh AA, Eisen MB, Davis RE, et al: Distinct types of diffuse large B-cell lymphoma identified by gene expression profiling. Nature 403:503-511, 2000 [DOI] [PubMed] [Google Scholar]
- 5.Hayes DN, Monti S, Parmigiani G, et al: Gene expression profiling reveals reproducible human lung adenocarcinoma subtypes in multiple independent patient cohorts. J Clin Oncol 24:5079-5090, 2006 [DOI] [PubMed] [Google Scholar]
- 6.Golub TR, Slonim DK, Tamayo P, et al: Molecular classification of cancer: Class discovery and class prediction by gene expression monitoring. Science 286:531-537, 1999 [DOI] [PubMed] [Google Scholar]
- 7.Yeoh EJ, Ross ME, Shurtleff SA, et al: Classification, subtype discovery, and prediction of outcome in pediatric acute lymphoblastic leukemia by gene expression profiling. Cancer Cell 1:133-143, 2002 [DOI] [PubMed] [Google Scholar]
- 8.Siebel NL, Steinherz PG, Sather HN, et al: Early postinduction intensification therapy improves surivival for children and adolescents with high-risk acute lymphoblastic leukemia: A report from the Children's Oncology Group. Blood 111:2548-2555, 2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Helman P, Veroff R, Atlas SR, et al: A Bayesian network classification methodology for gene expression data. J Comput Biol 11:581-615, 2004 [DOI] [PubMed] [Google Scholar]
- 10.Tibshirani R, Hastie T, Narasimhan B, et al: Diagnosis of multiple cancer types by shrunken centroids of gene expression. Proc Natl Acad Sci U S A 99:6567-6572, 2002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Benjamini Y, Hochberg Y: Controlling the false discovery rate: A practical and powerful approach to multiple testing. J R Statist Soc B 57:289-300, 1995 [Google Scholar]
- 12.Hosmer D, Lemeshow S: Applied Logistic Regression (ed 2). Hoboken, NJ, John Wiley and Sons Inc, 2000
- 13.Martin SB, Mosquera-Caro MP, Potter JW, et al: Gene expression overlap affects karyotype prediction in pediatric acute lymphoblastic leukemia. Leukemia 21:1341-1344, 2007 [DOI] [PubMed] [Google Scholar]
- 14.Holleman A, Cheok MH, den Boer ML, et al: Gene-expression patterns in drug-resistant acute lymphoblastic leukemia cells and response to treatment. N Engl J Med 351:533-542, 2004 [DOI] [PubMed] [Google Scholar]
- 15.Kamps WA, Bokkerink JP, Hakvoort-Cammel FG, et al: BFM-oriented treatment for children with acute lymphoblastic leukemia without cranial irradiation and treatment reduction for standard risk patients: Results of DCLSG protocol ALL-8 (1991-1996). Leukemia 16:1099-1111, 2002 [DOI] [PubMed] [Google Scholar]
- 15a.Ashburner M, Ball CA, Blake JA, et al: Gene ontology: Tool for the unification of biology—The Gene Ontology Consortium. Nature Genetics 25:25-29, 2000 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Newman JC, Weiner AM: L2L: A simple tool for discovering the hidden significance in microarray expression data. Genome Biol 6:R81, 2005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Nachman JB, Sather HN, Sensel MG, et al: Augmented post-induction therapy for children with high-risk acute lymphoblastic leukemia and a slow response to initial therapy. N Engl J Med 338:1663-1671, 1998 [DOI] [PubMed] [Google Scholar]
- 18.Flotho C, Coustan-Smith E, Pei D, et al: A set of genes that regulate cell proliferation predicts treatment outcome in childhood acute lymphoblastic leukemia. Blood 110:1271-1277, 2007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Flotho C, Coustan-Smith E, Pei D, et al: Genes contributing to minimal residual disease in childhood acute lymphoblastic leukemia: Prognostic significance of CASP8AP2. Blood 108:1050-1057, 2006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Brisco MJ, Sykes PJ, Dolman G, et al: Early resistance to therapy during induction in childhood acute lymphoblastic leukemia. Cancer Res 60:5092-5096, 2000 [PubMed] [Google Scholar]
- 21.Borowitz MJ, Pullen DJ, Shuster JJ, et al: Minimal residual disease detection in childhood precursor-B-cell acute lymphoblastic leukemia: Relation to other risk factors—A Children's Oncology Group study. Leukemia 17:1566-1572, 2003 [DOI] [PubMed] [Google Scholar]
- 22.Holleman A, den Boer ML, Cheok MH, et al: Expression of the outcome predictor in acute leukemia 1 (OPAL1) gene is not an independent prognostic factor in patients treated according to COALL or St Jude protocols. Blood 108:1984-1990, 2006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Lugthart S, Cheok MH, den Boer ML, et al: Identification of genes associated with chemotherapy cross resistance and treatment response in childhood acute lymphoblastic leukemia. Cancer Cell 7:375-386, 2005 [DOI] [PubMed] [Google Scholar]
- 24.Fan C, Oh DS, Wessels L, et al: Concordance among gene-expression-based predictors for breast cancer. N Engl J Med 355:560-569, 2006 [DOI] [PubMed] [Google Scholar]
- 25.Staal FJ, Cario G, Cazzaniga G, et al: Consensus guidelines for microarray gene expression analyses in leukemia from three European leukemia networks. Leukemia 20:1385-1392, 2006 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.