

Abstract
This paper describes the substrate scope and mechanism of Pd-catalyzed ligand-directed C–H arylation with diaryliodonium salts. This transformation was applied to the synthesis of a variety of different biaryl products, using directing groups including pyridines, quinolines, pyrrolidinones, and oxazolidinones. Electronically and sterically diverse aryl groups (Ar) were transferred in high yield using iodine(III) reagents of general structure [Mes–I–Ar]BF4. Mechanistic investigations have been conducted that establish the kinetic order of the catalytic reaction in each component, determine the resting state of the catalyst and the iodine(III) reagent, quantify the electronic influence of the arylating reagent on the reaction rate, and establish the intra- and intermolecular 1º H/D kinetic isotope effect. On the basis of these studies, this transformation is proposed to proceed via turnover limiting oxidation of the Pd dimer [Pd(N∼C)(OAc)]2 (N∼C = 3-methyl-2-phenylpyridine) by [Mes–I–Ph]BF4. This mechanism implicates a bimetallic high oxidation state Pd species as a key catalytic intermediate. The significance of this and other aspects of the proposed mechanism are discussed in detail.
Introduction
Biaryls are important synthetic targets, as they serve as key structural motifs of diverse natural products, pharmaceuticals, agrochemicals, and conjugated materials. The most common method for forming biaryl linkages involves transition metal-catalyzed cross coupling (e.g., Suzuki-Miyaura, Stille, Hiyama, Sonogashira, Kumada, and Negishi reactions).1 While these transformations have found widespread application, they suffer from the disadvantage that they require the use of two pre-functionalized starting materials. As such, each coupling reagent must be independently prepared, which adds one or more additional steps to a synthetic sequence.
More recent efforts have begun to explore metal-catalyzed C–H arylation as an alternative strategy for the construction of biaryls.2,3,4 This approach is attractive because it allows direct replacement of an Ar–H bond with an Ar–Ar’ bond, eliminating the need for a pre-installed functional group. In addition, appropriate functional groups can be used to direct highly site selective C–H arylation within complex organic molecules. In recent years, there has been a flurry of activity in this field, and ligand-directed C–H arylation methods have been reported with various late transition metal catalysts.2–4
Our group has been particularly interested in developing Pd-catalyzed ligand-directed C–H arylation reactions using [Ar–IIII–Ar’]X reagents.5,6 These iodine(III) compounds were selected on the basis of significant evidence that they could promote biaryl formation via an unusual PdII/IV catalytic cycle.7,8 We anticipated that a PdII/IV pathway for C–H arylation would be highly complementary to related reactions in the literature, particularly those involving more common Pd0/II mechanisms. For example, PdII/IV sequences should tolerate important functional groups (e.g., aryl halides and enolizable ketones) that can be reactive with the low valent Pd intermediates and/or strong bases often required in Pd0/II processes. In addition, high oxidation state Pd species are known to be stable towards air and moisture,7,8 obviating the need for special glassware and/or for rigorous purification of solvents and reagents. Finally, since PdII/IV-catalyzed reactions remain relatively rare,2,4–8 we reasoned that mechanistic studies could provide novel insights that might be broadly applicable to this class of transformations.
We report herein a detailed investigation of the substrate scope and mechanism of Pd(OAc)2-catalyzed ligand-directed C–H arylation with [Ar–I–Ar’]BF4.5a Studies of reaction order in each component, Hammett analysis, kinetic isotope effect data, and equilibrium investigations have all been used to interrogate the resting state of the catalyst and the oxidant under the reaction conditions. These investigations provide strong evidence for oxidation as the turnover-limiting step and suggest the formation of a dimeric high oxidation state Pd complex as a key catalytic intermediate. The potential implications of these findings are discussed.
Results and Discussion
Synthetic Scope
Initial studies examined the Pd(OAc)2-catalyzed ortho-phenylation of 3-methyl-2-phenylpyridine (1) with the hypervalent iodine reagent [Ph2I]BF4.5a This reaction afforded the mono-phenylated product 2 in a variety of common solvents, with the optimal conditions being 1.1 equiv of [Ph2I]BF4 and 5 mol % of Pd(OAc)2 at 100 °C in AcOH (eq. 1). Notably, this reaction proceeded efficiently in the presence of ambient moisture and air, and no special purification of solvents or reagents was required.
 |
(1) |
Numerous directing groups, including pyridines, quinolines, pyrrolidinones, and oxazolidinones, were also effective at promoting both aromatic and benzylic C–H phenylation with [Ph2I]BF4 (Table 1).5a This transformation tolerated the presence of enolizable ketones, aldehydes, ethers, amides, benzylic hydrogens, and aryl halides within the organic substrate. Good to excellent yields were obtained regardless of the electronic nature of the aromatic compound. Further, excellent (>20: 1) site selectivity was observed when a meta-substituent was present on the arene undergoing functionalization.9
Table 1.
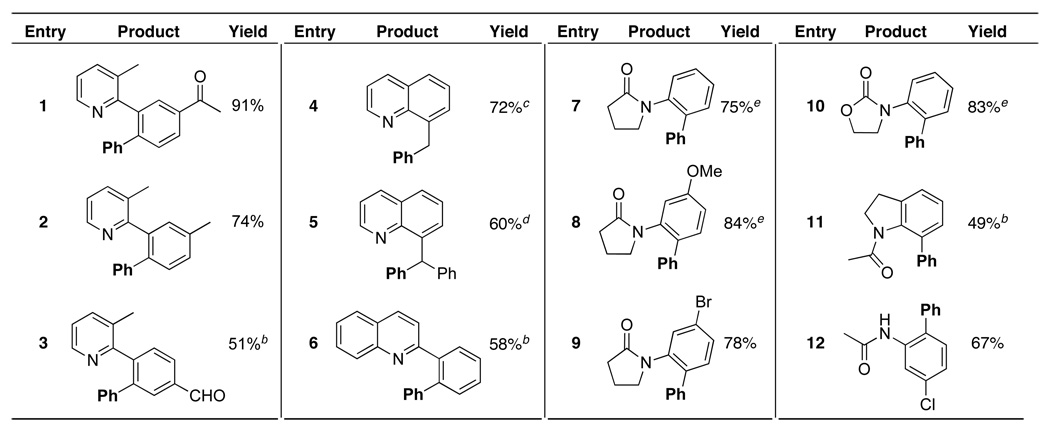
|
Conditions: 1 equiv of substrate, 1.1–2.5 equiv of [Ph2I]BF4, 5 mol % of Pd(OAc)2 in AcOH, AcOH/Ac2O, benzene, or toluene, 100 ºC, 8–24 h.
The balance of material was starting material (entry 11) or a mixture of starting material and diarylated product (entries 3 and 6).
Conditions: 2 equiv of 8-methylquinoline, 1 equiv of [Ph2I]BF4
Approximately 16% of the product in entry 5 was formed in the absence of Pd(OAc)2
1.2–2 equiv of NaHCO3 were added.
To further expand the utility of this methodology, we explored the installation of diverse arenes (Ar). We initially reasoned that selective transfer of different aryl groups might be achieved based on electronic preferences, and thus, a series of electronically unsymmetrical iodine(III) reagents [Ph–I–p-XC6H4]BF4 were examined. As summarized in Table 2, these reagents underwent preferential transfer of the most electron deficient aromatic group; however, the levels of selectivity were only modest, with ratios of 2: 2-Ar ranging from 2.6: 1 to 1: 0.3.
Table 2.
Arylation of 3-Methyl-2-Phenylpyridine with [Ar–I–Ar]BF4
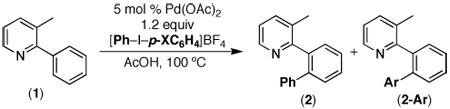 | ||
|---|---|---|
| Entry | X | 2: 2-Ar |
| 1 | CF3 | 2.6: 1 |
| 2 | F | 1: 0.33 |
| 3 | Cl | 1: 0.83 |
| 4 | CH3 | 1: 0.71 |
| 5 | OMe | 1: 0.31 |
We next sought to sterically differentiate the two Ar groups on iodine(III), reasoning that smaller groups should transfer more rapidly. Gratifyingly, the mesityl/aryl substituted reagents [Mes–I–Ar]BF4 reacted to transfer the smaller Ar group with high selectivity. As summarized in Table 3 and Table 4, pyridine and pyrrolidinone products containing both electron rich and electron deficient Ar could be prepared in high yield using these reagents. Many substituents were tolerated on the aryl ring being transferred, including enolizable ketones, ethers, benzylic hydrogens, and halogens. Further, these reagents were effective for the selective installation of relatively large aryl groups like ortho-tolyl.
Table 3.
Pd-Catalyzed Arylation of 3-Methyl-2-Arylpyridines with [Mes–I–Ar]BF4
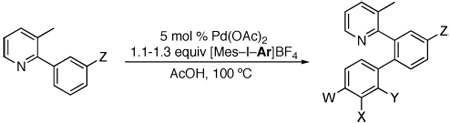 | |||||
|---|---|---|---|---|---|
| Entry | W | X | Y | Z | Yield |
| 1 | H | H | H | H | 85% |
| 2 | CF3 | H | H | H | 87% |
| 3 | F | H | H | H | 88% |
| 4 | Cl | H | H | H | 83% |
| 5 | Me | H | H | H | 84% |
| 6 | OMe | H | H | H | 81%a |
| 7 | C(O)Me | H | H | H | 81% |
| 8 | H | C(O)Me | H | H | 81% |
| 9 | H | CHO | H | H | 88% |
| 10 | H | H | Me | H | 72% |
| 11 | CF3 | H | H | OMe | 81%b |
| 12 | Cl | H | H | OMe | 79%b |
| 13 | F | H | H | OMe | 79%b |
| 14 | Me | H | H | OMe | 78%b |
| 15 | OMe | H | H | OMe | 56%b,c |
Reaction carried out at 120 ºC.
Traces of the Mes addition product were observed, but selectivity was generally >12: 1. See supporting information for further details.
With 2 equiv of [Mes–I–(p-MeOC6H4)]BF4.
Table 4.
Pd-Catalyzed Arylation of 2-Aryl Pyrrolidinones with [Ar–I–Ar]BF4
 | ||||
|---|---|---|---|---|
| Entry | X | Y | Z | Yield |
| 1 | Me | H | MeO | 83% |
| 2 | H | CF3 | MeO | 84% |
| 3 | Me | H | H | 81% |
Mechanistic Investigations
We next sought to gain a detailed mechanistic understanding of this Pd-catalyzed C–H arylation. Our previous investigations showed that the reaction is unaffected by the addition of 500 equiv of Hg0 or 25 mol % of the free radical inhibitors MEHQ and galvinoxyl.5a These results suggest against mechanisms involving palladium nanoparticles10 or free radical intermediates.11 In addition, palladacycle 3 was shown to be an effective catalyst, with a comparable kinetic profile to Pd(OAc)2. Further, the stoichiometric reaction between 3 and [Ph2I]BF4 afforded the ortho-arylated product 2 in high yield (eq. 2).5a In contrast, more traditional Pd0/II electrophiles like PhI and PhOTf were ineffective phenylating reagents under both catalytic and stoichiometric reaction conditions. Finally, Canty and Malinakova have both demonstrated the stoichiometric oxidation of PdII model complexes with [Ar2I]X reagents to form observable monomeric PdIV species.7 On the basis of all of these results, the general catalytic cycle shown in Scheme 1 was proposed, involving: (i) ligand directed C–H activation to afford palladacycle A, (ii) oxidation of A with [Ph2I]BF4 to generate a PdIV intermediate of general structure B, and finally, (iii) C–C bond forming reductive elimination to afford the arylated product 2.
Scheme 1.

Originally Proposed Catalytic Cycle for Pd-Catalyzed C–H Arylation5a
 |
(2) |
These preliminary studies left several key outstanding questions regarding the C–H arylation mechanism. For example, the turnover-limiting step of this catalytic cycle remained to be established. In addition, detailed information about the ligand environment of the proposed PdIV intermediate B was not available from the initial investigations. Finally, the effect of electronic modification of the oxidant on the rates of these reactions was poorly understood. To gain further mechanistic insights, we have undertaken detailed studies to establish: (1) the kinetic order in each component of the catalytic reaction, (2) the resting state of the catalyst and iodonium reagent, (3) the electronic influence of the iodonium reagent on the reaction rate, and (4) the intra- and intermolecular 1º H/D kinetic isotope effects.
Kinetic order of the reaction
We first sought to determine the turnover limiting step of this transformation by establishing the kinetic order of C–H arylation in each reaction component. These studies focused on the Pd(OAc)2-catalyzed ortho-phenylation of 3-methyl-2-phenylpyridine (1) with [Mes–I–Ph]BF4 (4).12 We first examined the order of this transformation in the IIII reagent by combining 3-methyl-2-phenylpyridine (250 mM), Pd(OAc)2 (5 mM), and varying concentrations of 4 (75.2–175.2 mM) in AcOH at 110 °C. The reactions were monitored by gas chromatography, and the initial rates method was used to determine rate at each [IIII]. As shown in Figure 1, a plot of initial reaction rate (Δ[2]/Δt) versus [IIII] was linear, indicating a first order dependence on the IIII reagent.13 This result suggests that the iodine(III) oxidant is involved in the turnover-limiting step. Notably, this is in contrast to most other Pd-catalyzed ligand-directed C–H functionalization reactions, where cyclopalladation is typically rate-limiting.14
Figure 1.


Plot of Initial Rate (Δ[2]/Δt) versus [IIII] Showing 1st Order Kinetics in IIII Reagent
The order in [Pd] was next determined using an analogous procedure, with 3-methyl-2-phenylpyridine (250 mM), [Mes–I–Ph]BF4 (100 mM), and varying concentrations of [Pd(OAc)2] (3.8–10 mM) in AcOH at 110 °C.12 The data from these experiments was subjected to an unweighted nonlinear least squares fit to the equation: f(x) = a[Pd]n. This fit provided an order (n) of 2.09 ± 0.08 (supplementary Figure S4), which was also reflected in a linear plot of initial rate versus [Pd]2 (Figure 2). This second order dependence on [Pd] is consistent with a monomeric catalyst resting state that is in equilibrium with a dimeric active catalyst (vide infra).
Figure 2.

Plot of Initial Rate (Δ[2]/Δt) versus [Pd]2 Showing 2nd Order Kinetics in [Pd]
Finally, the order of the reaction in 3-methyl-2-phenylpyridine (1) was examined, using [Mes–I– Ph]BF4 (100 mM) and Pd(OAc)2 (5 mM) along with [1] ranging from 202–304 mM in AcOH at 110 °C.12 Qualitative experiments revealed that the reaction rate slowed dramatically with increasing [1], suggesting an inverse order dependence on the substrate. The quantitative rate data (initial rate versus [1]) was subjected to an unweighted non-linear least squares fit to the equation: f(x) = a[1]n, which revealed an inverse 3rd order dependence on 1 (n = –3.1 ± 0.2, supplementary Figure S10). As shown in Figure 3, this is also reflected in the linear fit of the plot of initial rate versus [1]−3. This result indicates that 3 equiv of 1 must be lost in order to progress from the resting state to the transition state.
Figure 3.

Plot of Initial Rate (Δ[2]/Δt) versus [1]−3 Showing Inverse 3rd Order Kinetics in 1
Catalyst resting state
We next sought to probe the resting state of the [Pd] under the reaction conditions. Since complex 3 was previously shown to serve as an effective catalyst for C–H arylation and underwent stoichiometric reaction with [Ph2I]BF4 to afford 2 (eq. 2),5a we hypothesized that 3 might be a catalytic intermediate. When dimer 3 was combined with 20 equiv of 1 at 110 ºC in AcOH (mimicking the catalytic reaction conditions, but in the absence of oxidant), only the corresponding monomer 7 (which shows a diagnostic broad resonance at 6.2 ppm) was observed by 1H NMR spectroscopy (Scheme 2 and Figure S19).15 This is consistent with a literature report by Ryabov showing that the closely related dimer 5 reacts with 2-phenylpyridine to form monomer 6 with Keq of 33 ± 2 at 70 ºC in AcOH (eq. 3).15 When the catalytic reaction between Pd(OAc)2 (0.0027 mmol), 1 (0.05 mmol), and 4 (0.025 mmol) was monitored by 1H NMR spectroscopy at 110 ºC, an identical broad signal at 6.2 ppm was observed throughout this transformation (Scheme 3 and Figure S19). These experiments collectively suggest that the resting state of the Pd during the catalytic cycle is monomer 7 (Scheme 2).
Scheme 2.

Observation of 7 Under the Catalytic Reaction Conditions
Scheme 3.

Proposed Mechanism for C–H Arylation of 3-Methyl-2-phenylpyridine
 |
(3) |
Oxidant resting state
Several literature reports have demonstrated that pyridine derivatives can coordinate to diaryliodonium salts.16 In a particularly relevant example, Ochiai has shown by 1H NMR spectroscopy that [Ph2I]BF4 forms a 1: 1 complex with pyridine (Keq = 20.2 M−1 at 24 ºC in CH2Cl2).16d Discrete “bound” and “free” adducts were not observed due to fast exchange on the NMR timescale. Instead, the binding interaction was monitored by changes in the chemical shifts associated with pyridine resonances at varying relative concentrations of pyridine and [Ph2I]BF4.
We conducted an analogous set of studies to examine the interaction between 1 and [Mes–I–Ph]BF4 (4) under our catalytic conditions (110 ºC in AcOH). First, we used a Job plot to establish the stoichiometry of complexation. A series of solutions with a constant total concentration ([1] + [4] = 23.6 mM in CD3CO2D), but varied mole fractions of the two components were examined by 1H NMR spectroscopy at 110 ºC.17 The chemical shift of H(4) of 1 moved downfield as the mole fraction of 4 increased, and the change in chemical shift relative to free 1 (ΔδH(4)) was used to construct a Job plot (Figure 4). This plot showed a maximum at χ = 0.5, indicating that complexation between 1 and 4 occurs with a 1: 1 stoichiometry. 18,19 This data implicates the formation of iodine(III) adduct 8 under the catalytic reaction conditions (eq. 4).
Figure 4.

Job plot of Δδ•χ (mole fraction of substrate) versus χ at 110 °C
 |
(4) |
The equilibrium constant for the reaction in eq. 4 was next established by measuring δH(4) while titrating in increasing [Mes–I–Ph]BF4 in CD3CO2D at 110 ºC (Figure 5). The resulting curve was fitted to a 1: 1 binding model,20 and the unweighted non-linear least squares fit afforded an equilibrium constant of 111 ± 18 M−1. This value of Keq is consistent with complex 8 (eq. 4) as the resting state of the oxidant under the catalytic conditions.
Figure 5.

δ as a Function of [4] at 110 ºC
Hammett study
The rate of the Pd(OAc)2-catalyzed reaction between 3-methyl-2-phenylpyridine (1) and [Mes–I–p-XC6H4]BF4 was examined as a function of the substituent X. Substrate 1 (100 mM) was combined with Pd(OAc)2 (5 mM) and the appropriate oxidant [Mes–I–(p-XC6H4)]BF4 (200 mM) in AcOH at 80 °C, and the initial reaction rate was measured with each iodine(III) reagent. A Hammett plot of the resulting data provided a ρ value of +1.7 ± 0.2 (Figure 6). This indicates that C–H arylation is strongly accelerated by electron withdrawing groups on the iodine(III) reagent.21
Figure 6.


Hammett for Pd-Catalyzed C–H Arylation of 1 with [Mes–I–Ar]BF4
Kinetic isotope effect
Finally, the 1º intra- and intermolecular kinetic isotope effects (kH/kD) for this C–H phenylation reaction were determined. As shown in eq. 5, the intramolecular 1º kH/kD for substrate 1-d4 was 2.5 ± 0.2, based on 1H NMR spectroscopic analysis of the isolated product. This is similar to that seen in other Pd-catalyzed ligand-directed C–H functionalization reactions, which typically show 1º intramolecular KIE values ranging from 2.2 to 6.7.22
The intermolecular kinetic isotope effect was determined by comparing the initial reaction rate of 3-methyl-2-phenylpyridine (1) to that of 3-methyl-2-(d5)-phenylpyridine (1-d5) (eq. 6). In this system, kH/kD was determined to be 1. The observed lack of an intermolecular KIE implies that C–H bond cleavage occurs after the rate-determining step of the reaction.
 |
(5) |
 |
(6) |
Summary of mechanistic data
The data assembled above allows us to formulate a complete mechanistic picture of the catalytic C–H arylation reaction. As shown in Scheme 3, we propose that the resting state of the catalyst is the monomeric PdII species 7, and the resting state of the oxidant is the pyridine coordinated iodine(III) reagent 8. This proposal is consistent both with the values of Keq from eq. 3 and Figure 5, as well as with the direct observation of 7 under the catalytic reaction conditions (Scheme 2). To enter the catalytic cycle, free oxidant 4 is generated by dissociation of 1 equiv of 1 from 8. In addition, the Pd dimer 3 is formed by the combination of 2 equiv of monomer 7, with concomitant dissociation of 2 equiv of 1. The reaction between 3 and 4 to afford high oxidation state dimeric intermediate 9 is then proposed to be the turnover limiting step of the catalytic cycle, which is followed by C–C bond-formation, ligand exchange, and cyclometalation to return to the catalyst resting state. The rate equation shown in eq. 7 can be derived on the basis of the proposed mechanism, and application of the pre-equilibrium approximation for [3] and [4] affords the rate expression shown in eq. 8.
| (7) |
| (8) |
The proposed mechanism is supported by all of the data presented above. For example, eq. 8 predicts that the reaction will be 1st order in [IIII], 2nd order in [Pd], and inverse 3rd order in 1, consistent with the experimental observations. In addition, the Hammett ρ value of 1.7 ± 0.2 is consistent with oxidative addition as the turnover limiting step. This value is comparable to that observed in Pd0/II oxidative addition reactions.23 Finally, the observation of an intra- but not an intermolecular kinetic isotope effect is consistent with C–H activation occurring during the catalytic cycle but after the turnover limiting step.
There are several notable features of the mechanism proposed in Scheme 3. First, the observed 2nd order dependence on [Pd] implicates bimetallic acetate-bridged Pd complex 9 as a key intermediate in this process. This dimer can be viewed as a PdIV species attached to a bridging PdII complex (as drawn in Scheme 3 and also 9a in Figure 7). Alternatively, if there is significant bonding between the two Pd centers, this may be considered a PdIII-PdIII dimer (9b, Figure 7). This latter possibility is particularly interesting given the proximity of the palladium centers in crystal structures of 5.24 In addition, Cotton and more recently Ritter have reported the formation of related symmetrical PdIII-PdIII dimers 11 and 13 by oxidation of acetate bridged PdII complexes with PhICl2 (Figure 8).25,26,27 Similar PtIII-PtIII dimers have also been formed by reaction of monomeric PtII starting materials with hypervalent iodine(III) reagents, including PhICl2,28 PhI(OAc)2 (10),29 and [Ph–I–R]OTf (R = CCSi(Me)3) (12) (Figure 8).30
Figure 7.

Two Alternative Formulations for Intermediate 9: PdIV/PdII (9a) versus PdIII∼PdIII (9b)
Figure 8.

Related PdIII∼PdIII and PtIII∼PtIII Complexes from the Literature
Notably, if intermediate 9 is a PdIII-PdIII dimer (9b), this complex could undergo direct C–C bond-forming reductive elimination (similar to that depicted in Scheme 3)27 or could disproportionate to PdIV and PdII species (either acetate bridged or unbridged) prior to C–C coupling from PdIV.28,30 In both formulations of the proposed intermediate (9a and 9b), the second Pd center effectively serves as an ancillary ligand to the metal complex that mediates C–C bond-formation. As such, an attractive strategy for the development of 2nd generation C–H arylation catalysts with improved activity and selectivity would involve tuning this organometallic ligand.
It is also interesting to compare the turnover limiting step of C–H arylation with [Ar2I]BF4 to that of C–H acetoxylation with PhI(OAc)2.14h Remarkably, although both reactions utilize an iodine(III) oxidant and proceed under nearly identical conditions (catalytic Pd(OAc)2, 100–110 ºC in AcOH), the turnover limiting steps are different. As described above, C–H arylation involves turnover-limiting oxidation. In contrast, Pd-catalyzed C–H acetoxylation shows a zero order dependence on PhI(OAc)2 and a large intermolecular 1º H/D kinetic isotope effect, suggesting that cyclopalladation is the turnover limiting step.31 These results have important implications for the application of iodine(III) reagents in other Pd-catalyzed reactions. For example, PhI(OAc)2 has been used to intercept Pd-alkyl intermediates formed via aminopalladation,32 acetoxypalladation,33 or carbopalladation34 to generate diverse products. A key feature of these transformations is that the oxidation of PdII by PhI(OAc)2 is faster than competing β-hydride elimination. The current results suggest that analogous oxidations of PdII-alkyl species with [Ar2I]BF4 will be significantly slower, which may result in undesired competing processes. Thus, we anticipate that new and more reactive arylating reagents will be required to access chemistry similar to that of PhI(OAc)2.
Conclusions
In summary, this paper describes detailed synthetic and mechanistic investigations of Pd-catalyzed C–H arylation reactions. The turnover limiting step of the catalytic cycle involves oxidation of Pd dimer 3 with [Mes–I–Ph]BF4 (4) to form a dimeric high oxidation state Pd intermediate (9), which can potentially be viewed as a mixed valent PdIV/PdII species or as a PdIII∼PdIII dimer. The observed kinetics have important implications for future applications of this chemistry; for example, the 2nd order dependence on catalyst and inverse 3rd order dependence on substrate suggest that these transformations will be highly sensitive to reaction concentration and stoichiometry, an important consideration upon scale up or down. In addition, the intermediacy of the dimeric species 3 and 9 suggests that catalyst activity and selectivity may be tuned by modification of the tethered metal center. Finally, these investigations raise the larger question of whether similar dimeric intermediates may be involved in other Pd-catalyzed C–H functionalization reactions. Ongoing investigations in our laboratory are seeking to address all of these issues, and the results will be reported in due course.
Supplementary Material
ACKNOWLEDGMENT
We acknowledge the NIH NIGMS (GM-073836) for support of this research. In addition, we thank Dr. Eugenio Alavarado for assistance with NMR spectroscopy and Jim Windak for assistance with mass spectrometry.
Footnotes
SUPPORTING INFORMATION PARAGRAPH. Experimental details and spectroscopic and analytical data for new compounds. This material is available free of charge via the Internet at http://pubs.acs.org.
REFERENCES
- 1.(a) Stille JK. Angew. Chem., Int. Ed. Engl. 1986:508. [Google Scholar]; (b) Farina V, Krishnamurthy V, Scott WJ. Org. React. 1997;50:1. [Google Scholar]; (c) Miyaura N, Suzuki A. Chem. Rev. 1995;95:2457. [Google Scholar]; (d) Suzuki AJ. Organomet. Chem. 1999;576:147. [Google Scholar]; (e) Beletskaya IP, Cheprakov AV. Chem. Rev. 2000;100:3009. doi: 10.1021/cr9903048. [DOI] [PubMed] [Google Scholar]; (f) Kotha S, Lahiri K, Kashinath D. Tetrahedron. 2002;58:9633. [Google Scholar]; (g) Denmark SE, Sweis RF. Acc. Chem. Res. 2002;35:835. doi: 10.1021/ar020001r. [DOI] [PubMed] [Google Scholar]; (h) Muci AR, Buchwald SL. Top. Curr. Chem. 2002;219:131. [Google Scholar]; (i) Negishi E, Anastasia L. Chem. Rev. 2003;103:1979. doi: 10.1021/cr020377i. [DOI] [PubMed] [Google Scholar]
- 2.For recent reviews that discuss C–H arylation, see: Campeau LC, Fagnou K. Chem. Commun. 2006:1253. doi: 10.1039/b515481m. Daugulis O, Zaitsev VG, Shabashov D, Pham QN, Lazareva A. Synlett. 2006:3382. Yu JQ, Giri R, Chen X. Org. Biomol. Chem. 2006;4:4041. doi: 10.1039/b611094k. Godula K, Sames D. Science. 2006;312:67. doi: 10.1126/science.1114731. Alberico D, Scott ME, Lautens M. Chem. Rev. 2007;107:174. doi: 10.1021/cr0509760. Beccalli EM, Broggini G, Martinelli M, Sottocornola S. Chem. Rev. 2007;107:5318. doi: 10.1021/cr068006f. Ackermann L. Synlett. 2007:507. Fairlamb IJS. Chem. Soc. Rev. 2007;36:1036. doi: 10.1039/b611177g. Campeau LC, Fagnou K. Chem. Soc. Rev. 2007;36:1058. doi: 10.1039/b616082d. Catellani M, Motti E, Della Ca N. Acc. Chem. Res. 2008;41:1512. doi: 10.1021/ar800040u. Kakiuchi F, Kochi T. Synthesis. 2008:3013. Li BJ, Yand SD, Shi ZJ. Synlett. 2008:949. McGlacken GP, Bateman LM. Chem. Soc. Rev. 2009 doi: 10.1039/b805701j.
- 3.For examples of ligand-directed Ar–H oxidative coupling, see: Hull KL, Lanni EL, Sanford MSJ. Am. Chem. Soc. 2006;128:14047. doi: 10.1021/ja065718e. Xia JB, You SL. Organometallics. 2007;26:4869. Hull KL, Sanford MSJ. Am. Chem. Soc. 2007;129:11904. doi: 10.1021/ja074395z. Li BJ, Tian LS, Fang Z, Shi ZJ. Angew. Chem., Int. Ed. 2008;47:1115. doi: 10.1002/anie.200704092. Brasche G, García-Fortanet J, Buchwald SL. Org. Lett. 2008;10:2207. doi: 10.1021/ol800619c.
- 4.For other proposals of Pd-catalyzed ligand-directed C–H arylation via a PdII/IV mechanism, see ref. 5a, and: Shabashov D, Daugulis O. Org. Lett. 2005;7:3657. doi: 10.1021/ol051255q. Zaitsev VG, Shabashov D, Daugulis OJ. Am. Chem. Soc. 2005;127:13154. doi: 10.1021/ja054549f. Reddy BVS, Reddy LR, Corey EJ. Org. Lett. 2006;8:3391. doi: 10.1021/ol061389j. Giri R, Maugel N, Li JJ, Wang DH, Breazzano SP, Saunders LB, Yu JQ. J. Am. Chem. Soc. 2007;129:3510. doi: 10.1021/ja0701614. Chiong HA, Pham QN, Daugulis O. J. Am. Chem. Soc. 2007;129:9879. doi: 10.1021/ja071845e. Thirunavukkarasu VS, Parthasarathy K, Cheng CH. Angew. Chem., Int. Ed. 2008;47:9462. doi: 10.1002/anie.200804153. Yang F, Wu Y, Zhu Z, Zhang J, Li Y. Tetrahedron. 2008;64:6782. Yang F, Wu Y, Li Y, Wang B, Zhang J. Tetrahedron. 2009;65:914.
- 5.(a) Kalyani D, Deprez NR, Desai LV, Sanford MSJ. Am. Chem. Soc. 2005;127:7330. doi: 10.1021/ja051402f. [DOI] [PubMed] [Google Scholar]; (b) Deprez NR, Kalyani D, Sanford MS. J. Am. Chem. Soc. 2006;128:4972. doi: 10.1021/ja060809x. [DOI] [PubMed] [Google Scholar]
- 6.For other examples of the use of [Ar2I]X in C–H arylation, see: Daugulis O, Zaitsev VG. Angew. Chem., Int. Ed. 2005;44:4046. doi: 10.1002/anie.200500589. Phipps RJ, Grimster NP, Gaunt MJ. J. Am. Chem. Soc. 2008;130:8172. doi: 10.1021/ja801767s. Phipps RJ, Gaunt M. J. Science. 2009;323:1593. doi: 10.1126/science.1169975.
- 7.(a) Bayler A, Canty AJ, Ryan JH, Skelton BW, White AH. Inorg. Chem. Commun. 2000;3:575. [Google Scholar]; (b) Canty AJ, Rodemann T. Inorg. Chem. Commun. 2003;6:1382. [Google Scholar]; (c) Canty AJ, Patel J, Rodemann T, Ryan JH, Skelton BW, White AH. Organometallics. 2004;23:3466. [Google Scholar]; (d) Canty AJ, Rodemann T, Skelton BW, White AH. Inorg. Chem. Commun. 2005;8:55. [Google Scholar]; (e) Canty AJ, Rodemann T, Skelton BW, White AH. Organometallics. 2006;25:3996. [Google Scholar]; (f) Chaudhuri PD, Guo R, Malinakova HC. J. Organomet. Chem. 2007;693:567. [Google Scholar]
- 8.For other examples of the oxidation of PdII to PdIV with an iodine(III) reagent, see: Lagunas M-C, Gossage RA, Spek AL, van Koten G. Organometallics. 1998;17:731. Dick AR, Kampf JW, Sanford MS. J. Am. Chem. Soc. 2005;127:12790. doi: 10.1021/ja0541940. Whitfield SR, Sanford MS. J. Am. Chem. Soc. 2007;129:15142. doi: 10.1021/ja077866q. Racowski JM, Dick AR, Sanford MS. J. Am. Chem. Soc. 2009 doi: 10.1021/ja9014474. ASAP article.
- 9.Kalyani D, Sanford MS. Org. Lett. 2005;7:4149. doi: 10.1021/ol051486x. [DOI] [PubMed] [Google Scholar]
- 10.Heck reactions catalyzed by palladacycles were originally proposed to proceed via a PdII/IV catalytic cycle; however, more recent studies have shown the reactions are most likely catalyzed by Pd0 nanoparticles. For a review, see: Weck M, Jones CW. Inorg. Chem. 2007;46:1865. doi: 10.1021/ic061898h.
- 11.Kraatz HB, van der Boom ME, Ben-David Y, Milstein DIsr. J. Chem. 2001;41:163. [Google Scholar]
- 12.All of the order studies were conducted under conditions where [1] > ([4] + [Pd]). When [1] < ([4] + [Pd]), the kinetics become much more complex, and the order in [1] deviates from inverse 3rd order (see Supporting Information p. S16). This is expected based on the proposed mechanism, since under these conditions, the resting state of the oxidant is a mixture of 8 and 4.
- 13.Further confirmation of a first order dependence was provided by an unweighted non-linear least squares fit to the eqn: f(x) = a[4]n. This provided n = 1.12 ± 0.08 and a = 6 ± 1 × 10−4
- 14.(a) Boele MDK, van Strijdonck GPF, de Vries AHM, Kamer PCJ, de Vries JG, van Leeuwen PMWN. J. Am. Chem. Soc. 2002;124:1586. doi: 10.1021/ja0176907. [DOI] [PubMed] [Google Scholar]; (b) Zaitsev VG, Daugulis OJ. Am. Chem. Soc. 2005;127:4156. doi: 10.1021/ja050366h. [DOI] [PubMed] [Google Scholar]; (c) Giri R, Chen X, Yu JQ. Angew. Chem., Int. Ed. 2005;44:2112. doi: 10.1002/anie.200462884. [DOI] [PubMed] [Google Scholar]; (d) Wang DH, Ho XS, Wu DF, Yu JQ. Org. Lett. 2006;8:3387. doi: 10.1021/ol061384m. [DOI] [PubMed] [Google Scholar]; (e) Chen X, Goodhue CE, Yu JQJ. Am. Chem. Soc. 2006;128:12634. doi: 10.1021/ja0646747. [DOI] [PubMed] [Google Scholar]; (f) Chiong HA, Pham QN, Daugulis OJ. Am. Chem. Soc. 2007;129:9879. doi: 10.1021/ja071845e. [DOI] [PubMed] [Google Scholar]; (g) Xia JB, You SL. Organometallics. 2007;26:4869. [Google Scholar]; (h) Desai LV, Stowers KJ, Sanford MS. J. Am. Chem. Soc. 2008;130:13285. doi: 10.1021/ja8045519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Ryabov AD. Inorg. Chem. 1987;26:1252. [Google Scholar]
- 16.(a) Weis R, Seubert J. Angew. Chem., Int. Ed. Engl. 1994;33:891. [Google Scholar]; (b) Zhdankin VV, Koposov AY, Yashin NV. Tetrahedron Lett. 2002;43:5735. [Google Scholar]; (c) Ochiai M, Suefuji T, Miyamoto K, Shiro M. Chem. Commun. 2003:1438. doi: 10.1039/b302579a. [DOI] [PubMed] [Google Scholar]; (d) Suefuji T, Shiro M, Yamaguchi K, Ochiai M. Heterocycles. 2006;67:391. [Google Scholar]
- 17.A Job plot at 80 ºC also displayed a maximum at 0.5 and is available in the Supporting Information.
- 18.Blanda MT, Horner JH, Newcomb MJ. Org. Chem. 1989:4626. [Google Scholar]
- 19.Job A. Ann. Chem. 1928;9:113. [Google Scholar]
- 20.(a) Funasaki N, Ishikawa S, Neya SBull. Chem. Soc. Jpn. 2002;75:719. [Google Scholar]; (b) Bisson AP, Hunter CA, Morales JC, Young K. Chem. J. Eur. 1998;4:485. [Google Scholar]; (c) Fielding L. Tetrahedron. 2000;56:6151. [Google Scholar]
- 21.Ansyln EK, Dougherty DA. Modern Physical Organic Chemisry; University Science Books. Sausalito, CA: 2006. p. 447. [Google Scholar]
- 22.For recent selected examples of intramolecular kinetic isotope effects in Pd-catalyzed ligand-directed C–H functionalization reactions, see ref 21b, d, e, and f, as well as: Cai G, Fu Y, Li Y, Wan X, Shi ZJ. Am. Chem. Soc. 2007;129:7666. doi: 10.1021/ja070588a. Shi Z, Li B, Wan X, Cheng J, Fang Z, Cao B, Qin C, Wang Y. Angew. Chem., Int. Ed. 2007;46:5554. doi: 10.1002/anie.200700590. Li JJ, Giri R, Yu JQ. Tetrahedron. 2008;64:6979. Kirchberg S, Vogler T, Studer A. Synlett. 2008:2841.
- 23.Stille JK, Lau KSY. Acc. Chem. Res. 1977:434. [Google Scholar]
- 24.(a) Thu HY, Yu WY, Che CM. J. Am. Chem. Soc. 2006;128:9048. doi: 10.1021/ja062856v. [DOI] [PubMed] [Google Scholar]; (b) Kim M, Taylor TJ, Gabbaï FP. J. Am. Chem. Soc. 2008;130:6332. doi: 10.1021/ja801626c. [DOI] [PubMed] [Google Scholar]
- 25.Cotton FA, Koshevoy IO, Lahuerta P, Murillo CA, Sanau M, Ubeda MA, Zhao QJ. Am. Chem. Soc. 2006;128:13674. doi: 10.1021/ja0656595. [DOI] [PubMed] [Google Scholar]
- 26.Cotton FA, Gu J, Murillo CA, Timmons DJ. J. Am. Chem. Soc. 1998;120:13280. [Google Scholar]
- 27.Powers DC, Ritter T. Nat. Chem. 2009 doi: 10.1038/nchem.246. article in press. [DOI] [PubMed] [Google Scholar]
- 28.Baxter LAM, Heath GA, Raptis RG, Willis AC. J. Am. Chem. Soc. 1992;114:6944. [Google Scholar]
- 29.Dick AR, Kampf JW, Sanford MS. Organometallics. 2005;24:482. [Google Scholar]
- 30.Canty AJ, Gardiner MG, Jones RC, Rodemann T, Sharma MJ. Am. Chem. Soc. 2009;131:7236. doi: 10.1021/ja902799u. [DOI] [PubMed] [Google Scholar]
- 31.Our initial report on the mechanism of Pd-catalyzed C–H acetoxylation used benzylpyridines as substrates (ref. 14h). However, recent studies have shown analogous order and KIE data with 2-phenylpyridine derivatives Stowers KJ, Sanford MS. manuscript in preparation.
- 32.(a) Alexanian EJ, Lee C, Sorensen EJ. J. Am. Chem. Soc. 2005;127:7690. doi: 10.1021/ja051406k. [DOI] [PubMed] [Google Scholar]; (b) Streuff J, Hövelmann CH, Nieger M, Muñiz KJ. Am. Chem. Soc. 2005;127:14586. doi: 10.1021/ja055190y. [DOI] [PubMed] [Google Scholar]; (c) Liu G, Stahl SS. J. Am. Chem. Soc. 2006;128:7179. doi: 10.1021/ja061706h. [DOI] [PubMed] [Google Scholar]; (d) Desai LV, Sanford MS. Angew. Chem., Int. Ed. 2007;46:5737. doi: 10.1002/anie.200701454. [DOI] [PubMed] [Google Scholar]; (e) Muñiz KJ. Am. Chem. Soc. 2007;129:14542. doi: 10.1021/ja075655f. [DOI] [PubMed] [Google Scholar]; (f) Muñiz K, Hövelmann CH, Streuff JJ. Am. Chem. Soc. 2008;130:763. doi: 10.1021/ja075041a. [DOI] [PubMed] [Google Scholar]
- 33.Li Y, Song D, Dong VMJ. Am. Chem. Soc. 2008;130:2962. doi: 10.1021/ja711029u. [DOI] [PubMed] [Google Scholar]
- 34.(a) Tong X, Beller M, Tse MK. J. Am. Chem. Soc. 2007;129:4906. doi: 10.1021/ja070919j. [DOI] [PubMed] [Google Scholar]; (b) Welbes LL, Lyons TL, Cychosz KA, Sanford MS. J. Am. Chem. Soc. 2007;129:4906. doi: 10.1021/ja071204j. [DOI] [PubMed] [Google Scholar]; (c) Liu H, Yu J, Wang L, Tong X. Tetrahedron Lett. 2008;49:6924. [Google Scholar]; (d) Yin G, Liu G. Angew. Chem., Int. Ed. 2008;47:5442. doi: 10.1002/anie.200801438. [DOI] [PubMed] [Google Scholar]; (e) Lyons TL, Sanford MS. Tetrahedron. 2009;65:3211. doi: 10.1016/j.tet.2008.10.107. [DOI] [PMC free article] [PubMed] [Google Scholar]; (f) Tsujihara T, Takenaka K, Onitsuka K, Hatanaka M, Sasai HJ. Am. Chem. Soc. 2009;131:3453. doi: 10.1021/ja809965e. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.