

Abstract

We describe a new oxaziridine-mediated approach to the amination of sp3-hybridized C–H bonds. In the presence of a copper(II) catalyst, N-sulfonyl oxaziridines participate in efficient intramolecular cyclization reactions to afford a variety of piperidine and tetrahydroisoquinoline structures. The aminal intermediates provide a convenient functional handle for further elaboration of these structures, demonstrating the utility of this new methodology for the rapid construction of structurally complex nitrogen-containing heterocycles.
Dioxiranes and oxaziridines are members of a class of three-membered heterocyclic oxidants that perform a variety of atom-transfer reactions.1,2 Among the most intriguing of these reactions is the dioxirane-mediated hydroxylation of unactivated sp3-hybridized C–H bonds.3 The intramolecular version of this reaction, first reported by Yang, exhibits high regioselectivity for the δ position of the dioxirane.4 Oxaziridines, which are significantly more stable than dioxiranes, also perform a variety of hydrocarbon oxidations; however, only oxaziridines that bear strongly electron-withdrawing substituents have previously been shown to be reactive enough to oxidize C–H bonds.5 In this communication, we report that activation of N-sulfonyloxaziridines using copper(II) induces the regioselective intramolecular amination of sp3-hybridized C–H bonds.
Our lab has been investigating novel reactions of oxaziridines that occur in the presence of transition metal catalysts.6 Recently, we discovered that the rate of the copper(II)-catalyzed aminohydroxylation developed in our lab exhibits a marked rate acceleration in the presence of anionic halide and pseudohalide additives.7 The increased reactivity of this system led us to consider whether these conditions might also enable the oxidative functionalization of more challenging substrates such as alkanes.
As a starting point for our investigations, we prepared N-sulfonyloxaziridine 1 bearing an ortho-alkyl substituent on the C-aryl group. Subjecting 1 to conditions optimized for the aminohydroxylation7 afforded low yields of two regioisomeric aminals (2 and 3) resulting from C–H bond amination (eq 1). This result was remarkable for several reasons. First, we were surprised to observe formation of a new C–N bond, as we had expected to observe oxygen atom transfer reactivity consistent with the characteristic oxenoid reactivity of oxaziridines.8 To the best of our knowledge, this represents the first example of formal nitrogen atom transfer from an N-sulfonyl oxaziridine. Second, there has been considerable interest in the development of methods for the amination of sp3-hybridized C–H bonds.9 Our approach is a mechanistically distinct complement to the metal nitrenoid chemistry that has enjoyed the most success in this field. Finally, despite the low regioselectivity observed in this initial experiment, we were intrigued by the observation that the major product arose from functionalization of the stronger, unactivated δ C–H bond, rather than the weaker neighboring benzylic γ C–H bond.
 |
(1) |
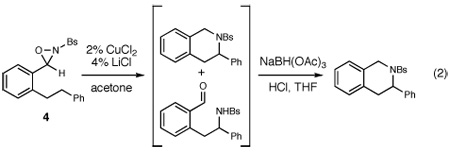
Given these results, we wondered whether an oxaziridine bearing electronically similar γ and δ methylene units might exhibit a stronger regiochemical preference. Indeed, under optimized conditions (2 mol% CuCl2, 4 mol% LiCl, 0.1 M in acetone), dihydrostilbene-derived oxaziridine 4 undergoes efficient intramolecular C–H amination with exclusive functionalization at the δ position (eq 2); no trace of reaction at the γ methylene could be observed by 1H NMR analysis of the unpurified reaction mixture. The product of the reaction is formed as an inseparable mixture of ring-opened and -closed isomers, which undergo efficient reductive amination to furnish a single tetrahydroisoquinoline product in 81% yield over two steps.
Table 1 summarizes experiments probing structural variation of the dihydrostilbene-derived oxaziridine. The reaction appears relatively insensitive to electronic perturbation of the tethering arene (entries 2 and 3). Electron-donating and -withdrawing substituents are also tolerated on the terminal arene (entries 4–8), although the reaction times in the latter case prove to be longer. A variety of functional groups are tolerated in the cyclization; the presence of ethers, esters, aryl halides, carbamates, and alcohols do not affect the efficiency of the reaction. In all cases, the amination is highly regioselective; functionalization of the γ position is observed only when the length of the tether is shortened by one carbon, although this modification results in somewhat longer reaction times (entry 9).
Table 1.
Aminations with dihydrostilbene-derived oxaziridines.a
 | |||||
|---|---|---|---|---|---|
| entry | oxaziridine | product | time | yieldb | |
| 1 | 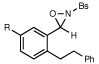 |
R=H | 75 min | 81% | |
| 2 | R=OMe | 75 min | 81% | ||
| 3 | R=Cl | 75 min | 87% | ||
| 4 | 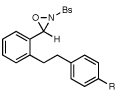 |
R=OMe | 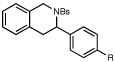 |
75 min | 76% |
| 5 | R=CO2Et | 1 h | 74% | ||
| 6 | R=NHBoc | 1 h | 72% | ||
| 7 | R=CH2OH | 1 h | 67% | ||
| 8 | R=CF3 | 3.5 h | 61% | ||
| 9 | 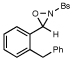 |
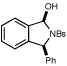 |
4 h | 84%c | |
See Supporting Information for full experimental details.
Isolated yields of the product after reductive amination.
Refers to the yield of the isolated aminal, which was not stable to the reductive amination conditions. Only the syn diastereomer was observed.
We also explored the reactions of substrates bearing aliphatic linkers (Table 2). Oxaziridine 5 undergoes efficient cyclization under our optimized conditions and eliminates upon treatment with acid to furnish the corresponding enamide in good yield (entry 1). The presence of substituents that bias the conformation of the chain towards cyclization proved to be essential for efficient amination; the corresponding substrate lacking gem-dimethyl substituents produced the cyclized product in only 13% yield (entry 2). On the other hand, dioxolane-linked oxaziridines are also suitable substrates for this method; acidic hydrolysis furnishes the corresponding 4-piperidones in good yield (entries 3–6). The amination is not limited to benzylic C–H bonds; alkynes are efficient activating groups as well (entry 7). Notably, cyclizations of oxaziridines with aliphatic tethers are δ-selective even when the functionalization occurs at unactivated methylene units (entry 8). This high positional selectivity is observed even when an adjacent benzylic methylene is present in the substrate (entry 9).10,11
Table 2.
Aminations with oxaziridines bearing aliphatic tethers.a
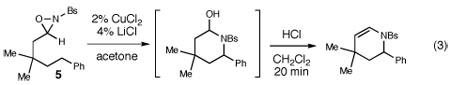 | |||||
|---|---|---|---|---|---|
| entry | oxaziridine | product | time | yieldb | |
| 1 | 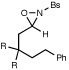 |
R = Me | 2 h | 74% | |
| 2 | R = H | 5 h | 13% | ||
| 3 | 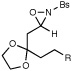 |
R = Ph | 2 h | 73% | |
| 4 | R = 4-MePh | 2 h | 92% | ||
| 5 | R = 4-ClPh | 75 min | 63% | ||
| 6 | R = 2-naph | 75 min | 69% | ||
| 7c | 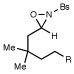 |
R = C≡CMe | 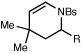 |
90 min | 69% |
| 8 | R = Et | 3 h | 37% | ||
| 9 | R = CH2Ph | 3 h | 40% | ||
See Supporting Information for full experimental details.
Isolated yields of the dihydropyridine product after treatment with acid.
Reaction conducted using 10 mol% CuCl2 at 40 °C.
To account for the C–N bond-forming process, we propose a mechanism consistent with our proposal for oxaziridine-mediated aminohydroxylation7 (Scheme 1). Upon coordination to the copper catalyst, the oxaziridine becomes activated towards substrate-induced homolysis; regioselective abstraction of the δ C–H bond followed by ring closure of the copper(III) sulfonamide onto the carbon-centered radical would produce the hemiaminal product of this process. The regioselectivity observed in this reaction is identical to that observed in intramolecular C–H bond oxidations mediated by dioxiranes.4 Thus, we deduce that the transition states of these two oxidative processes share a similar geometry. The regioselectivity of the hydrogen atom abstraction could be due to the combined influence of the stereoelectronic preferences of the oxaziridine and the geometrical constraints imposed by the cyclic conformation of the transition state.
Scheme 1.

Proposed mechanism.
While the N,O-aminal intermediates are generally not isolated, they are amenable to a variety of synthetic manipulations (Scheme 2). Treatment of the unpurified reaction mixture from 4 with a silyl enol ether in the presence of BF3•OEt2 produces the Mannich addition product 6 in 58% yield with excellent diastereocontrol (>10:1 trans:cis). Similarly, the same iminium intermediate can be intercepted by an allyl silane under the identical conditions to afford the allylated product 7 in 87% yield, again with good selectivity for the trans diastereomer (5:1 d.r.). The aminal-aldehyde equilibrium can also be exploited, and the open-chain aldehyde form can undergo Wittig olefination to produce alkene 8 in 49% yield. Finally, oxidation of the ring-closed aminal using IBX affords N-sulfonyl isoquinoline 9 in 58% yield.
Scheme 2a.

a Reagents and conditions: (a) 2-(trimethylsilyloxy)propene, BF3OEt2, CH2Cl2, −78→23 °C; (b) allyltrimethylsilane, BF3OEt2, CH2Cl2, −78→23 °C; (c) methyltriphenylphosphonium bromide, n-BuLi, −78→23 °C; (d) IBX, DMSO, 90 °C.
In summary, we have demonstrated that N-sulfonyl oxaziridines participate in efficient, regioselective intramolecular C–H bond amination reactions. This new reactivity is the first example of formal nitrogen atom transfer from N-sulfonyl oxaziridines and constitutes a fundamentally novel method for amination of sp3-hybridized C–H bonds. The ability to construct the structurally diverse heterocyclic compounds produced by this reaction should be useful in the synthesis of biologically active and medicinally relevant organic compounds.
Supplementary Material
Acknowledgment
Financial support for this research has been provided by an NSF CAREER Award (CHE-0645447) and the NIH (R01-GM084022). The NMR spectroscopy facility at UW-Madison is funded by the NSF (CHE-9629688).
Footnotes
Supporting Information Available: Experimental procedures and spectral data for all new compounds are provided (36 pages, PDF format). This material is available free of charge via the Internet at http://pubs.acs.org.
References
- 1.For reviews of dioxirane reactivity, see: Murray RW. Chem. Rev. 1989:1187–1201. Davis FA, Sheppard AC. Tetrahedron. 1989;45:5703–5742. Curci R, Dinoi A, Rubino MF. Pure Appl. Chem. 1995;67:811–822. Adam W, Mitchell CM, Saha-Möller CR, Weichold O. In: Structure and Bonding. Meunier B, editor. Vol. 97. Berlin: Springer-Verlag; 2000. pp. 237–285.
- 2.For reviews of oxaziridine reactivity, see: Davis FA, Sheppard AC. Tetrahedron. 1989;45:5703–5742. Davis FA, Chen BC. Chem. Rev. 1992;92:919–934. Petrov VA, Resnati G. Chem. Rev. 1996;96:1809–1824. doi: 10.1021/cr941146h. Adam W, Saha-Möller CR, Ganeshpure PA. Chem. Rev. 2001;101:3499–3548. doi: 10.1021/cr000019k.
- 3.For leading references, see: Murray RW, Jeyaraman R, Mohan L. J. Am. Chem. Soc. 1986;108:2470. doi: 10.1021/ja00269a069. Mello R, Fiorentino M, Fusco C, Curci R. J. Am. Chem. Soc. 1989;111:6749.
- 4.(a) Yang D, Wong M-K, Wang X-C, Tang Y-C. J. Am. Chem. Soc. 1998;120:6611. [Google Scholar]; (b) Wang M-K, Chung N-W, He L, Wang X-C, Yan Z, Tang Y-C, Yang D. J. Org. Chem. 2003;68:6321. doi: 10.1021/jo0347011. [DOI] [PubMed] [Google Scholar]; (c) Wong M-K, Chung N-W, He L, Yang D. J. Am. Chem. Soc. 2003;125:158. doi: 10.1021/ja028357l. [DOI] [PubMed] [Google Scholar]
- 5.(a) DesMarteau DD, Donadelli A, Montanari V, Petrov VA, Resnati G. J. Am. Chem. Soc. 1993;115:4897–4898. [Google Scholar]; (b) Arnone A, Foletto S, Metrangolo P, Pregnolato M, Resnati G. Org. Lett. 1999;1:281–284. [Google Scholar]; (c) Brodsky BH, Du Bois J. J. Am. Chem. Soc. 2005;127:15391–15393. doi: 10.1021/ja055549i. [DOI] [PubMed] [Google Scholar]
- 6.(a) Michaelis DJ, Shaffer CJ, Yoon TP. J. Am. Chem. Soc. 2007;129:1866–1867. doi: 10.1021/ja067894t. [DOI] [PubMed] [Google Scholar]; (b) Partridge KM, Anzovino ME, Yoon TP. J. Am. Chem. Soc. 2008;130:2920–2921. doi: 10.1021/ja711335d. [DOI] [PubMed] [Google Scholar]; (c) Michaelis DJ, Ischay MA, Yoon TP. J. Am. Chem. Soc. 2008;130:6610–6615. doi: 10.1021/ja800495r. [DOI] [PubMed] [Google Scholar]
- 7.Benkovics T, Du J, Guzei IA, Yoon TP. J. Org. Chem. 2009 doi: 10.1021/jo900902k. in press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.The ability of a Lewis acid to increase the oxygen atom transfer capability of an N-alkyl oxaziridine has been reported: Schoumacker S, Hamelin O, Téti S, Pécaut J, Fontecave M. J. Org. Chem. 2005;70:301–308. doi: 10.1021/jo048380k.
- 9.For recent reviews, see: Díaz-Requejo MM, Pérez PJ. Chem. Rev. 2008;108:3379–3394. doi: 10.1021/cr078364y. Davies HML, Manning JR. Nature. 2008;451:417–424. doi: 10.1038/nature06485. Müller P, Fruit C. Chem. Rev. 2003;103:2905–2920. doi: 10.1021/cr020043t. Halfen JA. Curr. Org. Chem. 2005;9:657–669. Davies HML, Manning JR. Nature. 2008;451:417–424. doi: 10.1038/nature06485.
- 10.No other products of C–H amination were observed; the mass balance in these experiments were the isomeric N-sulfonylated amides, analogous to the products of Aubé’s copper(I)-catalyzed rearrangements of oxaziridines. See: Aubé J. Chem. Soc. Rev. 1997;26:269–277.
- 11.Attempts to perform aminations of primary and tertiary C–H bonds have not been successful. We attribute these results to the greater bond strength of primary C–H bonds and the congested steric environment of tertiary C–H bonds.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.