

Abstract
Using the thermally stable salts of [Fe2(SR)2(CO)3(PMe3)(dppv)]BArF4, we found that the azadithiolates [Fe2(adtR)(CO)3(PMe3)(dppv)]+ react with high pressures of H2 to give the hydride [(μ-H)Fe2(adt)(CO)3(PMe3)(dppv)]BArF4. The related oxadithiolate and propanedithiolate complexes are unreactive toward H2. Molecular hydrogen is proposed to undergo heterolysis assisted by the amine followed by isomerization of an initially formed terminal hydride. Use of H2 and D2O gave the deuteride as well as the hydride, implicating protic intermediates.
Hydrogenases are enzymes that catalyze the interconversion of dihydrogen with protons and reducing equivalents.1 Understanding the reactivity of these enzymes via active site models remains topical,2 especially since these catalysts rely on inexpensive first-row transition metals.3 Significant progress has been made in [FeFe]-hydrogenases models,4,5 but nearly all studies to date have focused on proton reduction.6 The opposite reaction, hydrogen oxidation, has proven elusive. This lack of reactivity is surprising because [FeFe]-hydrogenases are exceptionally active towards H2 oxidation. The translation of models to applications in fuel cells requires progress on hydrogen oxidation. Herein we report the activation of dihydrogen by a model for the [FeFe]-hydrogenase, as well as the associated advances that have facilitated this progress.
Recently we reported [Fe2(pdt)(CO)3(PMe3)(dppv)]BF4 ([1]BF4, pdt = S2C3H6)‡, a paramagnetic (S = 1/2) spin-localized species that represents a useful structural model for the Hox state of the binuclear active site (dppv = cis-1,2-bis(diphenylphosphino)ethylene).7,8 With a vacant coordination site on the Fe(I) center, Hox and its models are poised to activate H2. Although [1]+ binds CO, it exhibits no discernable reactivity toward H2. The anticipated product of hydrogen activation, [Fe2(μ-H)(pdt)(CO)3(PMe3)(dppv)]BF4 ([1H]BF4), was prepared independently by protonation of the corresponding Fe(I)Fe(I) precursor; a terminal hydride is initially formed which rapidly isomerizes to bridging hydrides.9
The thermal sensitivity of [1]BF4 severely limits studies of its reactivity toward H2, but we found that the corresponding salt [1]BArF4 is stable in solution for days at room temperature (BArF4 = B(C6H3-3,5-(CF3)2)4−). The sensitivity of electrophilic Fe carbonyls towards fluorinated counterions is precedented.10 We confirmed that solutions of [1]BArF4 rapidly decomposed upon treatment with [Bu4N]BF4. The robust salt [1]BArF4, however, proved unreactive toward 1800 psi H2, even upon addition of the bulky base 2,6-(tBu)2pyridine. Other more basic reagents such as 2,6-dimethylpyridine are not compatible with [1]BArF4, causing it to decompose to unidentified products. Apparently H2 activation by Hox models is subject to a significant kinetic barrier.
The proposed azadithiolato cofactor has been shown to significantly affect the acid-base properties of diiron dithiolate complexes.11 Furthermore, pendant amine bases dramatically affect the rate of H2 uptake in Ni-based catalysts.12 We therefore investigated azadithiolato analogues of [1]BArF4. As described below, we present evidence that such mixed valence azadithiolato diiron complexes indeed activate H2.
Azadithiolate-containing analogues of [1]+ have proven particularly sensitive: attempts to generate [Fe2(adt)(CO)3(PMe3)(dppv)]BF4 ([2]BF4, adt = (SCH2)2NH) by oxidation of 2 with FcBF4 resulted in complex mixtures above −78 °C. We found, however, that toluene solutions of [2]BArF4, generated from treatment of 2 with FcBArF4,13 are stable for days at room-temperature. We also prepared the related benzyl derivative [Fe2[(SCH2)2NCH2Ph](CO)3(PMe3)(dppv)]BArF4 ([2′]BArF4), which was used for the majority of our studies for reasons described below. The spectroscopy for [1]+, [2]+, and [2′]+ are very similar (see Supporting Information),7 indicating that one carbonyl is semi-bridging and the apical site on the Fe(dppv) center is vacant. The unsaturated character of [2′]+ was confirmed by its rapid and reversible carbonylation to the adduct [2′ (CO)]+.
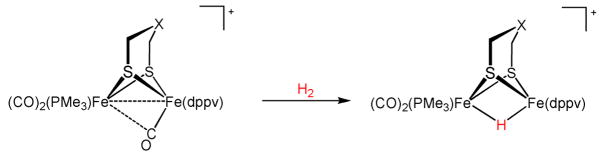
Treatment of [2]BArF4 with 1800 psi H2 for 26 h resulted in 30% conversion to the hydride [2(μ-H)]BArF4. The product [2(μ-H)]+ is spectroscopically identical to the hydride produced by protonation of 2 (see below). Analogous but more efficient reactivity was found for the tertiary amine [2′]BArF4, which gave [2′ (μ-H)]BArF4 in high yield. The mixed valence oxadithiolate [Fe2(odt)(CO)3(PMe3)(dppv)]+ ([3]+, odt = (SCH2)2O) was also found to be thermally stable, being comparable to [2′]BArF4. It was unreactive toward high pressures of H2. Collectively, these results point to the participation of the amine in the activation of H2 by the cationic diiron complex (Table 1). Under otherwise identical conditions but using argon in place of hydrogen, [2′]+ remain unchanged.
Table 1.
Reactivity of [Fe2[(SCH2)2X](CO)3(PMe3)(dppv)]+ Derivatives Toward H2. (10 mM toluene soln, 1800 psi H2, 26 h).
 | |
|---|---|
| complex (BArF4− salt) | yield of hydride |
| [Fe2(pdt)(CO)3(PMe3)(dppv)]+ ([1]+) | <5% |
| [Fe2(adt)(CO)3(PMe3)(dppv)]+ ([2]+) | ~30% |
| [Fe2(adt-Bn)(CO)3(PMe3)(dppv)]+ ([2′]+) | ~85% |
| [Fe2(odt)(CO)3(PMe3)(dppv)]+ ([3]+) | <5% |
A number of controls were conducted to verify the significance of our findings. 1H and 19F NMR analyses of reaction mixtures showed that BArF4− and ferrocene remained unaffected. The yield of the hydride was unaffected when the reaction was conducted in toluene-d7. When toluene solutions of [2′]BArF4 were stored in the absence of H2 for several days, only trace amounts of [2′ (μ-H)]+ formed, probably from a reaction involving adventitious water. This interesting process was confirmed by treatment of [2′]+ with D2O to give small amounts of [2′ (μ-D)]+. In the same way that the adt complexes [2]+ and [2′]+ exhibit greater reactivity toward H2 than does [1]+, we found that [Fe2(adt)(CO)2(dppv)2]+ is reactive towards H2 whereas the analogous propanedithiolate [Fe2(pdt)(CO)2(dppv)2]+ is not.
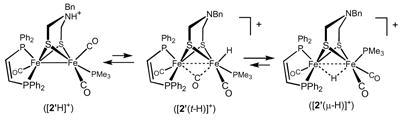
The formation of the μ-hydride [2′ (μ-H)]+ requires explanation since the mechanism for H2 activation should produce a terminal hydride adjacent to the adt.14,15 In an independent experiment, protonation of 2′ with H(OEt2)BAr 4F was found to afford the ammonium salt [2′H]+, which was characterized by IR and 31P NMR spectroscopies.16 This ammonium compound was found to convert to [2′ (μ-H)]+ via a first-order process (k = 3.9 × 10−4 s−1, 23 °C, CH2Cl2) (eq 1).
 |
(1) |
This tautomerization is assumed to occur via the terminal hydride ([2′ (t-H)]+), which is not observed. The related terminal hydride [1(t-H)]+, which is observable, isomerizes to [1(μ-H)]+ (see Supporting Info).14 The main point is that under the conditions of the hydrogenation (hours, 25 °C), [2′ (μ-H)]+ is the expected product.
The hydrogenation points to the stoichiometry shown in eq 2.
| (2) |
Although this reaction appears homolytic, the finding that it requires the azadithiolate indicates a heterolytic mechanism. A thermodynamic cycle highlights key steps – hydride binding, redox, and proton transfer (Figure 1). In the protein, the stoichiometry of course differs from eq 2, since the initial heterolysis is coupled to oxidation by an Fe-S cluster and one proton is released to the protein. In our system, the diiron complexes can interact, half of the [2′]+ serves sequentially as oxidant and then proton acceptor.
The proposed mechanism invokes the initial formation of the adduct [2′ (H2)]+. Since exogenous CO binds only weakly to [2′]+,8,17 the coordination of H2 is expected18 to be unfavorable. Binding of H2 initiates the heterolytic activation sequence as implied by the requirement for the amine-containing dithiolate. We have shown that the redox couple for CO adduct [Fe2[(SCH2)2X](CO)4(PMe3)(dppv)]+/2+ is ~250 mV more positive than the [Fe2[(SCH2)2X](CO)3(PMe3)(dppv)]0/+ couple.7 Thus, we anticipate that [2′ (H2)]+ would be unable to reduce [2′]+. The hydride [2′ (t-H)] is, however, expected19 to be a powerful reductant (~−1.6 V vs Fc0/+). Heterolysis of [2′ (H2)]+ is apparently irreversible, since [2′]+ was observed not to catalyze the formation of HD from H2 and D2O under the conditions of the experiment. We thus propose that heterolysis induces rapid proton- and electron-transfers, giving [2(t-H)]+, H+, and 2′. Being highly basic, 2′ protonates, initially at nitrogen.
When a solution of [2′]+ was treated with H2 (2000 psi) in the presence of D2O, we obtained [2′ (μ-D)]+ and [2′ (μ-H)]+ in a ratio of ~4:1. Comparable results were obtained for the corresponding reaction involving D2 and H2O, which gives both [2′ (μ-D)]+ and [2′ (μ-H)]+. Control experiments confirmed that [2′ (μ-H)]+ does not exchange with D2O.14 Proton transfer from an initial dihydrogen complex [2′ (H2)]+ would account for 50% of the exchange. The high degree of exchange is consistent with tautomerization between [2′ (H)]+ and [2′ (μ–H)]+ (see eq 1). We have shown that terminal hydrides are also inert in the absence of the amine cofactor.11
The results presented in this communication establish that biomimetic diiron dithiolato complexes can thermally activate hydrogen. The findings point to a mechanism that involves a coupling of the electron- and proton-transfers. Optimized models will require manipulation of these equilibria, as well as suppressing formation of the μ-hydride, which is a thermodynamic sink. The present results highlight the importance of the amine in the activation of dihydrogen by the diiron center.12,20
Supplementary Material
Scheme 1.

Thermodynamic cycle for the reaction 2 [Fe2(SR2)(CO)3(PR3)3]+ + H2 → 2[HFe2(SR2)(CO)3(PR3)3]+.
Scheme 2.

Proposed mechanism of H2 activation by [Fe2[(SCH2)2NR](CO)3(PMe3)(dppv)]+.
Acknowledgments
This research was supported by NIH. We thank Dr. D. L. DuBois (PNNL) for advice and Dr. M. Nilges for the EPR spectra. MTO is supported by a NIH-CBI fellowship.
Footnotes
Supporting Information Available. Preparative, spectroscopic details, and other supplementary information is available free of charge via …
References
- 1.Vincent KA, Parkin A, Armstrong FA. Chem Rev. 2007;107:4366–4413. doi: 10.1021/cr050191u. [DOI] [PubMed] [Google Scholar]; Fontecilla-Camps JC, Volbeda A, Cavazza C, Nicolet Y. Chem Rev. 2007;107:4273–4303. doi: 10.1021/cr050195z. [DOI] [PubMed] [Google Scholar]; Posewitz MC, Mulder DW, Peters JW. Curr Chem Biol. 2008;2:178–199. [Google Scholar]
- 2.Lewis NS, Nocera DG. Proc Natl Acad Sci USA. 2006;103:15729–15735. doi: 10.1073/pnas.0603395103. [DOI] [PMC free article] [PubMed] [Google Scholar]; Hu XL, Brunschwig BS, Peters JC. J Am Chem Soc. 2007;129:8988–8998. doi: 10.1021/ja067876b. [DOI] [PubMed] [Google Scholar]; Kluwer AM, Kapre R, Hartl F, Lutz M, Spek AL, Brouwer AM, van Leeuwen PWNM, Reek JNH. Proc Natl Acad Sci US. 2009:0000. doi: 10.1073/pnas.0809666106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Enthaler S, Junge K, Beller M. Angew Chem Int Ed. 2008;47:3317–3321. doi: 10.1002/anie.200800012. [DOI] [PubMed] [Google Scholar]
- 4.Artero V, Fontecave M. Coord Chem Rev. 2005;249:1518–1535. [Google Scholar]; Tard C, Liu X, Ibrahim SK, Bruschi M, De Gioia L, Davies SC, Yang X, Wang LS, Sawers G, Pickett CJ. Nature. 2005;433:610–614. doi: 10.1038/nature03298. [DOI] [PubMed] [Google Scholar]
- 5.Gloaguen F, Rauchfuss TB. Chem Soc Rev. 2009;38:100–108. doi: 10.1039/b801796b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Georgakaki IP, Darensbourg MY. In: In Comprehensive Coordination Chemistry II. McCleverty JA, Meyer TJ, editors. Elsevier; Amsterdam: 2004. pp. 549–568. [Google Scholar]; Zhao X, Chiang CY, Miller ML, Rampersad MV, Darensbourg MY. J Am Chem Soc. 2003;125:518–524. doi: 10.1021/ja0280168. [DOI] [PubMed] [Google Scholar]; Nehring JL, Heinekey DM. Inorg Chem. 2003;42:4288–4292. doi: 10.1021/ic034334b. [DOI] [PubMed] [Google Scholar]; Heiden ZM, Zampella G, De Gioia L, Rauchfuss TB. Angew Chem, Int Ed. 2008;47:9756–9759. doi: 10.1002/anie.200804400. [DOI] [PubMed] [Google Scholar]
- 7.Justice AK, De Gioia L, Nilges MJ, Rauchfuss TB, Wilson SR, Zampella G. Inorg Chem. 2008;47:7405–7414. doi: 10.1021/ic8007552. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Justice AK, Nilges M, Rauchfuss TB, Wilson SR, De Gioia L, Zampella G. J Am Chem Soc. 2008;130:5293–5301. doi: 10.1021/ja7113008. [DOI] [PMC free article] [PubMed] [Google Scholar]; Justice AK, Rauchfuss TB, Wilson SR. Angew Chem, Int Ed. 2007;46:6152–6154. doi: 10.1002/anie.200702224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Barton BE, Zampella G, Justice AK, De Gioia L, Rauchfuss TB, Wilson SR. 2009 Submitted for publication. [Google Scholar]
- 10.Landau SE, Morris RH, Lough AJ. Inorg Chem. 1999;38:6060–6068. doi: 10.1021/ic990876a. [DOI] [PubMed] [Google Scholar]
- 11.Barton BE, Olsen MT, Rauchfuss TB. J Am Chem Soc. 2008;130:16834–16835. doi: 10.1021/ja8057666. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Rakowski DuBois M, DuBois DL. Chem Soc Rev. 2009;38:62–72. doi: 10.1039/b801197b. [DOI] [PubMed] [Google Scholar]
- 13.Le Bras J, Jiao H, Meyer WE, Hampel F, Gladysz JA. J Organomet Chem. 2000;616:54–66. [Google Scholar]
- 14.Ezzaher S, Capon JF, Gloaguen F, Pétillon FY, Schollhammer P, Talarmin J, Pichon R, Kervarec N. Inorg Chem. 2007;46:3426–3428. doi: 10.1021/ic0703124. [DOI] [PubMed] [Google Scholar]; Ezzaher S, Capon JF, Gloaguen F, Kervarec N, Pétillon FY, Pichon R, Schollhammer P, Talarmin J. C R Chim. 2008;11:906–914. [Google Scholar]
- 15.Bruschi M, Greco C, Kaukonen M, Fantucci P, Ryde U, De Gioia L. Angew Chem Int Ed. 2009;48:3503–3506. doi: 10.1002/anie.200900494. [DOI] [PubMed] [Google Scholar]
- 16.Eilers G, Schwartz L, Stein M, Zampella G, de Gioia L, Ott S, Lomoth R. Chem Eur J. 2007;13:7075–7084. doi: 10.1002/chem.200700019. [DOI] [PubMed] [Google Scholar]
- 17.Thomas CM, Liu T, Hall MB, Darensbourg MY. Chem Commun. 2008:1563–1565. doi: 10.1039/b719559a. [DOI] [PubMed] [Google Scholar]
- 18.Kubas GJ. Metal Dihydrogen and s-Bond Complexes. Kluwer Academic/Plenum; New York: 2001. [Google Scholar]
- 19.Barton BE, Rauchfuss TB. Inorg Chem. 2008;47:2261–2263. doi: 10.1021/ic800030y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Henry RM, Shoemaker RK, DuBois DL, Rakowski DuBois M. J Am Chem Soc. 2006;128:3002–3010. doi: 10.1021/ja057242p. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.