

Abstract
Our recent unbiased functional screen of 54 chemotherapeutic drugs unveiled striking heterogeneity in their effects on dendritic cells (DCs). Most notably, vinblastine (VBL) was found to induce phenotypic and functional maturation of DCs in vitro. Here we sought to determine whether VBL exhibits “dual” therapeutic efficacy in living animals by directly killing tumor cells and by boosting host immunity via DC maturation. Local injection of VBL in a low dose into the skin of C57BL/6 mice induced in situ maturation of epidermal Langerhans cells. When co-injected with a model antigen, ovalbumin (OVA), VBL enhanced OVA-specific cellular and humoral immune responses. When injected directly into the OVA cDNA-transduced E.G7 tumors, VBL augmented clonal expansion of OVA-reactive CD8 T cells and CTL activities. In B16 melanoma model, intra-tumor VBL injection induced apoptosis of melanoma cells, phenotypic maturation of tumor-infiltrating DCs, and significant CTL activities. Although complete clearance was never achieved, growth kinetic of B16 melanoma was markedly reduced in C57BL/6 mice by intra-tumor VBL injection. Importantly, the same treatment was far less efficacious in immuno-compromised SCID mice, indicating the requirement of intact host immunity. Our results introduce a new concept that VBL may be used to design “immuno-stimulatory” chemotherapy regimens.
Keywords: chemotherapy, vinblastine, host immunity, dendritic cell
INTRODUCTION
Chemotherapy is an important mode of treatment for cancer patients, despite the fact that repeated administrations of cytotoxic agents at high doses often impair host immunity. Most of the currently available chemotherapeutics are immunosuppressive because they inhibit the division of not only cancer cells, but also effector leukocytes in the bone marrow and peripheral lymphoid tissues (1, 2). DCs play crucial roles in host immune responses to cancer cells (3). Although varying numbers of DCs are found in the cancer tissue, they remain immature due to immunosuppressive microenvironment associated with cancer cells and infiltrating leukocytes (4-6). Cytotoxic chemotherapy causes release of tumor-associated antigens by directly killing cancer cells. If tumor-infiltrating DCs can efficiently incorporate and cross-present these antigens to CD8 T cells, it would then enable the host immunity to combat with the remaining cancer cells. This may be achievable by delivering maturation signals to tumor-infiltrating DCs - selected anticancer drugs appear to trigger DC maturation (7-9), and DC maturation has been induced by co-administration of agonistic anti-CD40 mAb with a chemotherapeutic drug (10).
By testing 54 anticancer drugs for their impacts on DCs, we recently identified a unique class of drugs, termed “Type 1”, capable of inducing phenotypic and functional maturation of DCs. Briefly, a prototypic Type 1 drug vinblastine (VBL) was found to elevate CD40, CD80, CD86, and MHC class II expression by mouse bone marrow-derived DCs (BM-DCs), induce IL-1β, IL-6, and IL-12 production, and augment their ability to activate allogeneic T cells. Moreover, VBL-treated DCs were far more efficient than vehicle-treated DCs in dextran uptake and cross-presenting OVA protein to CD8 T cells. VBL induced all these changes at relatively low concentrations (0.1-1.0 μM) (Tanaka et al., accompanying paper).
Although direct cytotoxicity of VBL for tumor cells is generally attributed to its therapeutic effect, VBL has been reported to block angiogenesis when administered continuously at low doses, suggesting an additional mechanism of its action (11). VBL was shown to inhibit the expansion of “suppressor” T cells (12). Likewise, cyclophosphamide appears to potentiate immune responses against established tumors by eliminating regulatory T cells (13, 14). Paclitaxel promoted IL-12 production by macrophages (15), whereas doxorubicin augmented the cytostatic potential of macrophages against tumor cells (16). When combined with vaccination with whole tumor cells engineered to secrete GM-CSF, cyclophosphamide, paclitaxel, and doxorubicin all augmented the generation of anti-tumor immune responses (17). Gemcitabine enhanced T cell-mediated anti-tumor immune responses, while suppressing humoral responses (10, 18). Imatinib mesylate elevated antigen presenting capacities of DCs in the presence of LPS (19), promoted DC-dependent natural killer cell activation (20), and triggered in vivo expansion of a unique DC subset termed interferon-producing killer DCs when co-administered with IL-2 (21). These reports, although sporadic in nature, suggest that selected chemotherapeutics may boost host immunity against tumors (1).
Based on our in vitro findings that VBL induced maturation of DCs and augmented their antigen uptake and cross-presentation, we hypothesized that one might be able to kill small numbers of cancer cells and, at the same time, trigger maturation of tumor-infiltrating DCs by injecting VBL into the tumors locally at low doses. If so, maturing DCs would, in turn, incorporate tumor antigens released from dying cancer cells, migrate to draining lymph nodes, and then cross-present relevant antigens to CD8 T cells. The present study was conducted to test this hypothesis.
MATERIALS AND METHODS
Cell lines
The OVA-transduced EL4 tumor line, E.G7-OVA (22), kindly provided by Dr. Eli Gilboa (University of Miami), and the B16-F1 melanoma line purchased from the ATCC were maintained as before (23).
Reagents
VBL (Sigma) and cisplatin (CDDP, Acros Organics) were dissolved in DMSO at 2 mM. OVA was dissolved in PBS at 100 mg/ml and then passed through the polymixcin B column repeatedly until endotoxin became undetectable by the QCL-1000 system.
In vivo testing of immune-potentiating properties of VBL
All in vivo experiments were performed by injecting a 100 μM (or 90 μg/ml) VBL solution or vehicle alone (0.5% DMSO in PBS). C57BL/6 mice received subcutaneous (s.c.) injections of OVA (400 μg/animal) together with VBL (200 μl VBL solution or 18 μg VBL/animal) or vehicle alone at the base of the tail on days 0 and 7. On day 14, serum samples were examined for OVA-specific humoral responses by ELISA, and spleen cells and inguinal lymph node cells were tested for their proliferative responses to OVA by 3H-thymidine uptake (24, 25). To assess T cell cytokine profiles, draining lymph nodes harvested from immunized mice were incubated with OVA257-264 or OVA323-339 peptide and then analyzed for intracellular interferon-γ (IFNγ) and IL-4 in CD8+ or CD4+ T cell populations, respectively. To examine the impact on Langerhans cells (LCs), VBL (40 μl solution or 3.6 μg/animal) and vehicle alone was s.c. injected to the right and left ears of the same C57BL/6 mice, respectively. Two days later, ear skin samples were harvested to examine MHC II and CD86 expression in epidermal sheet preparations (24). In some experiments, VBL was injected to the Langerin-EGFP-diphtheria toxin receptor (DTR)-knock-in mice (26), and phenotypic maturation was then examined within the EGFP+ epidermal populations. To assess mechanisms for accelerated migration, we measured CCR7 expression by BM-DCs and their chemotactic activities as before (23, 24).
Measurement of dynamic behaviors of epidermal LCs
VBL or vehicle alone was administered into the ear of EGFP-I-Aβ-knock-in mice (27) and 3D images of EGFP+ epidermal cells were recorded every 2 min by a Zeiss LSM510 META2P confocal microscope (28). The magnitudes of motile activities of dendritic processes and cell bodies were then assessed by calculating the dSEARCH index and the total traveled distance, respectively (28).
Assessment of therapeutic efficacy of VBL in tumor models
Tumor cells (1 × 106 cells/mouse) were s.c. injected into the back of mice, and VBL (50 μl solution or 4.5 μg/animal), CDDP (50 μl 0.8 mM or 240 μg/ml solution or 12 μg/animal), or vehicle alone was directly injected into the tumor. CTL activities were measured by a standard 4 h 51Cr release assay (23). Perpendicular tumor diameters were measured three times a week using a caliper and the tumor areas calculated by multiplying the two diameter values. All experiments were conducted according to the NIH guidelines and were approved by the Institutional Animal Care and Use Committee at UT Southwestern.
Effects of VBL on B16 melanoma cells
B16 melanoma cells were incubated with VBL for 24 h and then examined for apoptosis by propidium iodide (PI) and Annexin V staining, calreticulin expression by flow cytometry, and release of high-mobility-group box 1 (HMGB1) by ELISA.
In vivo assays for tumor cell apoptosis and DC activation
Cryostat sections of the B16 melanoma skin lesions were doubled-stained with the In Situ Cell Death Detection kit (Roche) and DAPI (Sigma) or for CD11c and CD86. The extent of apoptosis was measured by counting the percentage of TUNEL expression in DAPI-positive nuclei (2,800-3,500 nuclei/sample).
Statistical analysis
All in vitro measurements were made in triplicate samples and each test drug was compared to vehicle alone by a two-tailed student's t-test. Synergistic effects were analyzed by ANOVA and multiple comparison by Ryan's method. The data from CTL assays were analyzed by Mann-Whitney's U test with Bonferroni adjustment. The tumor growth data were analyzed by log-rank test using time-to-event as the readout.
Online supplemental material
Supplemental Table S1 (Statistical analyses for the datasets shown in Figs. 4A, 6C and 7A-C), Supplemental Figure S1 (Effects of VBL on CCR7 expression by DCs), Supplemental Figure S2 (In vitro impacts of VBL on B16 melanoma cells), and Supplemental Movie S1 (Time-lapse images of dynamic LC behaviors).
Figure 4. Intra-tumor injection of VBL elicits T cell responses to a model tumor antigen (OVA).
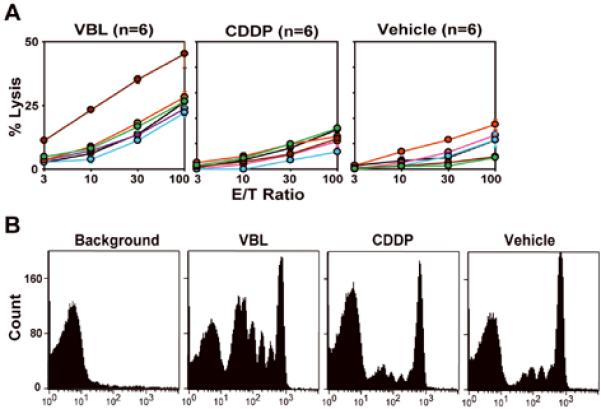
(A) C57BL/6 mice bearing the E.G7 tumor (6 mice/panel) received intra-tumor injection of VBL (4.5 μg/animal), CDDP (12 μg/animal), or vehicle alone on day 3 and sacrificed on day 11 to measure CTL activities against E.G7 tumor targets. Data shown the means ± SD (n = 3) of the % specific lysis observed with individual animals. Statistical significance is summarized in Supplemental Table S1. (B) C57BL/6 mice bearing the E.G7 tumor received intra-tumor injection of VBL, CDDP, or vehicle alone (day 3) and i.v. injection of CFSE-labeled OT-I CD8 T cells (3 × 106 cells/animal) (day 4). Two days later, the extent of OT-I T cell expansion in draining lymph nodes was assessed by measuring CFSE fluorescence intensity of the CD8+/TCR Vα2+ populations. Lymph node cells harvested from C57BL/6 mice without transfer of CFSE-labeled OT-I T cells were analyzed in parallel (left panel labeled as “background”).
Figure 6. Therapeutic efficacy of intra-tumor injection of VBL against B16 melanoma.

C57BL/6 mice (A, B) or SCID mice (C) bearing the B16 melanoma (10 mice/panel) received single injection on day 2 (A) or two repeated injections on days 2 and 7 (B, C) of VBL, CDDP, or vehicle alone into the tumors. In an additional control panel, VBL was injected subcutaneously twice at a distal anatomical location (VBL/normal skin) (B). Lines indicate the tumor sizes (mm2) measured with individual animals at the indicated time points. All the data shown are representative of at least two independent experiments. Statistical significance is summarized in Supplemental Table S1.
RESULTS
Local VBL injection triggers LC maturation and mobilization
To test the impact on LCs, tissue-resident immature DCs in skin (29), VBL or vehicle alone was s.c. injected VBL into right or left ears of BALB/c mice. In VBL injection sites, LCs visualized with anti-MHC II mAb showed a marked increase in size and irregular hyperelongation of dendrites (Fig. 1A), two characteristic changes observed with in vivo maturing LCs after topical application of reactive haptens (30). VBL injection also induced the expression of CD86 and ~50% reduction in LC densities, presumably reflecting LC migration to lymph nodes (Fig. 1B). We noticed no apparent inflammatory change in VBL injection sites, and the ear thickness remained unchanged (193 ± 5 μm, mean ± SEM, n = 5) compared to vehicle-injected ears (185 ± 7 μm). When VBL was injected into Langerin-EGFP-DTR knock-in mice (26), the EGFP+ epidermal cells (i.e., LCs) exhibited significantly upregulated expression of MHC II, CD40, and CD86 (Fig. 1C).
Figure 1. In vivo impact of VBL on LC maturation in mice.
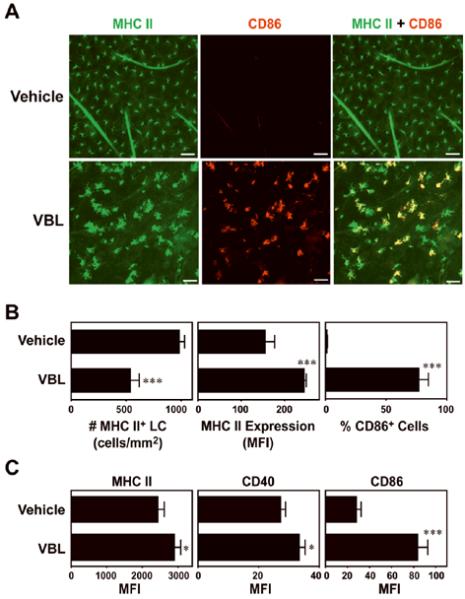
(A) VBL (3.6 μg/animal) or vehicle alone was s.c. injected into the right or left ears of C57BL/6 mice, respectively. Ear skin specimens were harvested 48 h later, and epidermal sheets were stained for MHC II (green) and CD86 (red). Scale bar, 50 μm. (B) The epidermal sheet preparations were examined for the number of MHC II+ LCs, the level of MHC II expression by LCs, and the frequency of CD86+ cells in MHC II+ LCs. Data represent the means ± SD (n = 5) measured by counting >10 independent fields in each specimen. (C) VBL or vehicle alone was s.c. injected into the Langerin-EGFP-DTR knock-in mice (n = 3), and epidermal cell suspensions were examined 48 h later for MHC II, CD40, and CD86 expression within the EGFP+ populations. Statistically significant differences compared with the vehicle controls are indicated with asterisks (***P < 0.001, *P<0.05).
Our recent imaging study revealed that in situ maturation of LCs is accompanied by dramatic changes in their motile behaviors (28). We next injected VBL into the ear of anesthetized I-Aβ-EGFP knock-in mice (27) and recorded motile activities of EGFP+ LCs. Local VBL injection significantly augmented the rhythmic extension and retraction of dendrites, a motion termed “dSEARCH” (Fig. 2A and B, and Supplemental Movie S1). VBL also induced amoeba-like lateral migration of cell bodies in the epidermal compartment (Fig. 2A and C). These results further support our conclusion that LCs undergo in situ maturation in response to local injection of a small amount of VBL.
Figure 2. VBL exacerbates in situ dynamic behaviors of epidermal LC.
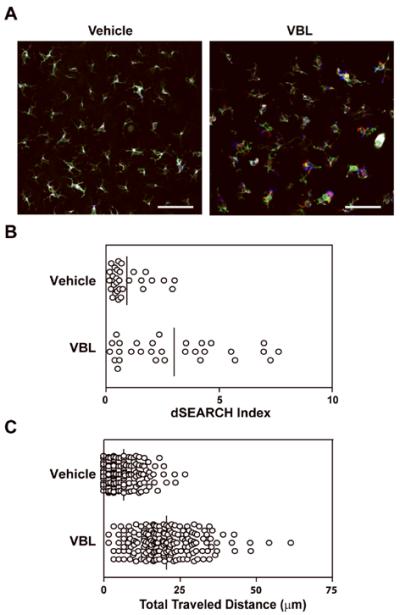
I-Aβ-EGFP knockin mice received s.c. injection of VBL (3.6 μg/animal) or vehicle alone into the right ear and anesthetized 24 h later to visualize dynamic behaviors of EGFP+ LCs in the injection sites. Actual behaviors are shown in Supplemental Movie S1. Data shown are the overlay of images of EGFP+ LCs recorded at time 0 (pseudo-colored in green), 30 min (pseudo-colored in red), and 60 min (pseudo-colored in blue) (A). Scale bar = 50 μm. Relative magnitude of dendrite movement is expressed by dSEARCH index (n = 30) (B). Extent of lateral migration of cell bodies is indicated by total traveled distance (n = 180) (C). Mean values among all the measured LCs are shown with bars. VBL significantly augmented the dSEARCH motion (P < 0.0003) and the lateral migration (P = 0) assessed by Mann-Whitney U test.
Because CCR7 reportedly mediates chemotactic migration of mature LCs from epidermis to lymph nodes (31), we examined whether VBL induces CCR7 expression in vitro. Mouse BM-DCs were treated with VBL (0.3 μM) or vehicle alone and then examined for CCR7 surface expression. VBL elevated CCR7 expression by BM-DCs and VBL-treated BM-DCs showed significantly augmented migration toward CCL19 (Supplemental Fig. S1). These in vitro observations were consistent with our in vivo finding that locally injected VBL reduced surface densities of epidermal LCs.
Local injection of VBL augments adaptive immune responses to a model protein antigen
The observed ability of VBL to trigger in situ maturation and mobilization of LCs implied that VBL might function as an adjuvant depending upon the routes and doses of administration. To test this, we s.c. injected VBL in a low dose together with a model antigen OVA. Mice immunized with OVA + VBL showed significantly higher concentrations of OVA-specific antibodies than did control mice immunized with OVA alone (Fig. 3A). Spleen cells and lymph node cells harvested from the mice immunized with OVA + VBL exhibited significantly augmented proliferative responses to OVA compared to controls immunized with OVA alone (Fig. 3B). In addition, immunization with VBL also resulted in a significant, albeit rather modest, increase in the percentage of IFNγ-producing CD4 T cells (Fig. 3C). These results demonstrated a previously unrecognized activity of VBL to augment both humoral and cellular immune responses.
Figure 3. Adjuvant activities of VBL to boost humoral and cellular responses to OVA.
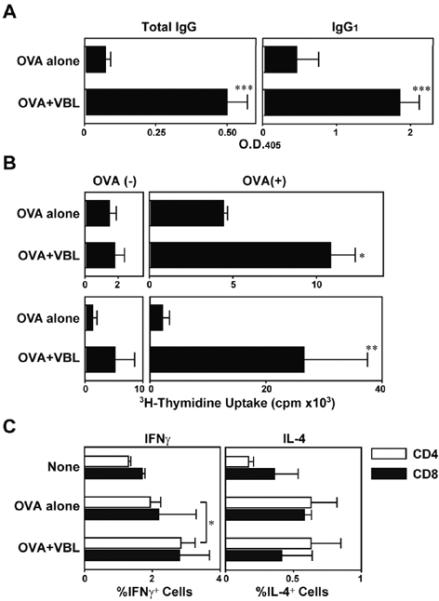
C57BL/6 mice (n = 3 each) were immunized twice by s.c. injections of OVA + VBL (18 μg/animal) or OVA + vehicle alone. (A) Serum samples collected 7 days after the second immunization were examined for OVA-specific total immunoglobulins and IgG of the indicated isotypes (means ± SD from three independent animals). (B) The spleen cells (top) and draining lymph node cells (bottom) were examined for their proliferative responsiveness in the presence or absence of OVA (100 μg/ml). Data shown are the means ± SD of 3H-thymidine uptake on day 4. (C) After two immunization (days 0 and 7) with vehicle alone, OVA alone, or OVA + VBL, draining lymph node cells were harvested on day 9. After in vitro re-stimulation with MHC I- or MHC II-restricted OVA peptide, CD8+ or CD4+ T cell populations were analyzed for intracellular cytokine profiles, respectively. Statistically significant differences compared to the control panel are indicated with asterisks (*P < 0.05, **P < 0.01, ***P<0.001).
Using the OVA-transduced tumor line, E.G7 (22), we next injected VBL directly into the tumors producing OVA as a model tumor antigen. Again, we injected vehicle alone to serve as a control. Because intra-tumor injection of VBL, but not vehicle alone, would kill some cancer cells, the second control panel received intra-tumor injection of a platinum agent cisplatin (CDDP), which failed to induce DC maturation, reduce DC viability, or inhibit DC growth at the tested concentrations (0.1-10 μM) (Tanaka et al., accompanying paper). Intra-tumor VBL injection significantly augmented CTL activities against E.G7 tumor targets compared to either control panel (Fig. 4A; statistical significance is summarized in Supplemental Table S1). To visualize the impact on T cell expansion, CD8 T cells purified from the OT-I transgenic mice were adoptively transferred into tumor-bearing mice. Marked expansion of OVA-reactive CD8 T cells was observed in those mice receiving intra-tumor injection of VBL, but not CDDP or vehicle alone (Fig. 4B). We interpret these results to suggest that OVA-specific CD8 T cell responses are readily inducible by injecting VBL directly into OVA-producing tumors.
Impact of intra-tumor injection of VBL on B16 melanoma
Since OVA is a highly immunogenic xeno-antigen, our observations with E.G7 tumor may have limited clinical relevance. Thus, we employed B16 melanoma as our second tumor model. First, we tested in vitro effects of VBL on B16 melanoma cells - VBL from 0.01 to 1 μM inhibited 3H-thymidine uptake and induced apoptosis in dose-dependent manners (Supplemental Fig. S2A and B). Recent studies have shown that tumor cells dying in response to chemotherapeutic drugs augment DC activation via releasing HMGB1 (9) and that selected anticancer drugs induce calreticulin surface expression by tumor cells, thereby facilitating their uptake by DCs (8). We observed that VBL induced HMGB1 release by B16 melanoma cells (Supplemental Fig. S2C) without triggering significant calreticulin expression (Supplemental Fig. S2D and E). VBL treatment of BM-DCs induced significantly augmented release of IL-12 (p40), consistent with our recent finding (Tanaka et al., accompanying paper). When co-cultured with VBL-pretreated, apoptotic B16 melanoma cells, BM-DCs released significant amounts of IL-12. Importantly, the two stimuli (i.e., co-culturing with apoptotic tumor cells and direct exposure to VBL) augmented IL-12 production in a synergistic manner (Supplemental Fig. S2F). Thus, we hypothesized that killing of B16 melanoma cells and maturation of tumor-infiltrating DCs might be both achievable by injecting a small amount of VBL directly into B16 melanoma.
VBL injection induced significant apoptosis of tumor cells (Fig. 5A). A small fraction (5.7 ± 1.0 %, n = 3) of the tumor cells were positive for TUNEL after vehicle injection, whereas significantly (P<0.01) higher numbers of TUNEL-positive tumor cells were observed after VBL injection (19.5 ± 2.7 %, n = 3), validating the first part of our hypothesis. To test the second part, we assessed phenotypic maturation of tumor-infiltrating DCs. The numbers of CD11c+ DCs appeared comparable between the two B16 melanoma lesions treated with VBL versus vehicle alone. Importantly, CD86-expressing CD11c+ DCs were detected only after VBL injection (Fig. 5B). Moreover, B16 melanoma-bearing mice that had received intra-tumor VBL injection showed significant CTL activities to kill 51Cr-labeled B16 melanoma targets (Fig. 5C). By contrast, control mice receiving intra-tumor injection of CDDP or vehicle alone showed only negligible CTL activities. Thus, in situ maturation of tumor-infiltrating DCs is inducible by local injection of VBL, but not CDDP.
Figure 5. Dual therapeutic efficacy of VBL to induce direct killing of B16 melanoma and to boost immune responses via DC maturation.
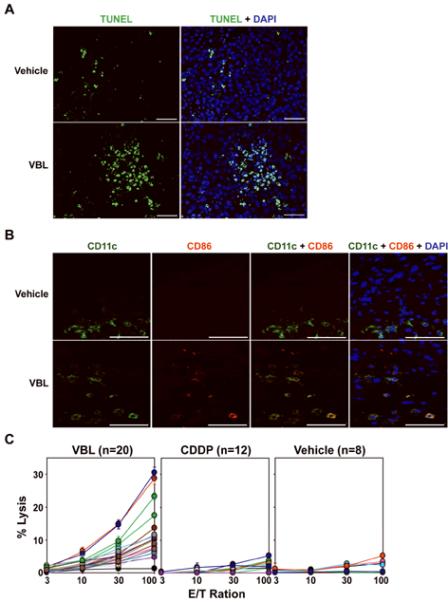
B16 melanoma cells were inoculated onto C57BL/6 mice on day 0, and VBL (4.5 μg/animal) or vehicle alone was injected into the tumors on day 2. The tumors were excised on day 4, and cryostat sections were then stained for apoptosis (TUNEL) and nuclei (DAPI) (A) or for CD11c (green) and CD86 (red) (B). Scale bar, 50 μm. The data shown are representative fields from three independent experiments. (C) C57BL/6 mice bearing the B16 melanoma received intra-tumor injection of VBL, CDDP, or vehicle alone (day 2) and sacrificed on day 11 to measure CTL activities against B16 melanoma targets. Data shown the means ± SD (n = 3) of the % specific lysis. Statistical significance is summarized in Supplemental Table S1.
To test the therapeutic efficacy, we injected VBL, CDDP, or vehicle alone directly into B16 melanoma and measured the subsequent tumor growth. For an ethical reason, all animals were euthanized when the tumor size exceeded 10 mm in diameter. Because of rapid and progressive growth of B16 melanoma, we had to sacrifice all animals in the vehicle-injected panel within 10 days. The second panel receiving intra-tumor CDDP injection showed statistically significant, although marginal, delay in B16 melanoma growth, most likely reflecting direct cytotoxicity of CDDP against B16 melanoma. Intra-tumor injection of VBL showed a more readily noticeable efficacy (Fig. 6A; statistical significance is summarized in Supplemental Table S1) - some (6/10) of the mice showed temporal tumor remission, although all mice were eventually euthanized at later time-points (14-43 days after tumor inoculation).
Two repeated intra-tumor VBL injections resulted in prolonged tumor remission (ranging from 10 to 40 days) in all animals, although complete regression was never observed (Fig. 6B). Two repeated CDDP injections showed statistically significant, but marginal, therapeutic efficacy over the vehicle injected panel. Importantly, two repeated injections of VBL into the abdominal skin did not affect the B16 melanoma growth in the back skin, indicating that VBL at the tested dose had no or limited systemic effects on tumor growth.
In both single and two repeated injection protocols, VBL showed more prominent therapeutic efficacies than did CDDP. The observed difference might reflect differential impact of the two anticancer drugs on host immunity or simply differential toxicity against B16 melanoma cells. To distinguish the two possibilities, we repeated the same experiments in SCID mice. Mice receiving two repeated intra-tumor VBL injections showed significantly delayed B16 melanoma growth compared to the vehicle injected control (Fig. 6C). The observed therapeutic efficacy for VBL in immune-deficient SCID mice, however, was rather modest compared to that observed in immune-competent C57BL/6 mice. By contrast, the efficacy of CDDP was comparable between C57BL/6 mice and SCID mice. Most importantly, CDDP was almost indistinguishable from VBL in their beneficial effects in the SCID mice, indicating comparable cytotoxicity against B16 melanoma between the two drugs. Thus, we concluded that intact host immunity is required for intra-tumor VBL injection to exhibit therapeutic advantages over CDDP.
DISCUSSION
Our results have unveiled previously unrecognized pharmacological activities of VBL. In situ maturation of LCs was readily induced by local injection of VBL in a low dose. VBL also functioned as an adjuvant when co-injected with a model antigen. Intra-tumor injection of VBL caused not only tumor cell apoptosis, but also phenotypic maturation of tumor-infiltrating DCs, significant CTL activities, and interrupted tumor growth. Thus, we conclude that VBL may be used to achieve dual therapeutic outcomes, direct killing of cancer cells and boosted host immunity via DC maturation.
Interactions between dying cancer cells and surrounding DCs determine the direction, type, and magnitude of host immune responses to the remaining cancer cells (32, 33). Like other phagocytes, DCs can recognize and captureng dying cancer cells occurring naturally or being induced by chemotherapy. Necrotic cell death is known to serve as a rich source of endogenous “danger” signals, thereby eliciting DC maturation, whereas apoptotic cell death is generally considered immunologically silent or even tolerogenic - DCs in the steady state induce peripheral immunological tolerance by capturing, processing, and presenting apoptotic cells that emerge during physiological tissue turnover (34, 35). We now know that apoptotic cells may induce DC maturation under certain conditions. For example, double-stranded RNA found in virally infected apoptotic cells induced DC maturation by a TLR3-dependent mechanism (36). Casares et al. reported that cancer cells killed by doxorubicin, a topoisomerase inhibitor, but not by mitomycin C, triggered phenotypic maturation of DCs (37). Moreover, they have successfully eradicated established CT26 tumors in mice by a single intra-tumor injection of doxorubicin, but not of mitomycin C. With regard to mechanisms, Obeid et al. reported that doxorubicin and its analogues (idarubicin and mitoxantrone) produce an immunogenic form of cancer cell apoptosis by triggering rapid translocation of calreticulin to the cell surface (8). Spisek et al. reported that myeloma cells killed by bortezomib, an inhibitor of 26S proteasome, but not by γ-irradiation or dexamethasone, expressed heat shock protein 90 (HSP90), thereby inducing DC maturation (38). Moreover, tumor cells dying in response to chemotherapeutic drugs have been shown to augment DC activation via releasing HMGB1 (9). These findings illustrate additional functional heterogeneity among chemotherapeutic drugs in terms of the modality (or immunogenicity) of resulting cancer cell death (2, 7). Interestingly, VBL induced HMGB1 release from B16 melanoma cells without causing calreticulin expression, and VBL-pretreated apoptotic melanoma cells synergized with VBL to augment DC maturation. These unique immune-stimulatory properties identified for selected anticancer drugs must be taken into consideration when combining multiple chemotherapeutics for ultimate clinical outcome.
The magnitude of immune responses observed after vaccination with OVA + VBL was rather modest compared with that inducible by OVA plus CpG oligonucleotides (data not shown). Although locally injected VBL significantly delayed the growth of B16 melanoma, all the mice showed relapses after temporal remission, representing a major limitation of our protocol to serve as a stand-alone therapy. Combination of VBL with additional immuno-stimulating agent(s) may produce markedly improved outcomes. In fact, VBL has been combined with IL-2 and/or interferon-α for the treatment of advanced cancer patients (39, 40).
Optimal phenotypic and functional maturation of DCs was induced by VBL at 1 μM or 0.9 μg/ml (Tanaka et al., accompanying paper), and this concentration was sufficient to cause optimal growth inhibition and apoptosis of B16 melanoma cells. Thus, we chose to administer a 100 μM (or 90 μg/ml) VBL solution in relatively small amounts. In standard clinical regimens, VBL is administered systemically and repeatedly by bolus i.v. infusions at maximal tolerated doses (MTDs) of 0.1-0.3 mg/kg body weight. In various tumor models in mice, VBL monotherapy produced only partial and temporal regression after a single systemic injection at the MTD (5-10 mg/kg) or even after repeated injections (0.5-6 mg/kg/day) (41-43). In our protocol, a significant delay in tumor growth was observed after a single or two intra-tumor injections of VBL at the dose of 4.5 μg/mouse (i.e., 0.18 mg/kg). Consistent with our observations in the SCID mice, weekly intra-tumor injections of VBL at 0.3 mg/injection/kg was reported to be ineffective for human melanomas implanted to the nude mice (44). Thus, immune-stimulatory outcomes appear to be inducible by VBL only when administered locally at relatively low doses and only in immune-competent hosts. Extravasation of Vinca alkaloids causes severe skin inflammation and necrosis. Although a 1 mg/ml VBL solution used for bolus i.v. infusion therapy, produces such local necrotic changes, a 0.2 mg/ml VBL solution has been injected directly to oral lesions of Kaposi's sarcomas without causing devastating complications (45). We observed no apparent inflammation after s.c. injection of a 90 μg/ml VBL solution, in agreement with the previous report that VBL produces dose-dependent local toxicity (46). These observations suggest relative safety of our protocol in which VBL is injected directly into tumors in relatively small amounts (causing minimal myelo-suppression) and at relatively low concentrations (causing minimal local toxicity).
Suppression of host immunity is a major adverse effect of chemotherapy, in which multiple agents with myelo-suppressive potentials are administered repeatedly and systemically at relatively high doses (1). We now know that selected anticancer drugs augment host immunity depending upon the routes and doses of administration and that cancer cells killed by certain drugs deliver maturation signals to DCs (2). Our results provide conceptual and technical frameworks for combining VBL with other FDA-approved drugs to formulate ultimate chemotherapy regimens designed to accomplish seemingly contradicting tasks of killing cancer cells and augmenting host immunity.
Supplementary Material
Acknowledgments
Financial Support: This work was supported by NIH grants to A.T.
REFERENCES
- (1).Lake RA, Robinson BW. Immunotherapy and chemotherapy--a practical partnership. Nat Rev Cancer. 2005;5:397–405. doi: 10.1038/nrc1613. [DOI] [PubMed] [Google Scholar]
- (2).Zitvogel L, Apetoh L, Ghiringhelli F, Kroemer G. Immunological aspects of cancer chemotherapy. Nat Rev Immunol. 2008;8:59–73. doi: 10.1038/nri2216. [DOI] [PubMed] [Google Scholar]
- (3).Steinman RM, Banchereau J. Taking dendritic cells into medicine. Nature. 2007;449:419–26. doi: 10.1038/nature06175. [DOI] [PubMed] [Google Scholar]
- (4).Smyth MJ, Dunn GP, Schreiber RD. Cancer immunosurveillance and immunoediting: the roles of immunity in suppressing tumor development and shaping tumor immunogenicity. Adv Immunol. 2006;90:1–50. doi: 10.1016/S0065-2776(06)90001-7. [DOI] [PubMed] [Google Scholar]
- (5).Melief CJ. Cancer immunotherapy by dendritic cells. Immunity. 2008;29:372–83. doi: 10.1016/j.immuni.2008.08.004. [DOI] [PubMed] [Google Scholar]
- (6).Finn OJ. Cancer immunology. N Engl J Med. 2008;358:2704–15. doi: 10.1056/NEJMra072739. [DOI] [PubMed] [Google Scholar]
- (7).van der Most RG, Currie AJ, Robinson BW, Lake RA. Decoding dangerous death: how cytotoxic chemotherapy invokes inflammation, immunity or nothing at all. Cell Death Differ. 2008;15:13–20. doi: 10.1038/sj.cdd.4402255. [DOI] [PubMed] [Google Scholar]
- (8).Obeid M, Tesniere A, Ghiringhelli F, et al. Calreticulin exposure dictates the immunogenicity of cancer cell death. Nat Med. 2007;13:54–61. doi: 10.1038/nm1523. [DOI] [PubMed] [Google Scholar]
- (9).Apetoh L, Ghiringhelli F, Tesniere A, et al. Toll-like receptor 4-dependent contribution of the immune system to anticancer chemotherapy and radiotherapy. Nat Med. 2007;13:1050–9. doi: 10.1038/nm1622. [DOI] [PubMed] [Google Scholar]
- (10).Nowak AK, Robinson BW, Lake RA. Synergy between chemotherapy and immunotherapy in the treatment of established murine solid tumors. Cancer Res. 2003;63:4490–6. [PubMed] [Google Scholar]
- (11).Klement G, Baruchel S, Rak J, et al. Continuous low-dose therapy with vinblastine and VEGF receptor-2 antibody induces sustained tumor regression without overt toxicity. J Clin Invest. 2000;105:R15–R24. doi: 10.1172/JCI8829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (12).Powderly WG, Pier GB, Markham RB. In vitro T cell-mediated killing of Pseudomonas aeruginosa. IV. Nonresponsiveness in polysaccharide-immunized BALB/c mice is attributable to vinblastine-sensitive suppressor T cells. J Immunol. 1986;137:2025–30. [PubMed] [Google Scholar]
- (13).Proietti E, Greco G, Garrone B, et al. Importance of cyclophosphamide-induced bystander effect on T cells for a successful tumor eradication in response to adoptive immunotherapy in mice. J Clin Invest. 1998;101:429–41. doi: 10.1172/JCI1348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (14).Ghiringhelli F, Larmonier N, Schmitt E, et al. CD4+CD25+ regulatory T cells suppress tumor immunity but are sensitive to cyclophosphamide which allows immunotherapy of established tumors to be curative. Eur J Immunol. 2004;34:336–44. doi: 10.1002/eji.200324181. [DOI] [PubMed] [Google Scholar]
- (15).Mullins DW, Burger CJ, Elgert KD. Paclitaxel enhances macrophage IL-12 production in tumor-bearing hosts through nitric oxide. J Immunol. 1999;162:6811–8. [PubMed] [Google Scholar]
- (16).Haskill JS. Adriamycin-activated macrophages as tumor growth inhibitors. Cancer Res. 1981;41:3852–6. [PubMed] [Google Scholar]
- (17).Machiels JP, Reilly RT, Emens LA, et al. Cyclophosphamide, doxorubicin, and paclitaxel enhance the antitumor immune response of granulocyte/macrophage-colony stimulating factor-secreting whole-cell vaccines in HER-2/neu tolerized mice. Cancer Res. 2001;61:3689–97. [PubMed] [Google Scholar]
- (18).Nowak AK, Robinson BW, Lake RA. Gemcitabine exerts a selective effect on the humoral immune response: implications for combination chemo-immunotherapy. Cancer Res. 2002;62:2353–8. [PubMed] [Google Scholar]
- (19).Wang H, Cheng F, Cuenca A, et al. Imatinib mesylate (STI-571) enhances antigen-presenting cell function and overcomes tumor-induced CD4+ T-cell tolerance. Blood. 2005;105:1135–43. doi: 10.1182/blood-2004-01-0027. [DOI] [PubMed] [Google Scholar]
- (20).Borg C, Terme M, Taieb J, et al. Novel mode of action of c-kit tyrosine kinase inhibitors leading to NK cell-dependent antitumor effects. J Clin Invest. 2004;114:379–88. doi: 10.1172/JCI21102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (21).Taieb J, Chaput N, Menard C, et al. A novel dendritic cell subset involved in tumor immunosurveillance. Nat Med. 2006;12:214–9. doi: 10.1038/nm1356. [DOI] [PubMed] [Google Scholar]
- (22).Boczkowski D, Nair SK, Snyder D, Gilboa E. Dendritic cells pulsed with RNA are potent antigen-presenting cells in vitro and in vivo. J Exp Med. 1996;184:465–72. doi: 10.1084/jem.184.2.465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (23).Kumamoto T, Huang EK, Paek HJ, et al. Induction of tumor-specific protective immunity by in situ Langerhans cell vaccine. Nat Biotechnol. 2002;20:64–9. doi: 10.1038/nbt0102-64. [DOI] [PubMed] [Google Scholar]
- (24).Mizumoto N, Gao J, Matsushima H, Ogawa Y, Tanaka H, Takashima A. Discovery of novel immunostimulants by dendritic cell-based functional screening. Blood. 2005;106:3082–9. doi: 10.1182/blood-2005-03-1161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (25).Matsue H, Matsue K, Walters M, Okumura K, Yagita H, Takashima A. Induction of antigen-specific immunosuppression by CD95L cDNA-transfected “killer” dendritic cells. Nat Med. 1999;5:930–7. doi: 10.1038/11375. [DOI] [PubMed] [Google Scholar]
- (26).Bennett CL, van Rijn E, Jung S, et al. Inducible ablation of mouse Langerhans cells diminishes but fails to abrogate contact hypersensitivity. J Cell Biol. 2005;169:569–76. doi: 10.1083/jcb.200501071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (27).Boes M, Cerny J, Massol R, et al. T-cell engagement of dendritic cells rapidly rearranges MHC class II transport. Nature. 2002;418:983–8. doi: 10.1038/nature01004. [DOI] [PubMed] [Google Scholar]
- (28).Nishibu A, Ward BR, Jester JV, Ploegh HL, Boes M, Takashima A. Behavioral responses of epidermal Langerhans cells in situ to local pathological stimuli. J Invest Dermatol. 2006;126:787–96. doi: 10.1038/sj.jid.5700107. [DOI] [PubMed] [Google Scholar]
- (29).Romani N, Holzmann S, Tripp CH, Koch F, Stoitzner P. Langerhans cells - dendritic cells of the epidermis. Apmis. 2003;111:725–40. doi: 10.1034/j.1600-0463.2003.11107805.x. [DOI] [PubMed] [Google Scholar]
- (30).Aiba S, Katz SI. Phenotypic and functional characteristics of in vivo-activated Langerhans cells. J Immunol. 1990;145:2791–6. [PubMed] [Google Scholar]
- (31).Dieu M-C, Vanbervliet B, Vicari A, et al. Selective recruitment of immature and mature dendritic cells by distinct chemokines expressed in different anatomic sties. J Exp Med. 1998;188:373–86. doi: 10.1084/jem.188.2.373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (32).Dhodapkar MV, Dhodapkar KM, Palucka AK. Interactions of tumor cells with dendritic cells: balancing immunity and tolerance. Cell Death Differ. 2008;15:39–50. doi: 10.1038/sj.cdd.4402247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (33).Zitvogel L, Casares N, Pequignot MO, Chaput N, Albert ML, Kroemer G. Immune response against dying tumor cells. Adv Immunol. 2004;84:131–79. doi: 10.1016/S0065-2776(04)84004-5. [DOI] [PubMed] [Google Scholar]
- (34).Liu K, Iyoda T, Saternus M, Kimura Y, Inaba K, Steinman RM. Immune tolerance after delivery of dying cells to dendritic cells in situ. J Exp Med. 2002;196:1091–7. doi: 10.1084/jem.20021215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (35).Steinman RM, Turley S, Mellman I, Inaba K. The induction of tolerance by dendritic cells that have captured apoptotic cells. J Exp Med. 2000;191:411–6. doi: 10.1084/jem.191.3.411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (36).Schulz O, Diebold SS, Chen M, et al. Toll-like receptor 3 promotes cross-priming to virus-infected cells. Nature. 2005;433:887–92. doi: 10.1038/nature03326. [DOI] [PubMed] [Google Scholar]
- (37).Casares N, Pequignot MO, Tesniere A, et al. Caspase-dependent immunogenicity of doxorubicin-induced tumor cell death. J Exp Med. 2005;202:1691–701. doi: 10.1084/jem.20050915. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (38).Spisek R, Charalambous A, Mazumder A, Vesole DH, Jagannath S, Dhodapkar MV. Bortezomib enhances dendritic cell (DC)-mediated induction of immunity to human myeloma via exposure of cell surface heat shock protein 90 on dying tumor cells: therapeutic implications. Blood. 2007;109:4839–45. doi: 10.1182/blood-2006-10-054221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (39).Pectasides D, Varthalitis J, Kostopoulou M, et al. An outpatient phase II study of subcutaneous interleukin-2 and interferon-alpha-2b in combination with intravenous vinblastine in metastatic renal cell cancer. Oncology. 1998;55:10–5. doi: 10.1159/000011829. [DOI] [PubMed] [Google Scholar]
- (40).Atzpodien J, Kirchner H, Jonas U, et al. Interleukin-2- and interferon alfa-2a-based immunochemotherapy in advanced renal cell carcinoma: a Prospectively Randomized Trial of the German Cooperative Renal Carcinoma Chemoimmunotherapy Group (DGCIN) J Clin Oncol. 2004;22:1188–94. doi: 10.1200/JCO.2004.06.155. [DOI] [PubMed] [Google Scholar]
- (41).Sersa G, Krzic M, Sentjurc M, et al. Reduced tumor oxygenation by treatment with vinblastine. Cancer Res. 2001;61:4266–71. [PubMed] [Google Scholar]
- (42).Hill SA, Lonergan SJ, Denekamp J, Chaplin DJ. Vinca alkaloids: anti-vascular effects in a murine tumour. Eur J Cancer. 1993;29A:1320–4. doi: 10.1016/0959-8049(93)90082-q. [DOI] [PubMed] [Google Scholar]
- (43).Chaplin DJ, Pettit GR, Hill SA. Anti-vascular approaches to solid tumour therapy: evaluation of combretastatin A4 phosphate. Anticancer Res. 1999;19:189–95. [PubMed] [Google Scholar]
- (44).Spruss T, Bernhardt G, Schonenberger H, Schiess W. Hyaluronidase significantly enhances the efficacy of regional vinblastine chemotherapy of malignant melanoma. J Cancer Res Clin Oncol. 1995;121:193–202. doi: 10.1007/BF01366962. [DOI] [PubMed] [Google Scholar]
- (45).Epstein JB, Scully C. Intralesional Vinblastine for Oral Kaposi Sarcoma in Hiv Infection. Lancet. 1989;2:1100–1. doi: 10.1016/s0140-6736(89)91114-8. [DOI] [PubMed] [Google Scholar]
- (46).Dorr RT, Alberts DS. Vinca Alkaloid Skin Toxicity - Antidote and Drug Disposition Studies in the Mouse. J Natl Cancer Inst. 1985;74:113–20. [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.