

Abstract
Lantibiotics are peptide antimicrobial compounds that are characterized by the thioether-bridged amino acids lanthionine and methyllanthionine. For lacticin 481, these structures are installed in a two-step post-translational modification process by a bifunctional enzyme, lacticin 481 synthetase (LctM). LctM catalyzes the dehydration of Ser and Thr residues to generate dehydroalanine or dehydrobutyrine, respectively, and the subsequent intramolecular regio- and stereospecific Michael-type addition of cysteines onto the dehydroamino acids. In this study, semi-synthetic substrates containing nonproteinogenic amino acids were prepared by expressed protein ligation and [3+2]-cycloaddition of azide and alkyne functionalized peptides. LctM demonstrated broad substrate specificity toward substrates containing β-amino acids, D-amino acids, and N-alkyl amino acids (peptoids) in certain regions of its peptide substrate. These findings showcase its promise for use in lantibiotic and peptide engineering applications, whereby nonproteinogenic amino acids may impart improved stability or modulated biological activities. Furthermore, LctM permitted the incorporation of an alkyne-containing amino acid that can be utilized for the site-selective modification of mature lantibiotics and used in target identification.
Keywords: lantibiotic, antibiotic, post-translational modification, peptoid, peptide ligation
Introduction
The lantibiotics are a class of ribosomally synthesized peptide antimicrobial agents produced by Gram-positive bacteria that have wide-ranging modes of action.[1] For example, nisin and mutacin 1140 exert their bacteriocidal activity through sequestration of the cell wall biosynthetic intermediate lipid II and targeted pore formation,[2,3] whereas cinnamycin inhibits the activity of phospholipase A2 by binding to phosphatidylethanolamine.[4] Lantibiotics are short peptides (19-38 amino acids) characterized by the cyclic thioether amino acids lanthionine (Lan) and/or methyllanthionine (MeLan) introduced by post-translational modifications. They typically also contain the unsaturated amino acids dehydroalanine (Dha) and Z-dehydrobutyrine (Dhb). Biosynthetic processing enzymes are responsible for first catalyzing the dehydration of serine or threonine residues to Dha or Dhb and the subsequent intramolecular Michael-type addition of Cys residues to the unsaturated amino acids to form Lan or MeLan rings. The conformational restraints afforded by cyclic lanthionine amino acids are responsible for the biological activity and stability of lantibiotics.[1] Due to the unique modes of action of these natural products and their interesting post-translational modifications, the enzymes involved in lantibiotic biosynthesis have received consider-able interest for use as general catalysts for applications in lantibiotic and peptide engineering.[5-11]
The understanding of the mode of action of lantibiotics and the relationships between structure and function has been increased greatly through investigations with analogs produced by in vivo mutagenesis.[1,12] However, this strategy is limited in scope due to possible cytotoxic or regulatory properties of expressed peptides and the restriction to proteinogenic amino acids. More recently, the activity of several lantibiotic biosynthetic enzymes has been reconstituted in vitro. These include the cyclase enzyme NisC[13] involved in nisin biosynthesis, and the bifunctional LanM enzymes involved in lacticin 481 (LctM)[14] and haloduracin (HalM1 and HalM2) biosynthesis.[15] The most extensively studied in vitro system is that of lacticin 481 synthetase (LctM, see Figure 1). Lacticin 481 is translated as a pre-peptide, LctA, containing an N-terminal leader peptide that is not modified and a C-terminal structural region that is dehydrated and cyclized by LctM. Enzymatic removal of the leader peptide provides mature lacticin 481.
Figure 1.
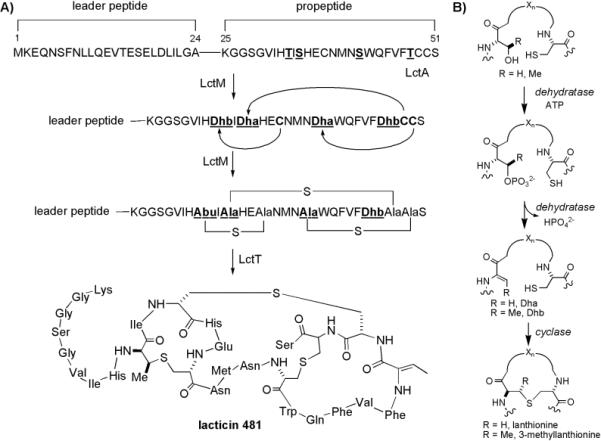
Post-translational modifications in lantibiotic biosynthesis. A) Biosynthesis of lacticin 481. The bifunctional enzyme LctM is responsible for both dehydration of Ser/Thr residues and the subsequent intramolecular cyclization of Cys residues onto the unsaturated amino acids Dha and Dhb in the structural region (also called propeptide). Subsequent proteolytic removal of the unmodified leader peptide by LctT provides bioactive lacticin 481. B) Formation of Lan and MeLan. Dehydration is catalyzed via an intermediate phosphorylation step, which targets Ser/Thr residues for elimination.
To date, the ability of LctM to process LctA prepeptides containing nonproteinogenic amino acids has not been fully explored. Such investigation is of significant interest both for its potential for creating novel lacticin 481 analogs with improved stability or biological activity and for utilizing LctM as a general dehydratase or cyclase enzyme for peptide engineering. Although (Me)Lan residues have been shown to provide enhanced proteolytic stability of peptides,[16-18] flexible regions linking the thioether rings in lantibiotics remain susceptible to degradation. For example, a nisin resistance (nsr) gene has been reported[19] that encodes an enzyme in the tail-specific protease family that inactivates nisin.[1] Similarly, lacticin 481 contains a flexible N-terminal tail of eight amino acids that is important for biological activity[20] but is prone to enzymatic proteolysis. The incorporation of protease-resistant nonproteinogenic amino acids, such as D-amino acids, β-amino acids,[21-24] and N-substituted glycine amino acids (peptoids)[22] might improve the stability of lantibiotics in vivo, thus increasing their therapeutic potential. Furthermore, α-amino acids containing reactive handles could be used for further elaboration of mature lantibiotics with fluorophores, sugars, or other biochemical probes to identify their molecular targets.
In this work, expressed protein ligation[25] was utilized to create a series of LctA analogues containing nonproteinogenic amino acids. In a second generation strategy to improve yields, 1,3-dipolar cycloaddition between azide- and alkyne-functionalized peptides was employed to generate the substrate analogs. Tests with these (semi)synthetic peptides demonstrate that LctM displays high tolerance of nonproteinogenic amino acid incorporation in certain regions of its substrate LctA. To the best of our knowledge, this study also represents the first example of enzymatic processing of oligopeptides containing engineered peptoid structures. The information gained herein can be applied to the design and synthesis of unnatural derivatives of lantibiotics or therapeutic peptides containing nonproteinogenic amino acids.
Results
Semi-synthesis of LctA analogs using Expressed Protein Ligation
Expressed protein ligation (EPL)[25] has been used previously to synthesize LctA derivatives containing phosphorylated amino acids and various unnatural cysteine and threonine analogs.[26-28] This methodology was utilized herein to synthesize substrates containing a wide array of nonproteinogenic amino acids at positions that are not involved directly in thioether ring formation (Figure 2, Table 1). For synthetic ease, truncated LctA substrates were chosen to survey the substrate promiscuity of LctM rather than the full length LctA peptide.
Figure 2.
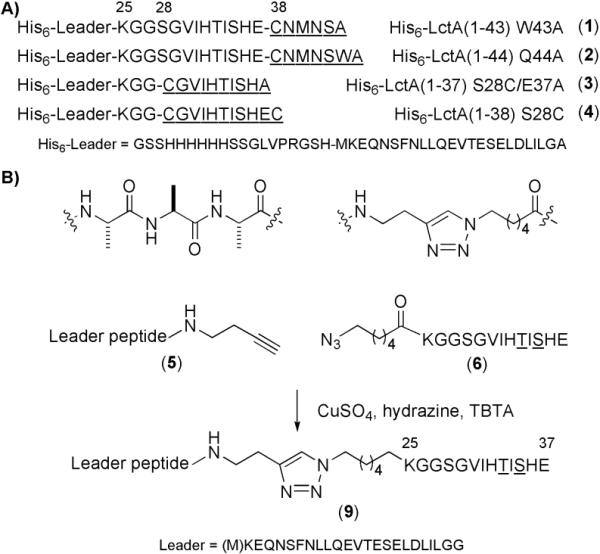
LctA substrate analogs used in this work. A) His6-tagged LctA substrates prepared using expressed protein ligation. Short synthetic peptides (underlined) were ligated to mercaptoethanesulfonic acid (MES) thioesters of His6-LctA(1-27) or His6-LctA(1-37). B) Strategy for the preparation of triazole-linked LctA substrates. Cu(I)-mediated 1,3-dipolar cycloaddition of alkyne (5) and azide (6) modified peptides was utilized for the preparation of triazole-linked truncated LctA substrates (9).
Table 1.
Truncated LctA mutants containing nonproteinogenic amino acids prepared by EPL and the results following incubation with LctM in the presence of Mg2+ and ATP, as analyzed by MALDI-TOF MS. Relative intensities (%) are denoted for LctM assay products containing multiple species. The structure of each nonproteinogenic amino acid is indicated. The substitutions that were tolerated by LctM are shown in bold whereas substitutions that resulted in lack of activity or incomplete dehydration of the peptide are shown in italics. NR, no reaction.
| Entry | Substrate | R1 = | R2 = | Assay Product | Maximum # of Expected Dehydrations |
|---|---|---|---|---|---|
| 1 | (2) Asn39Hse | CH2CH2OH | -- | - 3 H2O | 3 |
| 2 | (2) Asn39Nva | n-Pr | -- | - 3 H2O | 3 |
| 3 | (2) Asn39Cba | CH2CH2CN | -- | - 3 H2O | 3 |
| 4 | (1) Asn39D-Asn | CH2C(=O)NH2 | -- | - 2 H2O | 3 |
| 5 | (2) Met40Pra | CH2CCH | -- | - 3 H2O | 3 |
| 6 | (2) Met40Nle | n-Bu | -- | - 3 H2O | 3 |
| 7 | (2) Met40Nva | n-Pr | -- | - 3 H2O | 3 |
| 8 | (2) Met40N-Nle | -- | n-Bu | - 3 H2O | 3 |
| 9 | (1) Met40D-Met | CH2CH2SMe | -- | - 2 H2O | 3 |
| 10 | (2) Asn41Cba | CH2CH2CN | -- | - 3 H2O | 3 |
| 11 | (2) Trp43Nal | CH2-2-naphthyl | -- | - 3 H2O | 3 |
| 12 | (3) Ser28D-Cys | CH2SH | -- | - 2 H2O | 2 |
| 13 | (4) Gly29Sar | -- | CH3 | - 2 H2O | 2 |
| 14 | (4) Ile31Sar | -- | CH3 | NR | 2 |
| 15 | (4) Ile31N-Val | -- | i-Pr | NR | 2 |
| 16 | (4) His32Sar | -- | CH3 | - H2O (30%), +PO32- (100%) | 2 |
| 17 | (4) Ile34Sar | -- | CH3 | NR | 2 |
| 18 | (4) Glu37Sar | -- | CH3 | - H2O (30%), - H2O + PO32- (100%) | 2 |
Using Fmoc based solid-phase peptide synthesis (SPPS), suitably protected nonproteinogenic amino acids were incorporated into the peptides CGVIHTISHEC, CGVIHTISHEA, CNMNSWA, or CNMNSA (Figure 2A, underlined residues). The synthetic peptides were then ligated either to His6-LctA(1-27) or His6-LctA(1-37), bearing a C-terminal α-thioester. These latter peptides were obtained by intein-mediated thioester formation from the corresponding peptide-intein-CDB fusion proteins in the presence of 2-mercaptoethanesulfonic acid (MES) following established protocols.[27,28] Synthetic peptides were ligated with the thioester peptides, and the products purified by HPLC. Peptidic substrates thus obtained include His6-LctA(1-43)W43A (1), His6-LctA(1-44)Q44A (2), His6-LctA(1-37)S28C/E37A (3), and His6-LctA(1-38)S28C (4), as well as a series of derivatives containing nonproteinogenic amino acids, as shown in Table 1.
Synthesis of triazole-linked LctA analogs using 1,3-dipolar cycloaddition of azide- and alkyne-functionalized peptides
Although the EPL methodology has proven useful for the construction of LctA analogs, it suffered from low yields of LctA thioesters obtained by overexpression in E. coli (ca. 3 mg from 3 L of cell culture). Therefore, we sought to develop a fully synthetic ligation method that could provide larger quantities of LctA analogs. A recent report demonstrated that a substrate with three alanine residues inserted directly after Lys25 of LctA was accepted as a substrate by LctM, indicating a relaxed substrate specificity for mutation in the region of LctA between the leader and structural peptides.[5] This observation prompted the evaluation of non-peptidic linkers connecting the leader peptide and structural region. In particular, a triazole-linked substrate was shown to be tolerated by LctM resulting in dehydration and cyclization.[29] This fully synthetic method provided much higher yields of ligated substrates than the EPL methodology and was used in the later stages of the current work to synthesize truncated triazole-linked LctA substrates, as shown in Figure 2B. Two peptide components were required to prepare the triazole-linked substrates: an LctA leader peptide containing a C-terminal alkyne and a truncated propeptide containing an N-terminal azide. The azide-functionalized propeptide (6) was readily prepared by appending 6-azido hexanoic acid[30] in the last step of Fmoc-based SPPS, using HOBT and DIC as coupling reagents. Synthesis of the alkyne-functionalized leader peptide was carried out as shown in Scheme 1. LctA(1-24)Ala24Gly (7) was first synthesized on 2-chlorotrityl chloride resin using standard Fmoc SPPS protocols. Cleavage of the fully-protected peptide from the resin using acetic acid and trifluoroethanol, followed by coupling of 3-butyn-1-amine to the carboxy terminus of 7 using DIPEA, HOBT, and DIC gave fully-protected alkyne functionalized peptide 8. The Fmoc and side-chain protecting groups were removed using 20% piperidine and TFA, respectively, and the final product (5) was purified by HPLC. The synthesis of 5 provided relatively large quantities of peptide (30-40 mg from 0.1-0.12 mmol functionalized resin), a significant improvement compared to the yield of LctA-thioesters obtained from overexpression in E. coli. In later experiments, Met1 of LctA was omitted in the synthesis of LctA(2-24)Ala24Gly due to its propensity to oxidize and complicate MS spectral analysis.
Scheme 1.
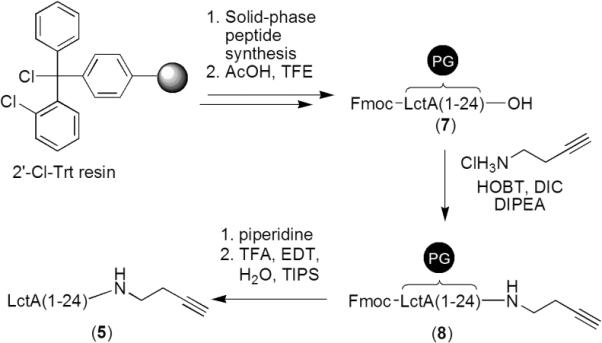
Synthesis of 3-butyn-1-amine functionalized LctA leader peptide. The peptide was elongated using Fmoc-based solid-phase peptide synthesis employing 2'-Cl-Trt resin pre-loaded with Gly. Following peptide synthesis, the Fmoc- and side-chain-protected peptide 7 was cleaved from the resin under mild acidic conditions. To this peptide was coupled 3-butyn-1-amine using HOBT, DIC, and DIPEA, yielding alkyne-modified protected peptide 8. Deprotection of the Fmoc- and side-chain-protecting groups (PG) provided peptide 5 following RP-HPLC purification.
Cu(I)-catalyzed 1,3-dipolar cycloaddition of azide-functionalized LctA(25-37) with alkyne-functionalized LctA(1-24) was attempted initially using CuSO4 with ascorbate as an in situ reductant.[31] However, the triazole-linked peptide product was isolated in very low yield following HPLC purification of the crude reaction mixture. Use of the recently reported tris-(benzyltriazolylmethyl)-amine (TBTA) ligand to enhance the catalytic activity of the catalyst[32] did not improve product formation. However, the use of anhydrous hydrazine as a reductant (K.B. Sharpless, personal communication), in combination with TBTA, resulted in complete conversion to the triazole-linked substrate 9 (Figure 2B), as judged by analytical HPLC.
LctM assay of the triazole-linked LctA(1-37) substrate in the presence of ATP and Mg2+ resulted in complete conversion to the expected two-fold dehydrated product, as evidenced by MALDI-TOF MS (see Supporting Information). This result prompted the synthesis of a series of analogues containing nonproteinogenic amino acids (Table 2).
Table 2.
Truncated LctA mutants containing nonproteinogenic amino acids prepared by Cu(I)-catalyzed triazole cycloaddition (C6 triazole) and the results following incubation with LctM in the presence of Mg2+ and ATP, as analyzed by MALDITOF MS. The side chains (R1) for α-amino acids in entries 19-25 are shown. Substrates that resulted in complete modification following LctM incubation are depicted in bold, whereas those that resulted in incomplete conversion or no reaction are shown in italics. NR, no reaction.
| Entry | Substrate | R1 = | Assay Product | Maximum # of Expected Dehydrations |
|---|---|---|---|---|
| 19 | (9) Gly29Acpc | CH2CH2 | - 2 H2O | 2 |
| 20 | (9) Val30Nva | n-Pr | - 2 H2O | 2 |
| 21 | (9)Val30D-Val | i-Pr | - 2 H2O | 2 |
| 22 | (9) His32D-His | CH2-Im | - H2O, + PO32- | 2 |
| 23 | (9) His36D-His | CH2-Im | - H2O | 2 |
| 24 | (9) Thr33D-Ser | CH2OH | NR | 2 |
| 25 | (9) Ser35D-Ser | CH2OH | - H2O | 2 |
| 26 | (9) Gly26βAla | - 2 H2O | 2 | |
| 27 | (9) Gly29βAla | - 2 H2O | 2 | |
| 28 | (9) Gly26/Gly27/Gly29βAla | - 2 H2O | 2 |
LctM Assays of LctA Analogues
The substrates in Tables 1 and 2 were incubated with LctM in the presence of Mg2+ and ATP for 2-3 h at 25 °C. Dehydration events were analyzed using MALDI-TOF MS. Two dehydrations at Thr33 and Ser35 are expected for LctA(1-37) substrates, whereas Ser42 may also be dehydrated for the longer substrates in Tables 1 and 2. LctM has been previously shown to activate Ser and Thr residues for dehydration through an intermediate phosphorylation step.[26,33] Therefore, in some instances partially processed phosphorylated species were also observed (Table 1). Assay results following incubation with LctM for each substrate are summarized in Tables 1 and 2, with mutants that were fully processed by LctM denoted in bold font, and those resulting in incomplete modification shown in italics. MALDI MS data for all substrates following treatment with LctM are provided in the Supplemental Information.
LctM fully processed a series of LctA peptides containing nonproteinogenic α- and β-amino acids synthesized by EPL. The mutations that were tolerated include homoserine (Hse), norvaline (Nva), and 4-cyano-2-aminobutyric acid (Cba) substitutions of Asn39, and propargyl glycine (Pra), norleucine (Nle) and Nva substitutions of Met40 (entries 1-3, 5-7, Table 1). Also fully processed were peptides containing Cba in place of Asn41, and naphthylalanine (Nal) in place of Trp43 (entries 10 and 11, Table 1). Triazole-linked LctA(1-37) substrates containing nonproteinogenic α- and β-amino acids in the N-terminal region of the structural peptide of LctA, including amino-cyclopropanoic acid (Acpc) in place of Gly29, Nva in place of Val30, and β-alanine at positions Gly26 and Gly29 were also good substrates for LctM (entries 19, 20, 26, and 27, Table 2). Furthermore, a peptide that contained three concurrent β-alanine substitutions of Gly26, Gly27, and Gly29 (entry 28, Table 2) was fully modified by LctM. These results demonstrate the relaxed substrate specificity of LctM toward substrates containing nonproteinogenic amino acids.
For LctA analogs containing D-amino acids, the position of incorporation proved critical with respect to full conversion by LctM. Peptides with a D-Cys at position 28 and D-Val at position 30 were fully processed (entries 12 and 21). However, substrates containing D-isomers of Asn39 and Met40 were only partially (two-fold) processed (entries 4 and 9, Table 1). Meanwhile, triazole-linked substrates (Table 2) containing D-Ser in place of Ser35 (entry 25) and D-His in place of His32 or His36 (entries 22 and 23) were poor substrates, and a peptide containing D-Ser in place of Thr33 (entry 24) was not a substrate at all. Overall, these results suggest that D-amino acids incorporated into LctA are tolerated by LctM as long as they are located in positions remote from the Ser and Thr residues targeted for dehydration.
N-Alkyl glycines or peptoids[34] are promising mimetics of peptides, as they have shown resistance to proteolysis in biomimetic polymers[34-36] as well as when introduced as single amino acid mimics in oligopeptides.[22] In addition, peptoid analogs of naturally occurring antibacterial peptides have increased selectivity for pore formation in bacterial membranes over hemolytic activity against human erythrocytes.[37-39] These advantageous pharmacological properties have resulted in screening efforts that have identified several novel antimicrobial peptoids.[40-46] To the best of our knowledge, peptoid containing substrate analogs that are acted upon by native enzymes have not been reported. The potential of lantibiotic synthetases to accept peptoid-containing substrates was probed through the incorporation of several peptoid monomers in substrate peptides prepared by EPL. These peptides contained sarcosine (N-methyl glycine, Sar) at positions Gly29, Ile31, His32, Ile34, and Glu37. Furthermore, Fmoc-N-isopropyl and Fmoc-N-butyl glycine were synthesized and incorporated in place of Ile31 and Met40, respectively. Each of the peptoid-containing substrates was subsequently incubated with LctM. The replacement of Gly29 with sarcosine resulted in full processing by LctM (entry 13, Table 1). Similarly, a substrate containing an N-butyl glycine residue in place of Met40 was fully processed by LctM (entry 8). However, no dehydrations were observed following incubation of the substrates containing sarcosine or N-isopropyl glycine in place of Ile31 or Ile34 (entries 14, 15 and 17). A substrate containing Sar in place of Glu37 was only partially accepted for dehydration by LctM, resulting in small amounts of singly- and doubly-dehydrated products (entry 18) with the major product consisting of phosphorylated peptide. Incubation of the mutant containing Sar in place of His32 also resulted in a product that was phosphorylated (entry 16). These results indicate that the substrates were recognized by LctM, but that elimination of phosphate did not occur. Overall, LctM was able to process several peptoid-containing LctA substrates, but incorporation of these nonproteinogenic amino acids in positions proximal to dehydratable amino acids had a negative effect on the activity of LctM.
Analysis of Cyclization of Selected LctA Analogues
For a subset of fully dehydrated LctA mutants, analysis of the cyclization of Cys38 onto Dhb33 was performed. The thiol modifying agent p-hydroxymercuribenzoic acid (PHMB) was used to probe for the presence of uncyclized cysteine residues[13,47] following treatment with LctM. For these studies, triazole-linked substrates were designed with a Ser35Ala mutation and synthesized. The Ser35Ala mutation was included to eliminate the possibility for non-enzymatic cyclization to Dha35 following dehydration by LctM.[27] Triazole cycloaddition using azide-functionalized peptides containing free sulfhydryl groups did not proceed; therefore Cys residues were protected as tert-butyl disulfides. Following Cu(I)-catalyzed triazole cycloaddition, the protecting groups were removed using dithiothreitol or 2-mercaptoethanol, yielding triazole-linked LctA(2-39) S35A substrates. PHMB treatment of singly-dehydrated triazole-linked peptides containing Val30Nva, His36DHis, or simultaneous β-Ala mutations at Gly26, Gly27, and Gly29 did not result in significant formation of a PHMB adduct, indicating cyclization had occurred (see Supplemental Information). When His32 was mutated to D-His, a small portion of dehydrated peptide was converted to a PHMB adduct, indicating that the substrate had not undergone complete cyclization.
Discussion
Lacticin 481 synthetase catalyzes a remarkable series of chemical transformations; it is able to dehydrate specific serine or threonine residues on a 51 amino acid peptide and subsequently guide the formation of three (Me)Lan cross-links in a regio- and stereospecific manner. Semi-synthetic procedures have been utilized to probe the permissiveness of LctM toward unnatural Cys and Thr analogs.[5,28] A comprehensive approach directed at investigating the permissiveness of LctM toward incorporation of nonproteinogenic amino acids at other positions has not been reported and is presented herein.
The results with peptides containing nonnatural α-amino acids as well as β-amino acids demonstrate the relaxed substrate specificity of LctM. The enzyme displayed a particularly strong promiscuity toward mutations in the N-terminal portion of the LctA structural region. For instance, LctM tolerates introduction of a non-peptidic triazole linker between the structural and leader peptides of LctA in substrate 9 that extends the distance between the leader peptide and the first dehydratable residue, Thr33, by approximately eleven atoms compared to wild-type LctA. Furthermore, a mutant containing concomitant β-alanine mutations at positions Gly26, Gly27, and Gly29 was also fully processed by LctM despite an increase in this distance by an additional three methylene units. These results suggest that alteration of the distance of Thr33 to the leader peptide does not appear to have an effect on the efficiency of LctM, consistent with previous mutagenesis studies ion the substrates for LctM[5] and NisB.[48] These results also suggest that LctM does not bind the N-terminal portion of the LctA structural peptide tightly during modification. This hypothesis is supported by previous studies where replacement of the GlySerGly sequence at positions 27-29 of LctA by an IleSerHis sequence, which has been shown to activate Ser residues for dehydration, did not result in modification of the inserted Ser by LctM.[5]
These results suggest that the residues in the N-terminal linear region of lacticin 481 (Lys25 through His32) might be good candidates for replacement with nonproteinogenic amino acids. It is encouraging that LctM accepted D-amino acids, peptoids and β-amino acid substitutions in this region (Gly26βAla, Ser28DCys, Gly29βAla, Gly29Acpc, Gly29Sar, Val30Nva, and Val30Dval) as these residues are well known to impart proteolytic stability onto α-peptides.[21-24] On the other hand, D-amino acids and peptoids were not tolerated at several other positions that were more proximal to Ser and Thr residues that are normally dehydrated, resulting in greatly decreased efficiency of dehydration. It has previously been demonstrated that flanking Gly residues deactivate Ser/Thr residues for dehydration;[5] peptoids located immediately N-terminal to Ser/Thr (entries 16 and 17, Table 1) may act in a similar manner because the lack of a substituent at the α-carbon imparts greater conformational flexibility to the substrate. This hypothesis is also consistent with recent findings, where a comprehensive bioinformatic analysis of known lantibiotic structures identified a weak consensus for large, hydrophobic residues flanking Ser/Thr positions that undergo dehydration.[8]
The region spanning Asn39 to Trp43 of LctA is also highly amenable to mutation with nonproteinogenic amino acids. Substrates containing a wide range of mutations in this portion of LctA were fully modified by LctM, including mutation of Asn39 with homoserine, norvaline and 4-cyano-2-aminobutyric acid (Cba), and mutation of Met40 with norvaline, norleucine, propargyl alanine, and N-butyl glycine. In addition, mutations of Asn41 to Cba and Trp43 to naphthyl alanine were tolerated by LctM. Therefore, the region spanning the B and C rings of lacticin 481 is also amenable to engineering of novel structural or functional features. The exception to these findings is that LctM cannot process substrates containing D-amino acids in this region, as substrates containing D-amino acids at positions Asn39 and Met40 were not fully processed. Lastly, a substrate containing an alkyne-bearing amino acid was readily accepted for modification by LctM (entry 5, Table 1), indicating that lantibiotics produced in vitro can be further elaborated with biochemical probes.
Conclusion
We report the synthesis of a series of peptides containing nonproteinogenic amino acids that were utilized to investigate the substrate specificity of lacticin 481 synthetase. LctM is highly tolerant toward incorporation of nonproteinogenic amino acids, including β-amino acids, D-amino acids, and unnatural α-amino acids, in the N-terminal tail region of lacticin 481. LctM also readily dehydrated mutants containing similar nonproteinogenic amino acids in the region spanning Asn39 to Trp43 of LctA. On the other hand, LctM did not dehydrate Ser or Thr residues with flanking peptoids or D-amino acids. These results indicate that lantibiotic biosynthetic enzymes, including LctM, have promising potential for use in peptide engineering applications, including the synthesis of tailor-made lantibiotics and post-translationally modified peptide-derived natural products containing structurally diverse architectures.
Experimental Section
General Methods and Materials
All chemicals, amino acids and resins used were obtained from Acros, Aldrich, Fisher, Novabiochem, Chem -Impex, or Advanced ChemTech. N,N'-Dimethylformamide (DMF) used for solid-phase peptide synthesis (SPPS) was high purity reagent grade and was used without further purification. All other reagent grade chemicals and solvents were used without further purification unless otherwise noted. Mass spectral data was collected at the Mass Spectrometry Laboratory, School of Chemical Sciences, University of Illinois by either fast atom bombardment (FAB) ionization, electrospray ionization (ESI), or matrix assisted laser desorption ionization (MALDI) techniques. Analytical RP-HPLC was performed on a Beckman Gold system using Vydac C4 or C18 analytical columns (5 micron, 0.46 cm × 25 cm) at a flow rate of 1 mL/min. Preparative RP-HPLC was performed on a Waters 600 system using a Waters PrepLC (25 mm module) column with a flow rate of 8 mL/min. For gradient HPLC, solution A was 0.1% TFA in H2O, and solution B was 80% MeCN/20% H2O with 0.086% TFA. For all HPLC purifications, detection was performed at 220 nm.
Solid-Phase Peptide Synthesis
Peptides were synthesized on solid-phase by standard Fmoc chemistry using an automated peptide synthesizer (Rainin PS3 or Advanced Chemtech Apex 396). Pre-loaded Wang resin (0.1 mmol) and Fmoc-amino acids (0.4 mmol) were used unless otherwise noted. All nonproteinogenic Fmoc-α-amino acids were purchased from Advanced Chemtech. Fmoc-amino acids were coupled using O-benzotriazole-N,N,N',N',-tetramethyl-uroniumhexafluorophosphate (HBTU, 0.4 mmol) as coupling reagent with N-methylmorpholine (NMM, 4.4% in DMF, 8 mL) as base. Fmoc deprotections were performed using piperidine (4 × 3 min, 20% in DMF, 8 mL). Resins were first swollen in DMF (8 mL) for 30 min. Couplings were performed from 45 min to 3 h. The completion of couplings was monitored by Kaiser test.[49] To minimize racemization during couplings, Fmoc-Cys derivatives (0.4 mmol) were coupled using 1-hydroxybenzotriazole (HOBT, 0.4 mmol) and N,N'-diisopropylcarbodiimide (DIC, 0.4 mmol) in DMF (8 mL).[50] For the synthesis of azide-functionalized peptide 6 and its analogues, 6-azido hexanoic acid[30] (0.4 mmol) was coupled using HOBT (0.4 mmol) and DIC (0.4 mmol). The terminal Fmoc group was deprotected prior to cleavage of peptides from the resin. Prior to cleavage, the resin was washed with DMF (5 × 10 mL), ethanol (5 × 10 mL), and CH2Cl2 (5 × 10 mL) and dried for 4-6 h under reduced pressure in a dessicator. Peptide cleavage from the resin was achieved by stirring the resin in a mixture of TFA (10 mL), water (150 μL), ethanedithiol (150 μL), and triisopropylsilane (TIPS, 150 μL) for 1.5 h at room temperature. The resin was then filtered and the filtrate concentrated under reduced pressure. The concentrated crude peptide was washed with cold diethyl ether (3 × 10 mL) and lyophilized from 10% aqueous acetic acid following filtration through a 0.45 μm filter. The lyophilized crude peptides were purified by preparative RP-HPLC. Fractions were lyophilized and those containing product analyzed by either ESI-TOF or MALDI-TOF MS.
General procedure for the synthesis of LctA leader peptide derivatized with 3-butyn-1-amine
The peptide was synthesized on pre-loaded H-Gly-2'-chlorotrityl resin followed by traditional Fmoc-based SPPS. The final N-terminal Fmoc group was not deprotected prior to cleavage from the resin. Cleavage of the fully-protected peptide was accomplished by stirring the resin in an 8:1:1 mixture of CH2Cl2:trifluoroethanol:acetic acid for 45 min. Solvents were removed via rotary evaporation and acetic acid was removed as an azeotrope with hexanes (5 × 5 mL) to yield the fully-protected peptide 7. The protected peptide was only partially soluble in all solvents evaluated, making MS analysis difficult. Therefore, the crude peptide was typically suspended in THF (10 mL) and to this was added a mixture of 3-butyn-1-amine hydrochloride (4 equiv), DIPEA (4 equiv), HOBT (4 equiv), and DIC (4 equiv), with stirring continuing for 4-6 h at 25 °C. At this time, solvent was removed by rotary evaporation and to the crude product was added a 20% piperidine/DMF mixture to remove the N-terminal Fmoc protecting group. Stirring was continued for 30 min, followed by in vacuo removal of piperidine and DMF to give the side-chain protected leader peptide. The side-chain protecting groups were removed to give 5 by stirring the crude product in a 87.5:5:5:2.5 mixture of TFA:water:ethanedithiol:TIPS for 1.5-2 h. Solvent was removed by rotary evaporation followed by trituration of the peptide in cold diethyl ether. The crude peptide was purified by preparative C18 RP-HPLC and the mass verified by MALDI-TOF or ESI MS analysis.
General procedure for 1,3-dipolar cycloadditions to give triazole-linked LctA substrates
The HPLC-purified peptides 5 and 6 or analogues thereof containing nonproteinogenic amino acids (0.2-1.5 mg each) were dissolved in a 50% mixture of dioxane and 5 mM TRIS buffer, pH 7.0 and sparged with N2. In a separate flask, 0.1 mg CuSO4 and 0.1 mg of tris(benzyltriazolylmethyl)amine (TBTA)[32] were dissolved in a 50% mixture of dioxane and 5 mM TRIS buffer, pH 7.0 and sparged with N2. To the cupric solution was added 10-20 μL of anhydrous hydrazine by syringe to form the active Cu(I) species. The cuprous solution was cannulated into the peptide solution and the mixture stirred for 45 min to 3 h, followed by analytical C4 or C18 RP-HPLC purification. The synthesis of substrates containing Cys residues required protection as StBu disulfides. Mass spectrometry data for each triazole-linked truncated LctA substrate is provided in the Supplemental Information.
Deprotection of tert-butylthio-protected triazole-linked LctA analogs
HPLC-purified triazole-linked truncated LctA substrates (10-100 μg) were resuspended in 50 μL of 100 mM TRIS buffer, pH 8.0 containing 50-100 mM dithiothreitol and maintained at 25 μC for 12-16 h. The crude product samples were acidified with 0.1% TFA and purified by analytical C4 RP-HPLC. The fractions containing product were analyzed by MALDI-TOF MS.
Peptoid Synthesis
Synthetic protocols and analytical data for Fmoc-N-butylglycine (norleucine peptoid) and Fmoc-N-isopropylglycine (valine peptoid) are presented in the accompanying Supplemental Information.
Overexpression and Purification of LctA Thioesters
E. coli BL21(DE3) cells carrying pET15b plasmids encoding either the His6-LctA(1-27)-intein-CDB fusion protein or His6-LctA(1-37)-intein-CBD fusion protein[14,27,28] were grown in 3 L LB with 100 μg/mL ampicillin at 37 °C until an OD600 of 0.5-0.7 was reached. Protein expression was induced by the addition of 0.65 mM isopropyl β-D-1-thiogalactopyranoside (IPTG), and the cells were grown for an additional 6 h at 25 °C. Cells were harvested by centrifugation at 18.5 kg for 20 min at 4 °C and the cell pellet (18 g) was resuspended in 15-20 mL of cell lysis buffer (20 mM Tris-HCl, pH 7.5, 500 mM NaCl, 1 mM EDTA, 0.1% Triton-X-100) and lysed by sonication at an amplitude of 70 (3.3 s on, 9.9 s off) for 10-15 min on ice. The lysate was centrifuged at 27 kg for 25 min, and the supernatant containing the fusion protein loaded onto a chitin column (15-20 mL) that was pre-equilibrated with column buffer A (20 mM HEPES, pH 7.2, 500 mM NaCl, 1 mM EDTA). The fusion protein was bound to the chitin column by shaking gently at 4 °C for 2-3 h. The column was then mounted upright and washed with twenty column volumes of column buffer A until the A280 of the eluent was less than 0.01. The column was subsequently washed with three column volumes of column buffer B (100 mM HEPES, pH 7.75, 200 mM NaCl, 1 mM EDTA). Intein-mediated cleavage of the His6-LctA thioesters was performed by gentle shaking of the chitin resin in 50 mL of column buffer B containing 50 mM MESNa for 12-16 h at 4 °C. At the end of this period, the His6-LctA thioester was eluted and fractions containing protein were concentrated by Amicon YM1 membrane (Millipore) and lyophilized. Crude thioesters were purified by preparative C4 RP-HPLC and the purified products analyzed by MALDI-TOF MS.
Expressed Protein Ligation
HPLC-purified synthetic peptides were redissolved in 200 μL of ligation buffer (100 mM HEPES, pH 7.75, 200 mM NaCl, and 50 mM MESNa) giving an approximate final concentration of 5-200 mM, depending upon the peptide used. The peptide solution was added directly to lyophilized His6-LctA MESNa thioester to obtain a final concentration of 1 mM of the thioester. The pH of the solution was adjusted to 7.6-7.8 and the ligation mixture was allowed to react for 12-16 h at 4 °C. The crude reaction products were analyzed by MALDI-TOF MS to judge the completeness of the reaction. The crude samples were then acidified using 0.1% TFA prior to purification by C4 analytical RP-HPLC. Fractions containing the desired ligation products were analyzed by MALDI-TOF MS.
LctM Assays
An analytical C4 RP-HPLC fraction containing the desired substrate (Abs220 ~ 0.5-1.6, 20-100 μg) was redissolved in 20 μL of sterile Millipore water. Typically, 2-5 μL of substrate was used for each assay. The total volume for the assays was 20 μL of buffer containing 50 mM Tris-HCl, pH 7.5, 10 mM MgCl2, 1 mM ATP, and 25 μg/mL BSA. His-LctM (0.5-2.0 μM) was added to the buffered peptide suspension and the assay was incubated for 1-5 h at 25 °C. Assay results were analyzed by MALDI-TOF MS. The samples were prepared as follows: 5 μL of assay was quenched with 1.0 μL of 0.1% TFA, then salts and small molecules that interfere with MS analysis were removed by C18 Zip-tip. The assay product was eluted directly using 4 μL of α-hydroxycinnamic acid matrix (prepared in 50-70% MeCN with 0.1% TFA). The sample was spotted on the MALDI target using 1.5 μL of eluent. For MS data see the Supporting Information.
p-Hydroxymercuribenzoic Acid (PHMB) Cyclization Assays
To test for the presence of free sulfhydryl groups, PHMB assays were performed as previously described.[13] Upon completion of the LctM assay of truncated LctA substrates containing Cys residues, the assays were concentrated to dryness. To the dry crude product was added 5 μL of 10 mM TCEP and 4 M guanidine hydrochloride and the sample was incubated at 25 °C for 15 min. Subsequently, 5 μL of a supersaturated solution of PHMB was added and the sample was incubated at 25 °C for 12-16 h. Thiol modification was monitored by MALDI-TOF MS. For MS results, see Supporting Information.
Supplementary Material
{kind=link}
Acknowledgements
This work was supported by the National Institutes of Health (GM58822 to WAV) and a Ruth L. Kirschstein National Research Service Award (T32 GM008276 to MRL).
Footnotes
Supporting information for this article is available on the WWW under http://www.chembiochem.org or from the author.
References
- [1].Chatterjee C, Paul M, Xie L, van der Donk WA. Chem. Rev. 2005;105:633. doi: 10.1021/cr030105v. [DOI] [PubMed] [Google Scholar]
- [2].Breukink E, Wiedemann I, van Kraaij C, Kuipers OP, Sahl H, de Kruijff B. Science. 1999;286:2361. doi: 10.1126/science.286.5448.2361. [DOI] [PubMed] [Google Scholar]
- [3].Hasper HE, Kramer NE, Smith JL, Hillman JD, Zachariah C, Kuipers OP, de Kruijff B, Breukink E. Science. 2006;313:1636. doi: 10.1126/science.1129818. [DOI] [PubMed] [Google Scholar]
- [4].Märki F, Hanni E, Fredenhagen A, van Oostrum J. Biochem. Pharmacol. 1991;42:2027. doi: 10.1016/0006-2952(91)90604-4. [DOI] [PubMed] [Google Scholar]
- [5].Chatterjee C, Patton GC, Cooper L, Paul M, van der Donk WA. Chem. Biol. 2006;13:1109. doi: 10.1016/j.chembiol.2006.08.015. [DOI] [PubMed] [Google Scholar]
- [6].Levengood MR, van der Donk WA. Bioorg. Med. Chem. Lett. 2008;18:3025. doi: 10.1016/j.bmcl.2008.01.062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Rink R, Kluskens LD, Kuipers A, Driessen AJ, Kuipers OP, Moll GN. Biochemistry. 2007;46:13179. doi: 10.1021/bi700106z. [DOI] [PubMed] [Google Scholar]
- [8].Rink R, Kuipers A, de Boef E, Leenhouts KJ, Driessen AJ, Moll GN, Kuipers OP. Biochemistry. 2005;44:8873. doi: 10.1021/bi050081h. [DOI] [PubMed] [Google Scholar]
- [9].Rink R, Wierenga J, Kuipers A, Kluskens LD, Driessen AJ, Kuipers OP, Moll GN. Appl. Environ. Microbiol. 2007;73:5809. doi: 10.1128/AEM.01104-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Rink R, Wierenga J, Kuipers A, Kluskens LD, Driessen AJ, Kuipers OP, Moll GN. Appl. Environ. Microbiol. 2007;73:1792. doi: 10.1128/AEM.02350-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Kuipers OP, Bierbaum G, Ottenwälder B, Dodd HM, Horn N, Metzger J, Kupke T, Gnau V, Bongers R, van den Bogaard P, Kosters H, Rollema HS, de Vos WM, Siezen RJ, Jung G, Götz F, Sahl HG, Gasson MJ. Antonie van Leeuwenhoek. 1996;69:161. doi: 10.1007/BF00399421. [DOI] [PubMed] [Google Scholar]
- [12].Lubelski J, Rink R, Khusainov R, Moll GN, Kuipers OP. Cell. Mol. Life Sci. 2007 doi: 10.1007/s00018-007-7171-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Li B, Yu JP, Brunzelle JS, Moll GN, van der Donk WA, Nair SK. Science. 2006;311:1464. doi: 10.1126/science.1121422. [DOI] [PubMed] [Google Scholar]
- [14].Xie L, Miller LM, Chatterjee C, Averin O, Kelleher NL, van der Donk WA. Science. 2004;303:679. doi: 10.1126/science.1092600. [DOI] [PubMed] [Google Scholar]
- [15].McClerren AL, Cooper LE, Quan C, Thomas PM, Kelleher NL, van der Donk WA. Proc. Natl. Acad. Sci. U.S.A. 2006;103:17243. doi: 10.1073/pnas.0606088103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Li P, Roller PP. Curr. Top. Med. Chem. 2002;2:325. doi: 10.2174/1568026023394209. [DOI] [PubMed] [Google Scholar]
- [17].Rew Y, Malkmus S, Svensson C, Yaksh TL, Chung NN, Schiller PW, Cassel JA, DeHaven RN, Taulane JP, Goodman M. J. Med. Chem. 2002;45:3746. doi: 10.1021/jm020108k. [DOI] [PubMed] [Google Scholar]
- [18].Tugyi R, Mezo G, Fellinger E, Andreu D, Hudecz F. J. Pept. Sci. 2005;11:642. doi: 10.1002/psc.669. [DOI] [PubMed] [Google Scholar]
- [19].Froseth BR, Herman RE, McKay LL. Appl. Environ. Microbiol. 1988;54:2136. doi: 10.1128/aem.54.8.2136-2139.1988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Uguen P, Hindre T, Didelot S, Marty C, Haras D, Le Pennec JP, Vallee-Rehel K, Dufour A. Appl. Environ. Microbiol. 2005;71:562. doi: 10.1128/AEM.71.1.562-565.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Aguilar MI, Purcell AW, Devi R, Lew R, Rossjohn J, Smith AI, Perlmutter P. Org. Biomol. Chem. 2007;5:2884. doi: 10.1039/b708507a. [DOI] [PubMed] [Google Scholar]
- [22].Stawikowski M, Stawikowska R, Jaskiewicz A, Zablotna E, Rolka K. ChemBioChem. 2005;6:1057. doi: 10.1002/cbic.200400412. [DOI] [PubMed] [Google Scholar]
- [23].Sadowsky JD, Murray JK, Tomita Y, Gellman SH. ChemBioChem. 2007;8:903. doi: 10.1002/cbic.200600546. [DOI] [PubMed] [Google Scholar]
- [24].Horne WS, Gellman SH. Acc. Chem. Res. 2008;41:1399. doi: 10.1021/ar800009n. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Muir TW, Sondhi D, Cole PA. Proc. Natl. Acad. Sci. U.S.A. 1998;95:6705. doi: 10.1073/pnas.95.12.6705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Chatterjee C, Miller LM, Leung YL, Xie L, Yi M, Kelleher NL, van der Donk WA. J. Am. Chem. Soc. 2005;127:15332. doi: 10.1021/ja0543043. [DOI] [PubMed] [Google Scholar]
- [27].Zhang X, Ni W, van der Donk WA. Org. Lett. 2007;9:3343. doi: 10.1021/ol071301h. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].Zhang X, van der Donk WA. J. Am. Chem. Soc. 2007;129:2212. doi: 10.1021/ja067672v. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].Levengood MR, Patton GC, van der Donk WA. J. Am. Chem. Soc. 2007;129:10314. doi: 10.1021/ja072967+. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].Grandjean C, Boutonnier A, Guerreiro C, Fournier JM, Mulard LA. J. Org. Chem. 2005;70:7123. doi: 10.1021/jo0505472. [DOI] [PubMed] [Google Scholar]
- [31].Rostovtsev VV, Green LG, Fokin VV, Sharpless KB. Angew. Chem. Int. Ed. 2002;41:2596. doi: 10.1002/1521-3773(20020715)41:14<2596::AID-ANIE2596>3.0.CO;2-4. [DOI] [PubMed] [Google Scholar]
- [32].Chan TR, Hilgraf R, Sharpless KB, Fokin VV. Org. Lett. 2004;6:2853. doi: 10.1021/ol0493094. [DOI] [PubMed] [Google Scholar]
- [33].You YO, van der Donk Biochemistry. 2007;46:5991. doi: 10.1021/bi602663x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [34].Simon RJ, Kania RS, Zuckermann RN, Huebner VD, Jewell DA, Banville S, Ng S, Wang L, Rosenberg S, Marlowe CK, et al. Proc. Natl. Acad. Sci. U.S.A. 1992;89:9367. doi: 10.1073/pnas.89.20.9367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [35].Miller SM, Simon RJ, Ng S, Zuckermann RN, Kerr JM, Moos WH. Bioorg. Med. Chem. Lett. 1994:2657. [Google Scholar]
- [36].Wender PA, Mitchell DJ, Pattabiraman K, Pelkey ET, Steinman L, Rothbard JB. Proc. Natl. Acad. Sci. U.S.A. 2000;97:13003. doi: 10.1073/pnas.97.24.13003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [37].Chongsiriwatana NP, Patch JA, Czyzewski AM, Dohm MT, Ivankin A, Gidalevitz D, Zuckermann RN, Barron AE. Proc. Natl. Acad. Sci. U.S.A. 2008;105:2794. doi: 10.1073/pnas.0708254105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [38].Patch JA, Barron AE. J. Am. Chem. Soc. 2003;125:12092. doi: 10.1021/ja037320d. [DOI] [PubMed] [Google Scholar]
- [39].Shankaramma SC, Moehle K, James S, Vrijbloed JW, Obrecht D, Robinson JA. Chem. Commun. 2003:1842. doi: 10.1039/b304310j. [DOI] [PubMed] [Google Scholar]
- [40].Garcia-Martinez C, Humet M, Planells-Cases R, Gomis A, Caprini M, Viana F, De La Pena E, Sanchez-Baeza F, Carbonell T, De Felipe C, Perez-Paya E, Belmonte C, Messeguer A, Ferrer-Montiel A. Proc. Natl. Acad. Sci. U.S.A. 2002;99:2374. doi: 10.1073/pnas.022285899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [41].Goodson B, Ehrhardt A, Ng S, Nuss J, Johnson K, Giedlin M, Yamamoto R, Moos WH, Krebber A, Ladner M, Giacona MB, Vitt C, Winter J. Antimicrob. Agents Chemother. 1999;43:1429. doi: 10.1128/aac.43.6.1429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [42].Humet M, Carbonell T, Masip I, Sanchez-Baeza F, Mora P, Canton E, Gobernado M, Abad C, Perez-Paya E, Messeguer A. J. Comb. Chem. 2003;5:597. doi: 10.1021/cc020075u. [DOI] [PubMed] [Google Scholar]
- [43].Ng S, Goodson B, Ehrhardt A, Moos WH, Siani M, Winter J. Bioorg. Med. Chem. 1999;7:1781. doi: 10.1016/s0968-0896(99)00132-7. [DOI] [PubMed] [Google Scholar]
- [44].Oh JE, Hong SY, Lee KH. Bioorg. Med. Chem. 1999;7:2509. doi: 10.1016/s0968-0896(99)00176-5. [DOI] [PubMed] [Google Scholar]
- [45].Ryge TS, Hansen PR. Bioorg. Med. Chem. 2006;14:4444. doi: 10.1016/j.bmc.2006.02.034. [DOI] [PubMed] [Google Scholar]
- [46].Song YM, Park Y, Lim SS, Yang ST, Woo ER, Park IS, Lee JS, Kim JI, Hahm KS, Kim Y, Shin SY. Biochemistry. 2005;44:12094. doi: 10.1021/bi050765p. [DOI] [PubMed] [Google Scholar]
- [47].Cooper LE, McClerren AL, Chary A, van der Donk WA. Chem. Biol. 2008;15:1035. doi: 10.1016/j.chembiol.2008.07.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [48].Lubelski J, Overkamp W, Kluskens LD, Moll GN, Kuipers OP. Appl. Environ. Microbiol. 2008;74:4680. doi: 10.1128/AEM.00112-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [49].Kaiser E, Colescott RL, Bossinger CD, Cook PI. Anal. Biochem. 1970;34:595. doi: 10.1016/0003-2697(70)90146-6. [DOI] [PubMed] [Google Scholar]
- [50].Han Y, Albericio F, Barany G. J. Org. Chem. 1997;62:4307. doi: 10.1021/jo9622744. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.