

Abstract
Methionine aminopeptidase (MetAP) is a promising target to develop novel antibiotics, because all bacteria express MetAP from a single gene that carries out the essential function of removing N-terminal methionine from nascent proteins. Divalent metal ions play a critical role in the catalysis, and there is an urgent need to define the actual metal used by MetAP in bacterial cells. By high throughput screening, we identified a novel class of catechol-containing MetAP inhibitors that display selectivity for the Fe(II)-form of MetAP. X-ray structure revealed that the inhibitor binds to MetAP at the active site with the catechol coordinating to the metal ions. Importantly, some of the inhibitors showed antibacterial activity at low micromolar concentration on Gram-positive and Gram-negative bacteria. Our data indicate that Fe(II) is the likely metal used by MetAP in the cellular environment, and MetAP inhibitors need to inhibit this metalloform of MetAP effectively to be therapeutically useful.
Introduction
Although methionine aminopeptidase (MetAP) is considered as a promising target for development of new antibiotics with novel mechanism of action 1, 2, current small molecule MetAP inhibitors with high potencies on purified enzymes failed to show any significant antibacterial activity 3–5. This is puzzling because MetAP carries out removal of the initiator methionine residue from newly synthesized proteins, and this removal is critical for activation, distribution and stability of many proteins 1. MetAP in bacteria is coded by a single gene and is essential for bacterial survival, because deletion of this gene in Escherichia coli or Salmonella typhimurium was shown to be lethal 6, 7. Divalent metal ions play a key role in the peptide hydrolysis catalyzed by MetAP, and purified apoenzyme of MetAP can be activated by several divalent metals, including Co(II), Mn(II), and Fe(II) 8, 9. Initially, MetAP was believed to be a Co(II) enzyme, because Co(II) is among the best activators and early X-ray structures of MetAP all contain two Co(II) ions at the active site 10. Most of the currently known MetAP inhibitors were discovered and characterized with MetAP in the Co(II)-form. However, we showed that inhibitors of the Co(II)-form may or may not inhibit other metalloforms of MetAP 9, 11. Thus, although there are many factors that an in vitro active compound may be inactive in vivo, such as absorption or metabolism, one explanation for the lack of antibacterial activities may be a disparity between the metalloform tested using a purified enzyme and the one that is important in cells. Walker and Bradshaw 12 suggested Zn(II) as a possible physiologically relevant metal because activity of Zn(II) substituted MetAP from Saccharomyces cerevisiae increased 1.7 fold under physiological concentration of reduced glutathione, while that of Co(II) substitution became inactive under the same condition. However, Yang et al. 13 concluded that Zn(II) is not the physiologically relevant metal in human type II MetAP and attributed the stoichiometric amount of Zn(II) associated with the enzyme to the Zn(II) that binds on protein surfaces. D’souza et al. 8 suggested that E. coli MetAP is a Fe(II) enzyme based on combination of whole cell metal analysis, enzyme activity measurements, and studies of substrate binding constants. Mn(II) is also a candidate, because the Mn(II)-form of E. coli MetAP is catalytically competent 14, and Mn(II) was suggested to be the physiological metal for human type II MetAP 15.
In the process of creating research tools to define the actual metal used by MetAP in cells, we have previously discovered two distinct classes of novel nonpeptidic MetAP inhibitors (e.g., 1 and 2 in Fig. 1) by screening a diverse chemical library of small organic compounds; each has a unique structural scaffold and each comprises several potent inhibitors highly selective for either the Mn(II) or the Co(II)-form of E. coli MetAP 11. Fe(II) is one of the best activators of MetAP besides Co(II) and the candidate metal for MetAP in E. coli cells 8. Now, we report the discovery of a new class of small molecule MetAP inhibitors, such as 3 (Fig. 1), by high throughput screening that showed high selectivity toward the Fe(II)-form. Some of these inhibitors clearly showed antibacterial activity, suggesting that Fe(II) is likely the physiologically relevant metal for MetAP in E. coli cells, and maybe also in other bacterial cells.
Figure 1.

Metalloform selective inhibitors of E. coli MetAP. Previously discovered 1 and 2 are selective for the Co(II)-form and the Mn(II)-form, respectively. The newly discovered 3 is selective for the Fe(II)-form. We synthesized 4–10 as derivatives of 3.
Results and Discussion
Discovery of MetAP inhibitors with selectivity for the Fe(II)-form by high throughput screening
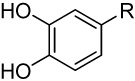
With no lead structures available for MetAP inhibitors that are selective for the Fe(II)-form, we turned to high throughput screening to identify such inhibitors with novel chemical structures. Activation of apoenzyme of E. coli MetAP by divalent metal ions is instantaneous 9, so that the screening was performed using purified apoenzyme with Fe(II) ion added immediately before monitoring hydrolysis of a fluorogenic substrate. The substrate Met-AMC is a methionine derivatized with 7-amino-4-methylcoumarin (AMC), and its hydrolysis produces AMC detectable by fluorescence 13. Fe(II) is an excellent activator, while Fe(III) cannot activate MetAP. Oxidation of Fe(II) to Fe(III) during the initial 12 min of the reaction was noticeable but not significant, because increase in fluorescence was nearly linear during this period. The screening assay measured the rate of substrate hydrolysis by MetAP in the presence or absence of screening compounds in kinetic mode by monitoring the increase in fluorescence for 12 min. A total of 74,976 compounds were screened at a single final concentration of 6.25 µg/mL. This screening campaign generated high-quality data with z’ factors 16 between 0.50 and 0.83 and their average at 0.67. Initial screening hits were further confirmed by determining their IC50 values using 6 serially diluted concentrations. Among confirmed hits is 3 with a catechol moiety. When 3 was characterized on the Co(II)-, Mn(II)- and Fe(II)-forms of E. coli MetAP, it showed low micromolar potency (13 µM) on the Fe(II)-form but showed no activity against the Co(II)-and Mn(II)-forms at the highest tested concentration of 100 µM (Table 1). Therefore, it has both significant inhibitory potency and considerable selectivity and represents an early lead structure for further development.
Table 1.
 | |||||||||
|---|---|---|---|---|---|---|---|---|---|
| Cmpd | R | Inhibition of enzymatic activity, IC50, µM | Inhibition of bacterial growth, IC50, µM c | ||||||
| Co(II) | Mn(II) | Fe(II) |
E. coli AS19 |
E. coli D22 |
B. subtilis |
B. megaterium |
|||
| 3 | 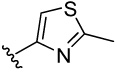 |
>100 | >100 | 13.0 | 117 (91) | 5.6 (4) | 67.0 (41) | 69.8 (73) | |
| 11 | 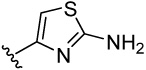 |
>100 | >100 | 6.6 | 145 (39) | 39.9 (11) | 28.6 (13) | 36.9 (26) | |
| 12 | 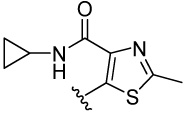 |
92.3 | 100 | 11.1 | 125 (138) | 166.8 (103) | 113 (51) | 75.5 (62) | |
| 13 | 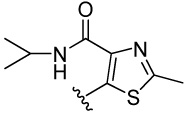 |
>100 | >100 | 15.4 | 116 (121) | 93.8 (73) | 186 (125) | 175 (122) | |
| 14 | 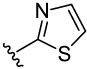 |
>100 | >100 | 16.6 | 235 (198) | 98.7 (66) | 397 (193) | 224 (112) | |
| 15 | 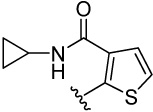 |
>100 | >100 | 7.8 | 116 (113) | 254 (151) | 109 (56) | 54.0 (79) | |
| 16 | 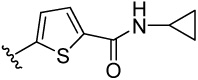 |
28.5 | 34.0 | 23.8 | 110 (78) | 238 (105) | 211 (122) | 133 (85) | |
| 17 | 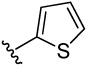 |
23.7 | 15.7 | 5.1 | 32.8 | 4.0 (3) | 16.1 (7) | 21.2 (9) | |
| 18 | 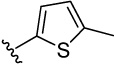 |
51.5 | 28.0 | 32.7 | 87.7 (60) | 39.1 (18) | 25.1 (10) | 26.0 (10) | |
| 19 | 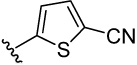 |
36.3 | 30.4 | 14.8 | 20.1 (11) | 44.2 (15) | 14.6 (13) | 15.3 (9) | |
| 20 | 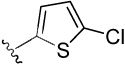 |
>100 | 44.8 | 12.8 | 15.5 (7) | 7.5 (3) | 7.1 (2) | 6.3 (2) | |
| 21 | 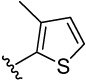 |
42.7 | 23.0 | 8.0 | 15.2 (7) | 9.0 (3) | 22.1 (8) | 15.3 (13) | |
| 22 | 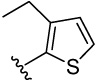 |
62.6 | 55.7 | 12.9 | 15.4 (5) | 11.8 (6) | 21.8 (7) | 27.8 (11) | |
| 23 | 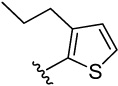 |
100 | 34.3 | 14.0 | 15.5 (4) | 43.0 (19) | 14.9 (4) | 15.5 (6) | |
| 24 | 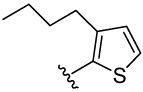 |
>100 | 48.2 | 9.2 | 12.4 (5) | 13.5 (5) | 7.8 (3) | 7.6 (3) | |
| 25 | 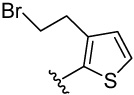 |
14.0 | 22.4 | 7.3 | 11.3 (5) | 86.0 (51) | 25.2 (10) | 21.1 (14) | |
| 26 | 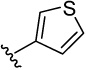 |
40.4 | 27.5 | 3.8 | 29.9 (15) | 2.0 (2) | 15.7 (9) | 19.2 (10) | |
| 27 | 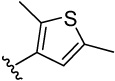 |
>100 | >100 | 7.4 | 18.5 (9) | 3.7 (4) | 16.5 (5) | 16.0 (5) | |
| 28 | 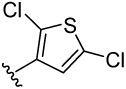 |
62.4 | 48.6 | 20.2 | 6.1 (3) | 7.3 (2) | 6.0 (2) | 5.0 (1) | |
| 29 | 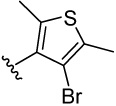 |
2.4 | 2.1 | 3.1 | 10.8 (5) | 7.7 (9) | 12.8 (8) | 17.5 (10) | |
Relative standard deviations are <20% in all values.
Compounds 4–10 were also tested for inhibition of E. coli AS19 strain, and their IC50 values in µM are 95.9, 158, 206, 63.3, 204, 117, and 125, respectively.
Numbers in parenthesis are MIC values in µg/ml.
Metalloform-selective inhibitors are both valuable research tools to define the physiologically relevant metal for MetAP and useful lead compounds to develop novel antibacterial, antifungal and anticancer agents. Discovery of the new class of nonpeptidic inhibitors with unique selectivity toward the Fe(II)-form, once again, demonstrated the power of high throughput screening technology in identifying compounds with novel activities.
Requirement of a catechol moiety for the inhibitory activity
The lead structure of 3 can be broken down to the catechol moiety and the thiazole moiety. We initially prepared 4–10 by the route outlined in Scheme 1, in which we substituted the dihydroxyl group on the phenyl ring of 3 with hydroxyl, methoxyl, amino, nitro, acetylamido, and aminocarbonyl groups. We noticed that none of them showed enzyme inhibition on the three metalloforms tested (up to 100 µM). This result underscores importance of the catechol moiety for activity, and the catechol possibly functions as a metal chelating group for the active site metal ions. The chelation by the adjacent hydroxyls with the metal likely contributes greatly to its activity because separating the hydroxyls (4), removing one of the hydroxyls (5), blocking the hydroxyls (6), or replacement with amino, nitro and other groups (7–10) all voided the activity. These results indicated that the adjacent hydroxyl groups on the phenyl ring is essential for effective inhibition of the Fe(II)-form of MetAP.
Scheme 1.

Syntheses of 4-phenylthiazolesa
a Reagents and conditions: (a) Ethanethioamide and X = Br for 4, 5, 6, 10; ethanethioamide and X = Cl for 7; thiourea and X = Cl for 11. MeOH, reflux, 80–90% yield. (b) For converting 7 to 8, 6 N HCl, MeOH, reflux, 50%. (c) For converting 8 to 9, H2, 10% Pd/C, MeOH, overnight, 70%.
Synthesis of thiazole and thiophene derivatives with a catechol moiety
To further understand the key structural scaffold, additional four thiazoles were designed, in which the catechol moiety is unchanged, while attachment of the thiazole moiety is varied, forming 2-phenylthiazole (14), 4-phenylthiazole (11) or 5-phenylthiazoles (12, 13). The syntheses of 11, 12 and 13, and 14 are shown in Scheme 1, Scheme 2, and Scheme 3, respectively. In scheme 2, the Michael addition reaction occurred in the Darzens condensation of methyl dichloroacetate and 30 17, and the resulted intermediate reacted with ethanethioamide to form 2-methylthiazole-4-carboxylate 31 18. Hydrolysis of 31 gave 32, followed by condensation with appropriate amines to afford 12a and 13a. Afterwards, the protective group of hydroxyls was removed to yield 12 and 13 (36%– 50% yield).
Scheme 2.

Syntheses of 5-phenylthiazolesa
a Reagents and conditions: (a) methyl dichloroacetate, Na, MeOH, 0°C; then ethanethioamide, THF, reflux, 43%. (b) LiOH, MeOH. (c) Amine, EDCI, DMAP, CH2Cl2, 63–70%. (d) BCl3, CH2Cl2, −78°C, 36–50%.
Scheme 3.

Syntheses of 2-phenylthiazoles and 2-thiophenes and 3-phenylthiophenesa
a Reagents and conditions: (a) Pd(PPh3)4, 2 N Na2CO3, RX (X = Br or I), DMF, 90 °C (R6 = Bn for 14–16; R6 = Me for 17–29), 31–90%. (b) For 14–16, 1 M BCl3, DCM, −78°C, 32–73%. (c) For 17–29, 1 M BBr3, DCM, −78°C, 13–90%.
2-Phenylthiazole 14 was synthesized by the Suzuki coupling reaction as outlined in Scheme 3. Coupling of appropriate phenylboronic acid with corresponding halogenated thiazole yielded 14a. The protective group of hydroxyls was removed by using BCl3, yielding 14.
Fifteen thiophene derivatives were also made and can be divided into 2-phenylthiophenes (15–25) or 3-phenylthiophenes (26–29) according to their attachment to the catechol moiety. Same Suzuki coupling was employed to make 15a–29a 19. Removal of the protective groups of hydroxyls produced 15 and 16 by using BCl3 or 17–29 by using BBr3.
Inhibition of the Co(II)-, Mn(II)- or Fe(II)-form of E. coli MetAP
We tested the synthesized phenylthiazoles and phenylthiophenes on the Co(II)-, Mn(II)- and Fe(II)-forms of E. coli MetAP to reveal the structural elements that are important in inhibitory potency and metalloform selectivity. All thiazoles 11–14, as well as the lead 3, showed low micromolar potency on the Fe(II)-form of MetAP and inactivity on the Co(II)- or Mn(II)-form, displaying considerable selectivity towards the Fe(II)-form (Table 1). It is interesting to note that although these thiazoles fused to the catechol with different positions (position 4 of the thiazole ring for 3 and 11, position 5 for 12 and 13, and position 2 for 14), neither potency nor selectivity was significantly affected.
Generally speaking, phenylthiophenes inhibited the Fe(II)-form of E. coli MetAP effectively with low micromolar potency. Some of the thiophenes, such as 17, 25, and 29, also displayed low micromolar potency on the Co(II)- and Mn(II)-forms, indicating that their selectivity for the Fe(II)-form was reduced. Nevertheless, 15 and 27 still showed good selectivity for the Fe(II)-form.
It is noticeable that all substitutions on the thiazole and thiophene rings were well tolerated for inhibition of the Fe(II)-form. MetAP as an exopeptidase has a shallow and mostly hydrophobic substrate-binding pocket with the catalytic metal sitting at the bottom of the pocket 10. Consistent with importance of the catechol moiety in the inhibitory activity and metalloform selectivity, a binding model is conceivable that the dihydroxyl group of catechol interacts with the catalytic metal ions, leaving the substituents on the thiazole or thiophene ring pointing towards the opening of the pocket. This arrangement for the substituents explains the observed inhibitory activity.
X-ray structure of E. coli MetAP in complex with inhibitor 22
To elucidate the exact binding mode of these newly discovered inhibitors on MetAP enzymes, an X-ray structure of the enzyme-inhibitor complex is desirable. However, the Fe(II)-form MetAP poses a significant challenge in crystallization due to the ease with which the ferrous ions oxidized and yielded undesirable precipitates of the protein in our initial crystallization experiments. To circumvent this problem, we attempted crystallization using the Mn(II)-form MetAP with several of these newly discovered inhibitors because we believe that the inhibitors bind to the two metalloforms of MetAP in the same mode. We succeeded in obtaining diffraction-quality crystals of E. coli MetAP in complex with 22 in the Mn(II)-form and solved the structure to 2.2 Å resolution (Fig. 2, Table 2).
Figure 2.

X-ray structure of E. coli MetAP in complex with 22. A. Stereo view of the ligand 22 at the binding pocket. The ligand and the residues at the binding site are shown as sticks (carbon yellow for 22 or grey for the protein residues, nitrogen blue, oxygen red, and sulfur orange), Mn(II) ions are shown as green spheres (Mn1 and Mn2), and a conserved water molecule is shown as a red sphere. Fobs − Fcalc difference map (before 22 was modeled in) is shown superimposed on the refined structure as blue meshes contoured at 2.5 σ. Interactions with the metal ions are shown as black dashed lines. B. Close-up view of 22, in comparison with 2, at the substrate binding pocket. The structure of the Fe(II)-form selective inhibitor 22 (carbon yellow) was superimposed with that of the Mn(II)-form selective inhibitor 2 (carbon grey). The pocket is shown as surface with colors coded by atom types. Mn(II) ions are shown as partially buried green spheres. C. Chemical structure of 22.
Table 2.
X-ray data collection and refinement statistics Cell Parameters
| Cell Parameters | |
|---|---|
| space group | P21 |
| a (Å) | 38.5 |
| b (Å) | 59.9 |
| c (Å) | 54.9 |
| β (deg) | 106.5 |
| X-ray Data Collection | |
| resolution range (Å) | 39.5–2.2 |
| collected reflections | 47,549 |
| unique reflections | 12,282 |
| completeness (%) (last shell) | 100 (99.7) |
| Redundancy (last shell) | 3.9 (3.8) |
| I/σ (I) (last shell) | 19.4 (5.4) |
| Rmerge (%) (last shell) | 7.0 (21.6) |
| Refinement Statistics | |
| resolution range (Å) | 26.9–2.2 |
| R (%) | 19.1 |
| Rfree (%) | 24.4 |
| Rmsd bonds (Å) | 0.007 |
| Rmsd angles (deg) | 1.065 |
| no. of solvent molecules | 139 |
| <B> overall (Å2) | 20.4 |
| <B> enzyme (Å2) | 20.2 |
| <B> Mn(II) ions (Å2) | 19.5 |
| <B> inhibitor (Å2) | 32.8 |
| <B> water (Å2) | 24.7 |
Consistent with the above prediction of the binding mode based on observed inhibitory activities of these unique inhibitors, inhibitor 22 indeed binds to MetAP at the substrate binding pocket with its hydroxyl groups of the catechol moiety chelating with two metal ions at the dinuclear metal site. One hydroxyl oxygen interacts only with the metal labeled as Mn1, which is the tighter bound metal ion 14, while the other hydroxyl oxygen interacts with both Mn1 and Mn2 in a monodentate bridging mode. The distance between the two metal ions is 3.38Å, and both metal ions exhibit octahedral coordination geometry. A strictly conserved water molecule (w1) found in many MetAP structures is also coordinated to Mn2. The phenyl ring of the inhibitor is not coplanar with the five-membered thiophene ring, probably to accommodate the steric bulk of an ortho-substituted ethyl group. The ethyl group is pointing towards inside of the pocket. This is consistent with our previous findings that small and non-polar side chains are tolerated in this pocket 20. The non-coplanar arrangement makes the conformation of the five-membered ring more flexible and provides one option of accommodating larger and more polar groups by rotating the bond joining the catechol and thiazole or thiophene to position the substituent towards the mouth of this binding pocket (Fig. 2B). This explains the tolerance of functional groups with different sizes on the five-membered rings.
The Fe(II)-form selectivity
It is intriguing how these inhibitors achieve metalloform selectivity, considering that X-ray structures of different metalloforms of MetAP are very similar overall and at the active site 11, 21. Comparing this structure (pdb code 3D27) with the previous MetAP structure in complex with the Mn(II)-form selective inhibitor 2 (pdb code 1XNZ), they are very similar because superimposition of the two structures gives a root mean square deviation (rmsd) value of 0.23Å for the backbone atoms of residues 4–256 (Fig. 2B). With overall similarity of the two structures, the major difference is the coordination of the two inhibitors to the metal ions. The Mn(II)-form selective inhibitor 2 coordinates with the metal ions using its carboxyl oxygens, while the Fe(II)-form selective inhibitor 22 uses two oxygens from the catechol for the coordination. It is likely that electronic properties and the geometry of the two catechol oxygens in 22 specifically satisfy the requirements for coordination to the Fe(II) ions, leading to the observed selectivity for the Fe(II)-form of MetAP.
The large number of compounds in the screening library were selected for structural diversity and drug-like properties. From these compounds with diverse structures, this screening campaign by chance identified compounds with a catechol moiety as inhibitors of the Fe(II)-form MetAP. However, this is not surprising retrospectively, because the catechol moiety is known for its affinity for Fe(II) and Fe(III) ions 22. Microbes use catechol-containing siderophores, such as enterobactin, for iron transport 23. Analysis of the structures of the active (3, 11–29) and inactive (4–10) compounds confirms the importance of the catechol moiety in their inhibitory activity. The two phenolic hydroxyl groups must be in adjacent positions, which is consistent with observation in the crystal structure that the two groups directly coordinate with the metal ions.
Inhibition of growth of E. coli and Bacillus bacterial strains
MetAP is an essential enzyme in bacteria 6, 7, and MetAP in E. coli was suggested to be a Fe(II) enzyme 8. Conceivably, the Fe(II)-form selective MetAP inhibitors will inhibit growth of bacterial cells. We selected two E. coli strains (Gram-negative) and two Bacillus strains (Gram-positive) for testing these MetAP inhibitors for antibacterial activity. While we did not see any antibacterial activity from the Co(II)- or Mn(II)-form selective inhibitors, such as 1 and 2, at the highest concentration tested (1 mM), we observed for the first time definite inhibition of cell growth at low micromolar concentrations of MetAP inhibitors (Table 1).
Both E. coli AS19 24 and D22 25 strains have genetic mutations so that their cell membranes are weakened in different ways for better penetration by small organic molecules. The two Bacillus strains are Gram-positive and believed to be more permeable to small molecules, and they are both wild type strains. Initial inhibitory activity on growth of E. coli D22 seen by 3 at 5.6 µM was encouraging. However, it inhibited growth of other three strains only modestly, and other thiazoles 11–14 did not improve the activity on growth inhibition, although they maintained potency and selectivity for the Fe(II)-form MetAP. With both inhibition of enzymatic activity on purified proteins and inhibition of bacterial growth as guidance, a series of thiophene derivatives were made and tested. Clearly, most of the thiophene derivatives showed much improved antibacterial activity, and the activity was seen on all four strains tested. Several of the thiophene derivatives have much improved antibacterial activity over the initial lead 3. The thiophene 28 is among the best with IC50 values of 6.1 µM for E. coli AS19, 7.3 µM for E. coli D22, 6.0 µM for B. subtilis, and 5.0 µM for B. megaterium.
Inhibition between E. coli AS19 and D22 strains showed bigger variations than that between two Bacillus strains. For example, 3, 17, and 26 had much better potency on E. coli D22 than on E. coli AS19, and 15, 16, 19, and 23 showed just the opposite. This observation may reflect the different mutations of E. coli AS19 and D22 strains, which render their membranes with different permeabilities to different inhibitors. The amide derivatives 12, 13, 15 and 16 all showed reduced cellular activity. The fact that they all inhibited the Fe(II)-form MetAP effectively indicates their possible difficulties in penetrating bacterial cell membranes due to the amide structure.
These bacterial strains provided the platforms to correlate inhibition of enzymatic activity and inhibition of bacterial cell growth. To evaluate the potential as an antibacterial therapeutics, we also tested two of these inhibitors on wild type E. coli strain ATCC25922, where its membrane is tougher to penetrate. We observed reduced but still measurable antibacterial activity with IC50 at 281 µM and 179 µM for 3 and 21, respectively. This result confirms the potential to develop MetAP inhibitors with effective inhibition of the Fe(II)-form MetAP as novel antibacterial drugs.
Bacterial MetAP enzymes belong to type I MetAP and are homologous to human type I MetAP in sequence. Although human type II MetAP was suggested to be a Mn(II) enzyme 15, the native metal for human type I MetAP is undefined. For therapeutic applications, MetAP inhibitors should block the activity of bacterial enzymes but spare the human counterparts. The metalloform-selective inhibition is a feature that could potentially be utilized to achieve such selectivity.
Conclusion
By high throughput screening, we discovered a novel class of inhibitors with potency and selectivity for the Fe(II)-form of E. coli MetAP. Structural modifications of the initial lead structure 3 and X-ray structural analysis of MetAP complexed with inhibitor 22 confirmed the requirement of a catechol moiety for the inhibitory activity and revealed their binding mode on the enzyme. Thiazole derivatives showed both excellent potency and selectivity of the Fe(II)-form on the purified enzyme and displayed considerable activity on growth inhibition of bacterial cells. Thiophene derivatives maintained effective inhibition of the Fe(II)-form of E. coli MetAP and some of them showed much improved inhibition on growth of all four tested Gram-positive and Gram-negative bacterial strains. The correlation between inhibition of the Fe(II)-form MetAP and antibacterial activity indicates that the Fe(II)-form is likely the physiologically relevant metalloform of MetAP and these unique inhibitors have potential to be developed as new antibiotics with novel mechanism of action.
Experimental Section
General Methods
All chemicals were reagent-grade or better and used as purchased. All reactions were performed under an inert atmosphere of dry argon or nitrogen using distilled dry solvents, and most reactions were not optimized. Where NMR data are presented, 1H spectra were obtained on either a Bruker Avance II 500 (500 MHz) or Varian Gemini 2000 (200 MHz), and 13C spectra were obtained on a Bruker Avance II 500 (125 MHz). Chemical shifts were reported in δ (ppm) using δ 7.26 signal of CDCl3 (1H NMR), δ 77.23 signal of CDCl3 (13C NMR), δ 3.31 signal of CD3OD (1H NMR), δ 49.15 signal of CD3OD (13C NMR), or δ 2.50 signal of DMSO-d6 (1H NMR) as the internal standard. High-resolution MS data were obtained on either a Thermo Finnigan MAT-95 XP or a Waters/Micromass LCT. LC-MS analyses were conducted using an Agilent system, consisting of a 1100 series HPLC connected to a diode array detector and a 1946D mass spectrometer configured for positive-ion/negative-ion electro-spray ionization. System A: column, Zorbax SB-C18, 4.6×150 mm, 5 µm; flow rate, 1.0 mL/min; UV wavelength, 254 nm; temperature, ambient; injection volume, 10 µL; A linear gradient using water with 0.1% TFA (solvent A) and acetonitrile with 0.1% TFA (solvent B); t = 0 min, 30% B, t = 15 min 100% B, t = 18 min, 100% B. System B: column, Supelco Ascentis C18, 4.6×150 mm, 5 µm; flow rate, 1.0 mL/min; UV wavelength, 254 nm; temperature, ambient; injection volume, 10 µL; A linear gradient using water with 0.1% TFA (solvent A) and methanol with 0.1% TFA (solvent B); t = 0 min, 25% B, t = 12 min 100% B, t = 17 min, 100% B.
The reaction mixture was generally poured into water, and the separated aqueous phase was then thoroughly extracted with the specified solvent. After the mixture was washed with 10% aqueous HCl and/or NaHCO3 (if required), water and brine, the combined organic phases were dried over anhydrous Na2SO4 or MgSO4 and then filtered and concentrated under reduced pressure to yield the crude reaction product. Separations by column chromatography were carried out using Dynamic Adsorbents Silica Flash 60A (40–63µ). Concentration and evaporation refer to removal of volatile material under reduced pressure on a Buchi rotavapor.
General Procedures
Procedure A
General Procedures for the Preparation of 5–10. To a stirred solution of N-(4-(2-chloroacetyl)-2-nitrophenyl)acetamide (1.283 g, 5 mmol) in EtOH (30 mL) was added thioactamide (6 mmol) and the mixture refluxed for 2 h. After cooling of the solvent, the precipitate was collected to yield 7 (1.18 g, 85%). TLC (SiO2, 30% EtOAc/Hexane): Rf = 0.32. 1H NMR (DMSO-d6, 200 MHz) δ 2.10 (s, 3H), 2.75 (s, 3H), 7.72 (d, J = 8.4 Hz, 1H), 8.14 (s, 1H), 8.24 (dd, J = 8.4 Hz, 2.2 Hz, 1H), 8.46 (d, J = 1.8 Hz, 1H), 10.33 (s, 1H). MS (ESI(+)) m/z 278 [M + H]+.
A solution of 6N HCl (3 mL) was added to a stirred suspension of 7 (554 mg, 2 mmol) in ethanol (3 mL) and the resulting mixture was refluxed for 4 h. The solvent was then vacuum-evaporated and the resulting aqueous layer was extracted with EtOAc (100 mL). The organic extracts were washed with saturated NaHCO3 solution, brine, dried (MgSO4) and concentrated to dryness. The residue was purified by column chromagraphy on silica gel, using a mixture of CH2Cl2: MeOH (100:3) as the eluent, to provide 8 (235 mg, 50%). 1H NMR (DMSO-d6, 200 MHz) δ 2.71 (s, 3H), 5.91 (br, 2H), 7.09 (d, J = 8.8 Hz, 1H), 7.81 (s, 1H), 7.96 (dd, J = 8.8 Hz, 2.2 Hz, 1H), 8.54 (d, J = 1.8 Hz, 1H). MS (ESI(+)) m/z 236 [M + H]+.
A solution of the nitro derivative 8 (200mg, 0.85 mmol) was hydrogenated in the presence of 10% Pd/C (30 mg) at room temperature for 4 h. The mixture was then filtered through a Celite pad, and the filtrate was evaporated to dryness to afford the amine 9 (596 mg, 70%). TLC (SiO2, 5% MeOH/CH2Cl2): Rf = 0.32. 1H NMR (CD3OD, 500 MHz) δ 2.72 (s, 3H), 6.72 (d, J = 8.0 Hz, 1H), 7.12 (dd, J = 8.0 Hz, 2.0 Hz, 1H), 7.20 (d, J = 2.0 Hz, 1H), 7.28 (s, 1H). MS (ESI(+)) m/z 206 [M + H]+.
4-(2-methylthiazol-4-yl)benzene-1,3-diol (4)
This compound was prepared by a procedure analogous to that of compound 7, Yield: 80%; TLC (SiO2, 30% EtOAc/Hexane): Rf = 0.33. 1H NMR (DMSO-d6, 500 MHz) δ 2.72 (s, 3H), 6.31(dd, J = 8.5 Hz, 4.0 Hz, 1H), 6.33 (d, J = 4.0 Hz, 1H), 7.72 (d, J = 8.5 Hz, 1H), 7.73 (s, 1H), 9.51 (s, 1H), 10.93 (s, 1H). MS (ESI(+)) m/z 208 [M + H]+.
3-(2-methylthiazol-4-yl)phenol (5)
This compound was prepared by a procedure analogous to that of compound 7, Yield: 85%; TLC (SiO2, 10% EtOAc/Hexane): Rf = 0.12. 1H NMR (CDCl3, 500 MHz) δ 2.77 (s, 3H), 5.69 (s, 1H), 6.81 (m, 1H), 7.26 (m, 2H), 7.38 (m, 2H). MS (ESI(+)) m/z 192 [M + H]+.
4-(3,4-dimethoxyphenyl)-2-methylthiazole (6)
This compound was prepared by a procedure analogous to that of compound 7, Yield: 87%; TLC (SiO2, 20% EtOAc/Hexane): Rf = 0.15. 1H NMR (CDCl3, 500 MHz) δ 2.77 (s, 3H), 3.92 (s, 3H), 3.97 (s, 3H), 6.90 (d, J = 8.5 Hz, 1H), 7.19 (s, 1H), 7.40 (dd, J = 8.0 Hz, 2.0 Hz, 1H), 7.45 (d, J = 2.0 Hz, 1H). MS (ESI(+)) m/z 236 [M + H]+.
2-hydroxy-5-(2-methylthiazol-4-yl)benzamide (10)
This compound was prepared by a procedure analogous to that of compound 7, Yield: 90%; TLC (SiO2, 30% EtOAc/Hexane): Rf = 0.25. 1H NMR (DMSO-d6, 200 MHz) δ 2.48 (s, 3H), 6.71 (d, J = 8.4 Hz, 1H), 7.52 (s, 1H), 7.74 (dd, J = 8.4 Hz, 2.2 Hz, 1H), 8.17 (d, J = 2.2 Hz, 1H); MS (ESI(+)) m/z 235 [M + H]+.
4-(2-aminothiazol-4-yl)benzene-1,2-diol (11)
This compound was prepared by a procedure analogous to that of compound 7, Yield: 90%; TLC (SiO2, 60% EtOAc/Hexane): Rf = 0.15. 1H NMR (CD3OD, 500 MHz) δ 6.78 (s, 1H), 6.86 (d, J = 8.5 Hz, 1H), 6.98 (dd, J = 8.5 Hz, 2.0 Hz, 1 H), 7.05 (d, J = 2.0 Hz, 1H); 13C NMR (CD3OD, 125 MHz) δ 100.64, 114.34, 117.01, 119.26, 121.33, 141.16, 147.33, 148.88, 172.63.
Procedure B
General procedure for the preparation of p12–13. A solution prepared from Na (40.3 mmol, 1 g) and MeOH (20 mL) was added to a stirred solution of 3,4-bis(benzyloxy)benzaldehyde (31mmol, 9.9 g) and methyl dichloroacetate (37.2 mmol, 3.8 mL) in THF (50 mL) at 0°C. After stirring for 15 min, the solution was warmed to room temperature and stirred for 3 h. The reaction was poured into brine under ice-bath cooling and extracted with ethyl acetate. The organic layer was dried over anhydrous sodium sulfate, and evaporated under reduced pressure. Methanol (20 mL), ethanethioamide (35 mmol, 2.66 g) were added to the residue and refluxed for 3 h. To above reaction mixture, methanesulfonic acid (35 mmol, 2.43 mL) was added and the reaction mixture was continuously refluxed overnight. The mixture was cooled to room temperature and the resulting precipitate 31 (8.2 g, 43%) was collected by filtration. MS (ESI(+)) m/z 447 [M + H]+.
Lithium hydroxide monohydrate (0.8 mmol, 35 mg) was added to a stirred solution of 31 (0.39 mmol, 200 mg) in MeOH/H2O (0.75 mL / 0.25 mL) at 0°C and stirred for 1 h with gradual warming to room temperature. TLC monitoring showed complete consumption of starting material. The solvents were removed and the residue was partitioned between EtOAc (10 mL) and H2O (5 mL). The organic phase was separated and the aqueous phase was acidified to pH = 2 with aqueous HCl (1 M) and then extracted with EtOAc. The combined organic phases were then processed in the usual way to 32 (190 mg). To the mixture of compound 32 (0.3 mmol, 150 mg), DMAP (0.12 mmol, 16 mg), EDC (0.45 mmol, 87 mg) and 4Å molecular sieves in appropriate DCM at room temperature were added to cyclopropyl amine (0.3 mmol, 38 µL), and the result mixture was stirred for 8 hours at room temperature. The reaction mixture was diluted with EtOAc (50 mL) and H2O (20 mL). The aqueous phase was extracted with EtOAc. The combined organic phases were then processed in the usual way and chromatographed (1/1 petroleum ether/EtOAc ) to yield 12a (100 mg, 63%). Then the intermediate 12a (0.19 mmol, 100 mg) was dissolved in CH2Cl2 at −80°C, following 1 M BCl3 (1 mL) in CH2Cl2 added dropwise with stirring. Stirring continued at −80°C for 3 hr and room temperature for 10 hr. The reaction was terminated by careful dropwise addition of water at 0°C, and the mixture was abstracted by EtOAc. The combined organic layers were washed with water and brine and dried with anhydrous magnesium sulfate. The resulting residue was chromatographed on silica gel (2/1 EtOAc/Hexane) to afford 12 (20mg, 36%). 1H NMR (CD3OD, 200 MHz) δ 0.56 (m, 2H), 0.77 (m, 2H), 2.64 (s, 3H), 2.72 (m, 1H), 6.82 (2H), 6.97 (d, J = 2.2 Hz, 1H); 13C NMR (CD3OD, 125 MHz) δ 6.56, 18.58, 23.45, 116.11, 117.97, 122.69, 122.97, 142.43, 143.66, 146.14, 147.61, 164.95, 166.50. MS (ESI(+)) m/z 291 [M + H]+; HRMS (CI) m/z 290.0726 (calcd for C14H14N2O3S 290.0725).
5-(3,4-dihydroxyphenyl)-N-isopropyl-2-methylthiazole-4-carboxamide (13)
was obtained from 32 according to procedure for 12 (two step 31%); TLC (SiO2, 65% EtOAc/Hexane): Rf = 0.43. 1H NMR (CD3OD, 500 MHz) δ 1.20 (d, J = 6.5 Hz, 3H), 1.25 (d, J = 7.5 Hz, 3H), 2.68 (s, 3H), 4.10 (m, 1H), 6.79 (d, J = 8.0 Hz, 1H), 6.86 (dd, J = 8.0 Hz, 2.0 Hz, 1 H), 6.98 (d, J = 2.0 Hz, 1H); 13C NMR (CD3OD, 125 MHz) δ 18.75, 22.65, 42.83, 116.32, 118.11, 122.82, 123.19, 143.10, 143.34, 146.35, 147.75, 164.34, 165.14. HRMS (CI) m/z 292.0883 (calcd for C14H16N2O3S 292.0882).
Procedure C
General procedure for the preparation of 14–29. To a solution of 2-bromothiazole (0.9 mmol, 146 mg), 3,4-bisbenzyloxyphenyl boronic acid 26 (600 mg, 1.78 mmol), and tetrakis(triphenylphosphine)-palladium (0) (0.09 mmol, 104 mg) in DMF (15 mL) was added 2M aqueous Na2CO3 (6 mmol, 3 mL). The reaction mixture was stirred at 80°C for 15 h. The reaction was quenched with water (100 mL) and extracted with EtOAc (300 mL). The combined organic layers were washed with water and brine and dried with anhydrous magnesium sulfate. The solvent was removed in vacuo. The resulting residue was chromatographed on silica gel (5% – 50% EtOAc/Hexane) to afford intermediates 14a. Yield 67%. 1H NMR (CDCl3, 200 MHz) δ 5.22 (s, 2H), 5.24 (s, 2H), 6.97 (d, J = 8.4 Hz, 1H), 7.31–7.51 (12H), 7.66 (d, J = 1.8 Hz, 1H), 7.80 (d, J = 3.2 Hz, 1H). MS (ESI(+)) m/z 374 [M+H]+.
Then the intermediate 14a (0.5 mmol, 187 mg) was dissolved in CH2Cl2 (3 mL) at −80°C, following 1 M BCl3 in CH2Cl2 (3 mL) added dropwise with stirring. Stirring continued at −80°C for 3 hr and room temperature for 5 hr. The reaction was terminated by careful dropwise addition of water (5 mL) at 0°C, and the mixture was abstracted by EtOAc (15 mL). The combined organic layers were then processed in usual way and chromatographed on silica gel (50% – 100% EtOAc/Hexane) to afford 14 (71mg, 73%). 1H NMR (CD3OD, 500 MHz) δ 6.98 (d, J = 8.0 Hz, 1H), 7.40 (d, J = 2.0 Hz, 1H), 7.41 (dd, J = 8.0 Hz, 2.0 Hz, 1H), 7.91 (d, J = 4.0 Hz, 1H), 8.10 (d, J = 3.5 Hz, 1H); 13C NMR (CD3OD, 125 MHz) δ 115.29, 117.57, 119.28, 122.04, 122.39, 135.52, 147.95, 153.16, 173.59; MS (ESI(−)) m/z 192 [M−H]−.
2-(3,4-bis(benzyloxy)phenyl)-N-cyclopropylthiophene-3-carboxamide (15a)
was obtained from 3,4-bisbenzyloxyphenyl boronic acid and 2-bromo-N-cyclopropylthiophene-3-carboxamide 27 according to the procedure described for 14a in 55%. TLC (SiO2, 50% EtOAc/Hexane): Rf = 0.21. 1H NMR (CDCl3, 500 MHz) δ 0.22 (m, 2H), 0.68 (m, 2H), 2.68 (m, 1H), 5.17 (s, 2H), 5.24 (s, 2H), 5.51 (brs, 1H), 6.98 (m, 2H), 7.06 (m, 1H), 7.21 (d, J = 5.5 Hz, 1H), 7.33 (m, 2H), 7.38 (m, 4H), 7.41 (d, J = 5.5 Hz, 1H), 7.47 (m, 4H).
N-cyclopropyl-2-(3,4-dihydroxyphenyl)thiophene-3-carboxamide (15)
was obtained from 15a according to the procedure described for 14 in 32%. TLC (SiO2, 60% EtOAc/Hexane): Rf = 0.32. 1H NMR (CD3OD, 500 MHz) δ 0.42 (m, 2H), 0.69 (m, 2H), 2.71 (m, 1H), 6.79 (m, 2H), 6.88 (m, 1H), 7.16 (d, J = 5.5 Hz, 1H), 7.29 (d, J = 5.5 Hz, 1H); 13C NMR (CD3OD, 125 MHz) δ 6.51, 23.74, 116.60, 117.23, 121.85, 125.04, 125.91, 129.37, 133.67, 146.26, 146.65, 147.44, 170.08. MS (ESI(+)) m/z 276 [M+H]+; HRMS (ESI(+)) m/z 276.0696 (calcd for C14H14NO3S 276.0694).
5-(3,4-bis(benzyloxy)phenyl)-N-cyclopropylthiophene-2-carboxamide (16a)
was obtained from 3,4-bisbenzyloxyphenyl boronic acid and 5-bromo-N-cyclopropylthiophene-2-carboxamide 27 according to the procedure described for 14a in 60%. TLC (SiO2, 25% EtOAc/Hexane): Rf = 0.12. 1H NMR (CDCl3, 200 MHz) δ 0.69 (m, 2H), 0.79 (m, 2H), 2.63 (m, 1H), 4.88 (s, 2H), 4.99 (s, 2H), 5.82 (s, 1H), 6.72 (m, 1H), 6.82 (m, 2H), 6.69 (m, 2H), 7.10–7.30 (10H). MS (ESI(+)) m/z 456 [M+H]+.
N-cyclopropyl-5-(3,4-dihydroxyphenyl)thiophene-2-carboxamide (16)
was obtained from 16a according to procedure for 14 in 40 %. TLC (SiO2, 50% EtOAc/Hexane): Rf = 0.19. 1H NMR (CD3OD, 500 MHz) δ 0.64 (m, 2H), 0.79 (m, 2H), 2.80 (m, 1H), 6.79 (d, J = 8.5 Hz, 1H), 7.01 (dd, J = 8.0 Hz, 2.0 Hz, 1H), 7.08 (d, J = 2.0 Hz, 1H), 7.15 (d, J = 4.0 Hz, 1H), 7.55 (d, J = 4.0 Hz, 1H); 13C NMR (CD3OD, 125 MHz) δ 6.72, 24.01, 114.32, 117.00, 119.15, 123.31, 127.16, 130.84, 137.23, 146.99, 147.62, 151.53, 166.24. MS (ESI(+)) m/z 276 [M+H]+; HRMS (ESI(−)) m/z 274.0545 (calcd for C14H12NO3S 274.0538).
2-(3,4-dimethoxyphenyl)thiophene (17a)
was obtained from commercial available compound 3,4-dimethoxyphenylboronic acid and 2-bromothiophene according to the procedure described for 14a in 50%. TLC (SiO2, 5% EtOAc/Hexane): Rf = 0.20. 1H NMR (CDCl3, 200 MHz) δ 3.91 (s, 3H), 3.94 (s, 3H), 6.88 (d, J = 8.4 Hz, 1H), 7.06 (dd, J =5.0 Hz, 3.6 Hz, 1H), 7.11 (d, J = 1.8 Hz, 1H), 7.18 (dd, J = 8.4 Hz, 1.8 Hz, 1H), 7.20 (dd, J =3.6 Hz, 1.0 Hz, 1H), 7.23 (dd, J = 5.0 Hz, 1.0 Hz, 1H). MS (ESI(+)) m/z 221 [M+H]+.
4-(thiophen-2-yl)benzene-1,2-diol (17)
was obtained from 17a in presence of BBr3 according to the procedure described for 14 in 57%. TLC (SiO2, 25% EtOAc/Hexane): Rf = 0.30. 1H NMR (CD3OD, 500 MHz) δ 6.76 (d, J = 8.5 Hz, 1H), 6.95 (dd, J = 8.0 Hz, 2.5 Hz, 1H), 7.00 (dd, J = 5.0 Hz, 3.5 Hz, 1H), 7.04 (d, J = 2.0 Hz, 1H), 7.15 (dd, J = 3.5 Hz, 1.0 Hz, 1H), 7.22 (dd, J = 5.5 Hz, 1.0 Hz, 1H); 13C NMR (CD3OD, 125 MHz) δ 114.24, 116.86, 118.79, 122.77, 124.39, 128.23, 128.89, 146.18, 146.44, 146.75. MS (ESI(+)) m/z 193 [M+H]+; HRMS (EI) m/z 192.0236 (calcd for C10H8O2S 192.0245). HPLC purity: system A, tR 6.29 min (99.12 %); system B, tR 9.90 min (99.61 %).
2-(3,4-dimethoxyphenyl)-5-methylthiophene (18a)
was obtained from commercial available compound 3,4-dimethoxyphenylboronic acid and 2-iodo-5-methylthiophene according to the procedure described for 14a in 64%. TLC (SiO2, 5% EtOAc/Hexane): Rf = 0.21. 1H NMR (CDCl3, 500 MHz) δ 2.49 (s, 3H), 3.89 (s, 3H), 3.92 (s, 3H), 6.70 (d, J = 3.5 Hz, 1H), 6.85 (d, J = 8.5 Hz, 1H), 6.99 (d, J = 3.5 Hz, 1H), 7.05 (d, J = 2.0 Hz, 1H), 7.10 (dd, J = 8.5 Hz, 2.0 Hz, 1H). MS (ESI(+)) m/z 235 [M+H]+.
4-(5-methylthiophen-2-yl)benzene-1,2-diol (18)
was obtained from 18a in presence of BBr3 according to the procedure described for 14 in 68%. TLC (SiO2, 25% EtOAc/Hexane): Rf = 0.31. 1H NMR (CD3OD, 500 MHz) δ 2.43 (s, 3H), 6.64 (dd, J = 3.0 Hz, 1.0 Hz, 1H), 6.75 (d, J = 8.0 Hz, 1 H), 6.88 (dd, J = 2.0 Hz, 8.0 Hz, 1H), 6.91 (d, J = 3.5 Hz, 1H), 6.99 (d, J = 2.0 Hz, 1H); 13C NMR (CD3OD, 125 MHz) δ 15.31, 113.85, 116.81, 118.36, 122.51, 127.11, 128.59, 138.98, 143.83, 146.05, 146.65. MS (ESI(+)) m/z 207 [M+H]+; HRMS (CI) m/z 206.0400 (calcd for C11H10O2S 206.0402). HPLC purity: system A, tR 7.57 min (95.67 %); system B, tR 10.93 min (97.44 %).
5-(3,4-dimethoxyphenyl)thiophene-2-carbonitrile (19a)
was obtained from commercial available compound 3,4-dimethoxyphenylboronic acid and 5-bromothiophene-2-carbonitrile according to the procedure described for 14a in 67%. TLC (SiO2, 10% EtOAc/Hexane): Rf = 0.20. 1H NMR (CDCl3, 200 MHz) δ 3.93 (s, 3H), 3.95 (s, 3H), 6.91 (d, J = 8.4 Hz, 1H), 7.06 (d, J = 1.8 Hz, 1H), 7.17 (d, J = 4.4 Hz, 1H), 7.19 (m, 1H), 7.57 (d, J = 4.4 Hz, 1H). MS (ESI(+)) m/z 246 [M+H]+.
5-(3,4-dihydroxyphenyl)thiophene-2-carbonitrile (19)
was obtained from 19a in presence of BBr3 according to the procedure described for 14 in 68%. TLC (SiO2, 25% EtOAc/Hexane): Rf = 0.14. 1H NMR (CD3OD, 500 MHz) δ 6.81 (d, J = 8.0 Hz, 1H), 7.04 (dd, J = 8.0 Hz, 2.5 Hz, 1H), 7.09 (d, J = 2.5 Hz, 1H), 7.26 (d, J = 4.0 Hz, 1H), 7.66 (d, J = 4.0 Hz, 1H); 13C NMR (CD3OD, 125 MHz) δ 107.11, 114.47, 115.54, 117.10, 119.60, 123.38, 125.67, 140.31, 147.21, 148.52, 154.34. MS (ESI(+)) m/z 218 [M+H]+; HRMS (CI) m/z 217.0197 (calcd for C11H7NO2S 217.0197). HPLC purity: system A, tR 6.25 min (99.30%); system B, tR 7.02 min (99.68%).
2-chloro-5-(3,4-dimethoxyphenyl)thiophene (20a)
was obtained from commercial available compound 3,4-dimethoxyphenylboronic acid and 2-bromo-5-chlorothiophene according to the procedure described for 14a in 70%. TLC (SiO2, 5% EtOAc/Hexane): Rf = 0.22. 1H NMR (CDCl3, 200 MHz) δ 3.90 (s, 3H), 3.92 (s, 3H), 6.857 (d, J = 4.0 Hz, 1H), 6.860 (d, J = 8.4 Hz, 1H), 6.95 (d, J = 3.6 Hz, 1H), 6.99 (d, J = 1.8 Hz, 1H), 7.06 (dd, J = 8.0 Hz, 2.0 Hz, 1H). MS (ESI(+)) m/z 255 [M+H]+.
4-(5-chlorothiophen-2-yl)benzene-1,2-diol (20)
was obtained from 20a in presence of BBr3 according to the procedure described for 14 in 45%. TLC (SiO2, 25% EtOAc/Hexane): Rf = 0.35. 1H NMR (CD3OD, 500 MHz) δ 6.77 (d, J = 8.5 Hz, 1H), 6.86 (d, J = 4.0 Hz, 1H), 6.88 (dd, J = 8.5 Hz, 2.5 Hz, 1H), 6.95 (d, J = 4.0 Hz, 1H), 6.97 (d, J = 2.0 Hz, 1H); 13C NMR (CD3OD, 125 MHz) δ 113.78, 116.93, 118.50, 122.07, 127.17, 128.36, 132.11, 145.18, 146.91, 146.95. MS (ESI(−)) m/z 225 [M−H]−; HRMS (CI) m/z 225.9852 (calcd for C10H7ClO2S 225.9855). HPLC purity: system A, tR 8.42 min (96.68%); system B, tR 11.90 min (99.60%).
2-(3,4-dimethoxyphenyl)-3-methylthiophene (21a)
was obtained from commercial available compound 3,4-dimethoxyphenylboronic acid and 2-bromo-3-methylthiophene according to the procedure described for 14a in 75%. TLC (SiO2, 5% EtOAc/Hexane): Rf = 0.20. 1H NMR (CDCl3, 200 MHz) δ 2.31 (s, 3H), 3.91 (s, 3H), 3.92 (s, 3H), 6.91 (d, J = 8.0 Hz, 1H), 6.92 (d, J = 5.8 Hz, 1H), 6.97 (d, J = 1.8 Hz, 1H), 7.02 (d, J = 8.0 Hz, 2.2 Hz, 1H), 7.17 (d, J = 5.2 Hz, 1H). MS (ESI(+)) m/z 235 [M+H]+.
4-(3-methylthiophen-2-yl)benzene-1,2-diol (21)
was obtained from 21a in presence of BBr3 according to the procedure described for 14 in 50%. TLC (SiO2, 25% EtOAc/Hexane): Rf = 0.33. 1H NMR (CD3OD, 500 MHz) δ 2.26 (s, 3H), 6.76 (dd, J = 8.0 Hz, 2.0 Hz, 1H), 6.80 (d, J = 8.5 Hz, 1H), 6.86 (d, J = 5.5 Hz, 1H), 6.87 (d, J = 2.0 Hz, 1H), 7.15 (d, J = 5.0Hz, 1H); 13C NMR (CD3OD, 125 MHz) δ 15.06, 116.53, 117.36, 121.91, 123.35, 128.12, 131.90, 133.28, 139.46, 146.10, 146.39. HRMS(CI) m/z 206.0399 (calcd for C11H10O2S 206.0402). HPLC purity: system A, tR 7.23 min (99.83%); system B, tR 10.59 min (99.01%).
2-(3,4-dimethoxyphenyl)-3-ethylthiophene (22a)
was obtained from 3,4-dimethoxyphenylboronic acid and 2-bromo-3-ethylthiophene 28 according to the procedure described for 14a in 81%. TLC (SiO2, 5% EtOAc/Hexane): Rf = 0.21. 1H NMR (CDCl3, 200 MHz) δ 1.23 (t, J = 7.8 Hz, 3H), 2.68 (q, J = 7.6 Hz, 2H), 3.91 (s, 3H), 3.92 (s, 3H), 6.88–7.02 (4H), 7.20 (d, J = 5.0 Hz, 1H). MS (ESI(+)) m/z 249 [M+H]+.
4-(3-ethylthiophen-2-yl)benzene-1,2-diol (22)
was obtained from 22a in presence of BBr3 according to the procedure described for 14 in 71%. TLC (SiO2, 25% EtOAc/Hexane): Rf = 0.33. 1H NMR (CD3OD, 500 MHz) δ 1.18 (t, J = 7.5 Hz, 3H), 2.64 (q, J = 7.5 Hz, 2H), 6.73 (dd, J = 8.0 Hz, 2.5 Hz, 1H), 6.81 (d, J = 8.0 Hz, 1H), 6.86 (d, J = 2.5 Hz, 1H), 6.93 (d, J = 5.0 Hz, 1H), 7.16 (d, J = 5.0 Hz, 1H); 13C NMR (CD3OD, 125 MHz) δ 16.01, 22.95, 116.50, 117.61, 122.16, 123.82, 127.99, 130.00, 139.09, 140.29, 146.15, 146.31. MS (ESI(−)) m/z 219 [M−H]−; HRMS(CI) m/z 220.0555 (calcd for C12H12O2S 220.0558). HPLC purity: system A, tR 8.71 min (99.72%); system B, tR 11.30 min (99.39%).
2-(3,4-dimethoxyphenyl)-3-propylthiopene (23a)
was obtained from 3,4-dimethoxyphenylboronic acid and 2-bromo-3-propylthiophene 28 according to the procedure described for 14a in 70%. TLC (SiO2, 5% EtOAc/Hexane): Rf = 0.22. 1H NMR (CDCl3, 200 MHz) δ 0.92 (t, J = 7.2 Hz, 3H), 1.65 (m, 2H), 2.62 (t, J = 7.8 Hz, 2H), 3.90 (s, 3H), 3.92 (s, 3H), 6.82–7.01 (4H), 7.19 (d, J = 5.4 Hz, 1H). MS (ESI(+)) m/z 263 [M+H]+.
4-(3-propylthiophen-2-yl)benzene-1,2-diol (23)
was obtained from 23a in presence of BBr3 according to the procedure described for 14 in 13%. TLC (SiO2, 25% EtOAc/Hexane): Rf = 0.33. 1H NMR (CD3OD, 500 MHz) δ 0.89 (t, J = 7.5 Hz, 3H), 1.59 (m, 2H), 2.60 (t, J = 7.5 Hz, 2H), 6.72 (d, J = 8.0 Hz, 1H), 6.80 (d, J = 8.0 Hz, 1H), 6.85 (s, 1H), 6.91 (d, J = 5.0 Hz, 1H), 7.17 (d, J = 4.5 Hz, 1H); 13C NMR (CD3OD, 125 MHz) δ 14.39, 25.32, 31.81, 116.47, 117.77, 122.29, 123.69, 128.07, 130.38, 138.77, 139.64, 146.19, 146.33. MS (ESI(+)) m/z 235 [M+H]+; HRMS(CI) m/z 234.0705 (calcd for C13H14O2S 234.0715). HPLC purity: system A, tR 9.19 min (99.77%); system B, tR 11.94 min (99.60%).
3-butyl-2-(3,4-dimethoxyphenyl)thiophene (24a)
was obtained from 3,4-dimethoxyphenylboronic acid and 2-bromo-3-butylthiophene 28 according to the procedure described for 14a in 72%. TLC (SiO2, 5% EtOAc/Hexane): Rf = 0.19. 1H NMR (CDCl3, 200 MHz) δ 0.68 (t, J = 7.4 Hz, 3H), 1.13 (m, 2H), 1.39 (m, 2H), 2.45 (t, J = 7.4 Hz, 2H), 3.70 (s, 3H), 3.71 (s, 3H), 6.68–6.77 (4H), 6.98 (d, J = 5.4 Hz, 1H). MS (ESI(+)) m/z 277 [M+H]+.
4-(3-butylthiophen-2-yl)benzene-1,2-diol (24)
was obtained from 24a in presence of BBr3 according to the procedure described for 14 in 15%. TLC (SiO2, 25% EtOAc/Hexane): Rf = 0.33. 1H NMR (CD3OD, 500 MHz) δ 0.87 (t, J = 7.5 Hz, 3H), 1.30 (m, 2H), 1.55 (m, 2H), 2.62 (t, J = 7.0 Hz, 2H), 6.71 (d, J = 8.0 Hz, 1 H), 6.79 (d, J = 8.0 Hz, 1H), 6.83 (d, J = 2.0 Hz, 1H), 6.91 (d, J = 5.0 Hz, 1H), 7.61 (d, J = 3.5 Hz, 1H); 13C NMR (CD3OD, 125 MHz) δ 14.31, 23.60, 29.38, 34.45, 116.47, 117.78, 122.29, 123.72, 128.06, 130.37, 138.93, 139.53, 146.20, 146.35. MS (ESI(+)) m/z 249 [M+H]+; HRMS(CI) m/z 248.0858 (calcd for C14H16O2S 248.0871). HPLC purity: system A, tR 10.25 min (99.96%); system B, tR 12.57 min (99.01%).
2-(2-(3,4-dimethoxyphenyl)thiophen-3-yl)ethanol (25a)
was obtained from 3,4-dimethoxyphenylboronic acid and 2-(2-bromothiophen-3-yl)ethanol 28 according to the procedure described for 14a in 50%. TLC (SiO2, 30% EtOAc/Hexane): Rf = 0.15. 1H NMR (CDCl3, 200 MHz) δ 2.95 (t, J = 6.2 Hz, 3H), 3.84 (t, J = 6.6 Hz, 2H), 3.91 (s, 3H), 3.92 (s, 3H), 6.93 (m, 1H), 7.00(m, 2H), 7.25 (m, 2H).
4-(3-(2-bromoethyl)thiophen-2-yl)benzene-1,2-diol (25)
was obtained from 25a in presence of BBr3 according to the procedure described for 14 in 38%. TLC (SiO2, 25% EtOAc/Hexane): Rf = 0.30. 1H NMR (CD3OD, 500 MHz) δ 3.17 (t, J = 7.5 Hz, 2H), 3.53 (t, J = 7.5 Hz, 2H), 6.74 (dd, J = 8.5 Hz, 2.0 Hz, 1H), 6.81 (d, J = 8.5 Hz, 1H), 6.84 (d, J = 2.0 Hz, 1H), 6.99 (d, J = 5.0 Hz, 1H), 7.23 (d, J = 5.0 Hz, 1H); 13C NMR (CD3OD, 125 MHz) δ 32.78, 33.50, 116.65, 117.71, 122.27, 124.32, 127.23, 130.00, 135.43, 141.62, 146.56, 146.64. MS (ESI(+)) m/z 299/301 [M+H]+; HRMS(CI) m/z 297.9653 (calcd for C12H11BrO2S 297.9663). HPLC purity: system A, tR 8.54 min (99.33%); system B, tR 11.42 min (98.10%).
3-(3,4-dimethoxyphenyl)thiophene (26a)
was obtained from commercial available compound 3,4-dimethoxyphenylboronic acid and 3-bromothiophene according to the procedure described for 14a in 64%. TLC (SiO2, 5% EtOAc/Hexane): Rf = 0.20. 1H NMR (CDCl3, 200 MHz) δ 3.91 (s, 3H), 3.94 (s, 3H), 6.90 (d, J = 8.4 Hz, 1H), 7.10 (d, J = 1.8 Hz, 1H), 7.15 (dd, J = 8.4 Hz, 1.8 Hz, 1H), 7.32–7.38 (3H). MS (ESI(+)) m/z 221 [M+H]+.
4-(thiophen-3-yl)benzene-1,2-diol (26)
was obtained from 26a in presence of BBr3 according to the procedure described for 14 in 57%. TLC (SiO2, 25% EtOAc/Hexane): Rf = 0.32. 1H NMR (CD3OD, 500 MHz) δ 6.77 (d, J = 8.0 Hz, 1H), 6.97 (dd, J = 8.0 Hz, 2.5 Hz, 1H), 7.06 (d, J = 2.5 Hz, 1H), 7.32 (dd, J = 5.0 Hz, 1.0 Hz, 1H), 7.37 (dd, J = 3.0 Hz, 1 Hz, 1H), 7.39 (dd, J = 5.0 Hz, 3.0 Hz, 1H); 13C NMR (CD3OD, 125 MHz) δ 114.68, 116.79, 119.16, 119.37, 126.86, 127.17, 129.74, 143.87, 145.90, 146.62. MS (ESI(+)) m/z 193 [M+H]+; HRMS (EI) m/z 192.0238 (calcd for C10H8O2S 192.0245). HPLC purity: system A, tR 6.07 min (97.24%); system B, tR 9.59 min (98.16%).
3-(3,4-dimethoxyphenyl)-2,5-dimethylthiophene (27a)
was obtained from 3,4-dimethylphenylboronic acid and 3-iodo-2,5-dimethylthiophene-2,5-dimethylthiophene 29 according to the procedure described for 14a in 90%. TLC (SiO2, 5% EtOAc/Hexane): Rf = 0.28. 1H NMR (CDCl3, 200 MHz) δ 2.43 (s, 3H), 2.44 (s, 3H), 3.90 (s, 3H), 3.91 (s, 3H), 6.68 (1H), 6.89–6.91 (3H). MS (ESI(+)) m/z 249 [M+H]+.
4-(2,5-dimethylthiophen-3-yl)benzene-1,2-diol (27)
was obtained from 27a in presence of BBr3 according to the procedure described for 14 in 90%. TLC (SiO2, 25% EtOAc/Hexane): Rf = 0.34. 1H NMR (CD3OD, 500 MHz) δ 2.36 (s, 3H), 2.37 (s, 3H), 6.59 (s, 1H), 6.66 (m, 1H), 6.78 (m, 2H); 13C NMR (CD3OD, 125 MHz) δ 14.31, 15.15, 116.38, 116.88, 121.37, 127.55, 128.49, 130.53, 136.15, 139.75, 145.26, 146.14. MS (ESI(+)) m/z 221 [M+H]+; HRMS(CI) m/z 220.0551 (calcd for C12H12O2S 220.0558). HPLC purity: system A, tR 9.61 min (98.52%); system B, tR 12.31 min (99.55%).
3-(3,4-bis(benzyloxy)phenyl)-2,5-dichlorothiophene (28a)
was obtained from 3,4-bis(benzyloxy)phenylboronic acid and 3-bromo-2,5-dichlorothiophene according to the procedure described for 14a in 68%. TLC (SiO2, 5% EtOAc/Hexane): Rf = 0.51. 1H NMR (CDCl3, 500 MHz) δ 5.198 (s, 2H), 5.202 (s, 2H), 6.81 (s, 1H), 6.97 (d, J = 8.0 Hz, 1H), 7.01 (dd, J = 8.0 Hz, 2.0 Hz, 1H), 7.12 (d, J = 2.0 Hz, 1H), 7.32 (m, 2H), 7.37 (m, 4H), 7.45 (m, 4H). MS (ESI(+)) m/z 463 [M+Na]+.
4-(2,5-dichlorothiophen-3-yl)benzene-1,2-diol (28)
was obtained from 28a in presence of BCl3 according to the procedure described for 14 in 57%. TLC (SiO2, 25% EtOAc/Hexane): Rf = 0.37. 1H NMR (CD3OD, 500 MHz) δ 6.81 (d, J = 8.0 Hz, 1H), 6.86 (dd, J = 8.0 Hz, 2.0 Hz, 1H), 6.97 (s, 1H), 6.99 (d, J = 2.0 Hz, 1H); 13C NMR (CD3OD, 125 MHz) δ 116.54, 116.69, 121.15, 121.48, 126.38, 127.03, 129.19, 140.10, 146.43, 146.84. MS (ESI(+)) m/z 259/261 [M−H]−; HRMS(CI) m/z 259.9459 (calcd for C10H6Cl2O2S 259.9466). HPLC purity: system A, tR 9.75 min (99.80%); system B, tR 12.72 min (99.00%).
3-bromo-4-(3,4-dimethoxyphenyl)-2,5-dimethylthiophene (29a)
was obtained from 3,4-dimethylphenylboronic acid and 3,4-dibromo-2,5-dimethylthiophene 30 according to the procedure described for 14a in 31%. TLC (SiO2, 5% EtOAc/Hexane): Rf = 0.30. 1H NMR (CDCl3, 200 MHz) δ 2.31 (s, 3H), 2.40 (s, 3H), 3.89 (s, 3H), 3.92 (s, 3H), 6.82 (m, 2H), 6.93 (d, J = 8.8 Hz, 1H). MS (ESI(+)) m/z 327/329 [M+H]+.
4-(4-bromo-2,5-dimethylthiophen-3-yl)benzene-1,2-diol (29)
was obtained from 29a in presence of BBr3 according to the procedure described for 14 in 40%. TLC (SiO2, 25% EtOAc/Hexane): Rf = 0.40. 1H NMR (CD3OD, 500 MHz) δ 2.26 (s, 3H), 2.34 (s, 3H), 6.53 (d, J = 8.0 Hz, 1H), 6.66 (s, 1H), 6.80 (d, J = 8.0 Hz, 1H); 13C NMR (CD3OD, 125 MHz) δ 14.58, 15.18, 112.26, 116.15, 118.57, 123.08,128.48, 129.07, 130.92, 132.87, 139.97, 146.00. MS (ESI(+)) m/z 299/301 [M+H]+; HRMS(CI) m/z 297.9655 (calcd for C12H11BrO2S 297.9663). HPLC purity: system A, tR 9.56 min (99.30%); system B,tR 12.29 min (99.42%).
MetAP enzymatic activity assay
The recombinant E. coli MetAP was purified from BL21(DE3) cells as apoenzyme 9. Met-AMC was purchased from Bachem Bioscience (King of Prussia, PA).Enzyme activity was monitored by following the hydrolysis of MetAMC (λex 360 nm, λem 460) on a SpectraMax Gemini XPS plate reader (Molecular Devices, Sunnyvale, CA), carried out on 384-well plates at room temperature. The assay consisted of an 80 µL assay mixture containing 50 mM MOPS (pH 7.0), 200 µM Met-AMC, 500 nM apoenzyme, metal ions (50 µM FeCl2 with 100 µM ascorbic acid, 10 µM CoCl2, or 20 µM MnCl2) and an inhibitor at serially diluted concentrations. The IC50 values were calculated from non-linear curve fitting procedures of percent inhibition as a function of inhibitor concentrations.
High throughput screening
A chemical library of 74,976 small organic molecules used for screening was purchased from ChemBridge and ChemDiv (both at San Diego, CA), and these compounds were selected from vendors’ much larger compound collections for structural diversity and drug-like properties. Reactive, unstable and potentially toxic compounds were eliminated in the selection process. All compounds have molecular weight between 150 and 480 and calculated LogP (cLogP) below 5. The assay was performed on 384-well flat-bottom polystyrene microplates on a SpectraMax Gemini XS microplate fluorescence reader (Molecular Devices Corp., Sunnyvale, CA). The screening compounds were distributed on 213 plates at a final compound concentration of 6.25 µg/mL. Each plate had 352 compound wells and 32 positive and negative control wells. The negative control wells contained no screening compounds, and the positive control wells contained no screening compounds and no apoenzyme. The assay mixture (80 µL final volume) contains 50 mM MOPS (pH 7.0), 1.0 µM apo-MetAP, 200 µM Met-AMC, and 4 µM FeCl2. Screening hits were ranked according to their potencies (percent inhibition), and the top ranked 320 hits with a cut-off at 84% inhibition were selected for confirmation of inhibitory activity by determining their IC50 values using 6 serially diluted concentrations. Liquid handling for hit-picking and serial dilutions was carried out on a Biomek FX liquid handling workstation (Beckman Coulter Inc., Fullerton, CA) and a Precision 2000 automated microplate pipetting system (BioTek Instruments Inc, Winooski, VT).
Bacterial growth inhibition assay
Bacillus megaterium, Bacillus subtilis, and E. coli ATCC 25922 were acquired from Fisher (Pittsburgh, PA). E. coli strain D22 25 was obtained from the E. coli Genetic Stock Center at Yale (http://cgsc.biology.yale.edu/). E. coli strain AS19 was obtained as a gift from Dr. Liam Good at Karolinska Institute. Strain AS19 has a severely depleted lipopolysaccharides (LPS) layer 24, but its exact mutation is unknown. Bacterial strains (E. coli AS19, E. Coli D22, Bacillus megaterium and B. subtilis) grown to exponential phase with 0.5 McFarland optical density 31 was diluted 1000 fold in Mueller Hinton media containing 100 mM Tris pH 7.5, which were then dispensed in 40 µL volumes into an opaque 384-well plate containing 20 µL of inhibitor at increasing concentrations, followed by addition of 20 µL of resazurin for a final concentration of 110 µM. Cell growth was monitored kinetically by fluorescence (λex 530 nm, λem 590) for 10 hours with 5 min intervals at 37°C. In the case of E. coli AS19, resazurin was omitted and the signal monitored by absorbance at 600 nm, because the absorbance signal was much more stable for E. coli AS19 than for the other strains tested. Fluorescence or absorbance values corresponding to 50–85% of total intensity of an uninhibited sample were averaged and converted to percent inhibition to obtain IC50 values as described above. MIC values were calculated as the concentration of compound resulting in 90% inhibition of cell growth. Assay was performed for E. coli ATCC 25922 in the presence of resazurin as described above, with the exception that Tris was not included in the culture medium.
Crystallization and data collection
Crystals of the enzyme-inhibitor complex were obtained by hanging-drop vapor-diffusion method at room temperature. Inhibitor 22 (100 mM in DMSO) was added to concentrated apoenzyme (10 mg/mL, 0.4 mM) in 50 mM Tris.HCl (pH 7.8), and the molar ratio of 22 to MetAP was 6:1. The enzyme/inhibitor mixture was mixed with the reservoir buffer (100 mM MES, pH 6.5, 2 mM MnCl2, and 17% PEG 20,000) in a 1:1 ratio. Crystals were seen after about 3 days. After one crystal was picked up from the mother liquor with a fiber loop, the loop was dipped into a cryo solution containing 23% glycerol plus the reservoir solution and then immediately frozen in a nitrogen liquid stream. Diffraction data to 2.2 Å were collected over 180° in 0.5° increments using an in-house Cu Kα radiation source at 100 K. Raw reflection data were indexed and integrated using MOSFLM 32 and merged and scaled using SCALA in CCP4 33 with CCP4i interface 34. The crystal belongs to space group P21. One molecule is in the asymmetric unit.
Structural solution and refinement
The structure was solved by molecular replacement with the program Phaser 35. The search model is the coordinates of an unligated E. coli methionine aminopeptidase (PDB code 2MAT) with metal ions and water molecules being removed. The structure was refined with REFMAC5 36 with iterative model building using the program WinCoot 37. The refinement was monitored using 5% of the reflections set aside for free R factor analysis throughout the whole refinement process. After MetAP protein, two Mn atoms and most of water molecules were modeled, an Fobs − Fcalc electron density difference map contoured at 2.5 σ level (Fig. 2) clearly showed the presence of the inhibitor 22. The inhibitor was then added according to the density map. The final structure was analyzed using the program PROCHECK 38. 99.6% of the main-chain angles of the residues are in the allowed region, and the residue in 0.4% disallowed regions is Asn-74, which remains the same conformation in other E. coli MetAP structures. The resulting map showed clear electron densities for most of the atoms except for a few side chain atoms on the molecular surface. Comparison of structures and generation of structural drawings were carried out by using PyMOL 39. Statistic parameters in data collection and structural refinement are shown in Table 2.
Acknowledgment
This research was supported by NIH grant AI065898 (Q.-Z. Y.). The High Throughput Screening Laboratory at University of Kansas was supported by NIH grant RR015563 from the COBRE program of the National Center for Research Resources, University of Kansas, and Kansas Technology Enterprise Corporation. We thank Dr. Liam Good for providing us E. coli AS19 strain for the testing.
Abbreviations
- MetAP
methionine aminopeptidase
- AMC
7-amino-4-methylcoumarin
- rmsd
root mean square deviation
- IC50
concentration that causes 50% inhibition
- MIC
minimum inhibitory concentration
- PDB
protein data bank
Footnotes
Coordinates and structure factors for E. coli methionine aminopeptidase complexed to 22 have been deposited in the Protein Data Bank under the access code 3D27.
References
- 1.Bradshaw RA, Brickey WW, Walker KW. N-terminal processing: the methionine aminopeptidase and N alpha-acetyl transferase families. Trends Biochem Sci. 1998;23:263–267. doi: 10.1016/s0968-0004(98)01227-4. [DOI] [PubMed] [Google Scholar]
- 2.Vaughan MD, Sampson PB, Honek JF. Methionine in and out of proteins: targets for drug design. Curr Med Chem. 2002;9:385–409. doi: 10.2174/0929867023371102. [DOI] [PubMed] [Google Scholar]
- 3.Luo QL, Li JY, Liu ZY, Chen LL, Li J, Qian Z, Shen Q, Li Y, Lushington GH, Ye QZ, Nan FJ. Discovery and structural modification of inhibitors of methionine aminopeptidases from Escherichia coli and Saccharomyces cerevisiae. J Med Chem. 2003;46:2631–2640. doi: 10.1021/jm0300532. [DOI] [PubMed] [Google Scholar]
- 4.Schiffmann R, Heine A, Klebe G, Klein CD. Metal ions as cofactors for the binding of inhibitors to methionine aminopeptidase: A critical view of the relevance of in vitro metalloenzyme assays. Angew Chem Int Ed Engl. 2005;44:3620–3623. doi: 10.1002/anie.200500592. [DOI] [PubMed] [Google Scholar]
- 5.Douangamath A, Dale GE, D'Arcy A, Almstetter M, Eckl R, Frutos-Hoener A, Henkel B, Illgen K, Nerdinger S, Schulz H, Mac Sweeney A, Thormann M, Treml A, Pierau S, Wadman S, Oefner C. Crystal structures of Staphylococcusaureus methionine aminopeptidase complexed with keto heterocycle and aminoketone inhibitors reveal the formation of a tetrahedral intermediate. J Med Chem. 2004;47:1325–1328. doi: 10.1021/jm034188j. [DOI] [PubMed] [Google Scholar]
- 6.Chang SY, McGary EC, Chang S. Methionine aminopeptidase gene of Escherichia coli is essential for cell growth. J Bacteriol. 1989;171:4071–4072. doi: 10.1128/jb.171.7.4071-4072.1989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Miller CG, Kukral AM, Miller JL, Movva NR. pepM is an essential gene in Salmonella typhimurium. J Bacteriol. 1989;171:5215–5217. doi: 10.1128/jb.171.9.5215-5217.1989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.D'Souza VM, Holz RC. The methionyl aminopeptidase from Escherichia coli can function as an iron(II) enzyme. Biochemistry. 1999;38:11079–11085. doi: 10.1021/bi990872h. [DOI] [PubMed] [Google Scholar]
- 9.Li JY, Chen LL, Cui YM, Luo QL, Li J, Nan FJ, Ye QZ. Specificity for inhibitors of metal-substituted methionine aminopeptidase. Biochem Biophys Res Commun. 2003;307:172–179. doi: 10.1016/s0006-291x(03)01144-6. [DOI] [PubMed] [Google Scholar]
- 10.Lowther WT, Matthews BW. Structure and function of the methionine aminopeptidases. Biochim Biophys Acta. 2000;1477:157–167. doi: 10.1016/s0167-4838(99)00271-x. [DOI] [PubMed] [Google Scholar]
- 11.Ye QZ, Xie SX, Huang M, Huang WJ, Lu JP, Ma ZQ. Metalloform-selective inhibitors of escherichia coli methionine aminopeptidase and X-ray structure of a Mn(II)-form enzyme complexed with an inhibitor. J Am Chem Soc. 2004;126:13940–13941. doi: 10.1021/ja045864p. [DOI] [PubMed] [Google Scholar]
- 12.Walker KW, Bradshaw RA. Yeast methionine aminopeptidase I can utilize either Zn2+ or Co2+ as a cofactor: a case of mistaken identity? Protein Sci. 1998;7:2684–2687. doi: 10.1002/pro.5560071224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Yang G, Kirkpatrick RB, Ho T, Zhang GF, Liang PH, Johanson KO, Casper DJ, Doyle ML, Marino JP, Jr, Thompson SK, Chen W, Tew DG, Meek TD. Steady-state kinetic characterization of substrates and metal-ion specificities of the full-length and N-terminally truncated recombinant human methionine aminopeptidases (type 2) Biochemistry. 2001;40:10645–10654. doi: 10.1021/bi010806r. [DOI] [PubMed] [Google Scholar]
- 14.D'Souza VM, Swierczek SI, Cosper NJ, Meng L, Ruebush S, Copik AJ, Scott RA, Holz RC. Kinetic and structural characterization of manganese(II)-loaded methionyl aminopeptidases. Biochemistry. 2002;41:13096–13105. doi: 10.1021/bi020395u. [DOI] [PubMed] [Google Scholar]
- 15.Wang J, Sheppard GS, Lou P, Kawai M, Park C, Egan DA, Schneider A, Bouska J, Lesniewski R, Henkin J. Physiologically relevant metal cofactor for methionine aminopeptidase-2 is manganese. Biochemistry. 2003;42:5035–5042. doi: 10.1021/bi020670c. [DOI] [PubMed] [Google Scholar]
- 16.Zhang JH, Chung TD, Oldenburg KR. A simple statistical parameter for use in evaluation and validation of high throughput screening assays. J Biomol Screen. 1999;4:67–73. doi: 10.1177/108705719900400206. [DOI] [PubMed] [Google Scholar]
- 17.Mee SP, Lee V, Baldwin JE, Cowley A. Total synthesis of 5,5’,6,6’-tetrahydroxy-3,3’-biindolyl, the proposed structure of a potent antioxidant found in beetroot (Beta vulgaris) Tetrahedron. 2004;60:3695–3721. [Google Scholar]
- 18.Barton A, Breukelman SP, Kaye PT, Denis Meakins G, Morgan DJ. The preparation of thiazole-4- and -5-carboxylates, and an infrared study of their rotational isomers. J. Chem. Soc. Perkin. Trans. 1. 1982:159–164. [Google Scholar]
- 19.Handy ST, Zhang Y, Bregman H. A modular synthesis of the lamellarins: total synthesis of lamellarin G trimethyl ether. J Org Chem. 2004;69:2362–2366. doi: 10.1021/jo0352833. [DOI] [PubMed] [Google Scholar]
- 20.Huang QQ, Huang M, Nan FJ, Ye QZ. Metalloform-selective inhibition: Synthesis and structure-activity analysis of Mn(II)-form-selective inhibitors of Escherichia coli methionine aminopeptidase. Bioorg Med Chem Lett. 2005;15:5386–5391. doi: 10.1016/j.bmcl.2005.09.019. [DOI] [PubMed] [Google Scholar]
- 21.Xie SX, Huang WJ, Ma ZQ, Huang M, Hanzlik RP, Ye QZ. Structural analysis of metalloform-selective inhibition of methionine aminopeptidase. Acta Crystallogr D Biol Crystallogr. 2006;62:425–432. doi: 10.1107/S0907444906003878. [DOI] [PubMed] [Google Scholar]
- 22.Jo DH, Chiou YM, Que L., Jr Models for extradiol cleaving catechol dioxygenases: syntheses, structures, and reactivities of iron(II)-monoanionic catecholate complexes. Inorg Chem. 2001;40:3181–3190. doi: 10.1021/ic001185d. [DOI] [PubMed] [Google Scholar]
- 23.Crosa JH, Walsh CT. Genetics and assembly line enzymology of siderophore biosynthesis in bacteria. Microbiol Mol Biol Rev. 2002;66:223–249. doi: 10.1128/MMBR.66.2.223-249.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Zorzopulos J, de Long S, Chapman V, Kozloff LM. Evidence for a net-like organization of lipopolysaccharide particles in the Escherichia coli outer membrane. FEMS Microbiol Lett. 1989;52:23–26. doi: 10.1016/0378-1097(89)90163-8. [DOI] [PubMed] [Google Scholar]
- 25.Normark S, Boman HG, Matsson E. Mutant of Escherichia coli with anomalous cell division and ability to decrease episomally and chromosomally mediated resistance to ampicillin and several other antibiotics. J Bacteriol. 1969;97:1334–1342. doi: 10.1128/jb.97.3.1334-1342.1969. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Cammidge AN, King ASH. Model studies towards liquid crystalline dendrimers with mesogenic repeat units throughout the structure. Tetrahedron Lett. 2006;47:5569–5572. [Google Scholar]
- 27.Luo QL, Li JY, Chen LL, Li J, Ye QZ, Nan FJ. Inhibitors of type I MetAPs containing pyridine-2-carboxylic acid thiazol-2-ylamide. Part 2: SAR studies on the pyridine ring 3-substituent. Bioorg Med Chem Lett. 2005;15:639–644. doi: 10.1016/j.bmcl.2004.11.035. [DOI] [PubMed] [Google Scholar]
- 28.Herland A, Nilsson KP, Olsson JD, Hammarstrom P, Konradsson P, Inganas O. Synthesis of a regioregular zwitterionic conjugated oligoelectrolyte, usable as an optical probe for detection of amyloid fibril formation at acidic pH. J Am Chem Soc. 2005;127:2317–2323. doi: 10.1021/ja045835e. [DOI] [PubMed] [Google Scholar]
- 29.Pu S, Liu G, Li G, Yang T. Photochromic thiophene pyrrole mixed linkage type dithienylethene compounds, and their synthesizing and use. Faming Zhuanli Shenqing Gongkai Shuomingshu. 2007 application number CN1978444. [Google Scholar]
- 30.Cook MJ, Jafari-Fini A. Phthalocyanine-related macrocycles: cross cyclotetramerisation products from 3,4-dicyanothiophenes, 2,3-dicyanothiophene and 3,6-dialkylphthalonitriles. Tetrahedron. 2000;56:4085–4094. [Google Scholar]
- 31.NCCLS. Methods for determining bactericidal activity of antimicrobial agents. Approved guideline. M26-A. Vol. 19. Payne, PA: National Committee for Clinical Laboratory Standards (Now Clinical and Laboratory Standards Institute); 1999. [Google Scholar]
- 32.Leslie AGW. Recent changes to the MOSFLM package for processing film and image plate data. Joint CCP4 + ESF-EAMCB Newsletter on Protein Crystallography No. 26. 1992 [Google Scholar]
- 33.Collaborative Computational Project Number 4. The CCP4 Suite: Programs for Protein Crystallography. Acta Cryst. 1994;D50:760–763. doi: 10.1107/S0907444994003112. [DOI] [PubMed] [Google Scholar]
- 34.Potterton E, Briggs P, Turkenburg M, Dodson E. A graphical user interface to the CCP4 program suite. Acta Crystallogr D Biol Crystallogr. 2003;59:1131–1137. doi: 10.1107/s0907444903008126. [DOI] [PubMed] [Google Scholar]
- 35.Read RJ. Pushing the boundaries of molecular replacement with maximum likelihood. Acta Crystallogr D Biol Crystallogr. 2001;57:1373–1382. doi: 10.1107/s0907444901012471. [DOI] [PubMed] [Google Scholar]
- 36.Murshudov GN, Vagin AA, Dodson EJ. Refinement of macromolecular structures by the maximum-likelihood method. Acta Crystallogr D Biol Crystallogr. 1997;53:240–255. doi: 10.1107/S0907444996012255. [DOI] [PubMed] [Google Scholar]
- 37.Emsley P, Cowtan K. Coot: model-building tools for molecular graphics. Acta Crystallogr D Biol Crystallogr. 2004;60:2126–2132. doi: 10.1107/S0907444904019158. [DOI] [PubMed] [Google Scholar]
- 38.Laskowski RA, MacArthur MW, Moss DS, Thornton JM. PROCHECK: a program to check the stereochemical quality of protein structures. J Appl Cryst. 1993;26:283–291. [Google Scholar]
- 39.DeLano WL. The PyMOL Molecular Graphics System. 2002 on World Wide Web http://www.pymol.org. [Google Scholar]