

Abstract
We have developed a stable RNA interference (RNAi) delivery system that is based on the Frog Prince transposable element. This plasmid-based vector system combines the gene silencing capabilities of H1 polymerase III promoter-driven short hairpin RNAs (shRNA) with the advantages of stable and efficient genomic integration of the shRNA cassette mediated by transposition. We show that the Frog Prince-based shRNA expressing system can efficiently knock down the expression of both exogenous as well as endogenous genes in human cells. Furthermore, we use the Frog Prince-based system to study the effect of knockdown of the DNA repair factor Ku70 on transposition of the Sleeping Beauty transposon. Transposon-mediated genomic integration ensures that the shRNA expression cassette and a selectable marker gene within the transposon remain intact and physically linked. We demonstrate that a major advantage of our vector system over plasmid-based shRNA delivery is both its enhanced frequency of intact genomic integration as well as higher target suppression in transgenic human cells. Due to its simplicity and effectiveness, transposon-based RNAi is an emerging tool to facilitate analysis of gene function through the establishment of stable loss-of-function cell lines.
KEYWORDS: RNA interference, short hairpin RNA, Frog Prince, Sleeping Beauty, nonviral gene transfer, stable gene knockdown, transposon-based gene delivery
INTRODUCTION
The recent discovery and development of RNA interference (RNAi) to knock down the expression of a gene-of-interest has brought a widely applicable tool into the toolbox of the molecular or developmental biologist. RNAi is a mechanism of post-transcriptional gene silencing mediated by double-stranded RNA (dsRNA) (reviewed in Fire, 1999). While introduction of long dsRNA works well in such organisms as C. elegans and Drosophila, it induces a strong cytotoxic effect in mammalian somatic cells (Gil and Esteban, 2000). Synthetic, 21-23-nucleotide short interfering RNAs (siRNAs) were shown to circumvent this response (Caplen et al, 2001; Elbashir et al, 2001). However, despite their efficient and specific suppression of gene expression, siRNAs are costly to manufacture, the silencing effect is short-lived, and generation of stable knock-down cell lines (or organisms) is not possible. A refinement of this technique demonstrated that vector-based siRNAs allowed efficient gene silencing in transgenic cell derivatives due to stable chromosomal integration. These vectors express siRNAs through either convergent or divergent transcription (Miyagishi and Taira, 2002; Tran et al, 2003), or by expression from an RNA polymerase III (pol III) promoter, such as U6 or H1, of a hairpin-containing inverted repeat, called short-hairpin RNA (shRNA) (Brummelkamp et al, 2002; Paul et al, 2002; Yu et al, 2002).
Initially, plasmid DNA was used as the vector, but this is inefficient in chromosomal integration. Stable cell lines containing and expressing shRNAs can be generated by cotransfection of a selectable marker or by placing the selectable marker on the shRNA expression plasmid (Brummelkamp et al, 2002). However, expression of the selectable marker does not guarantee that the shRNA expression cassette is integrated and that it is expressed at a level high enough to elicit the desired silencing effect in either of these cases. This is because chromosomal integration of plasmid constructs is probably preceded by random breakage in the plasmid, which could prevent linkage of the marker and the shRNA expression cassette (Bishop, 1996). Viral vectors, such as retroviruses and lentiviruses, were developed to alleviate these problems (Abbas-Terki et al, 2002; Devroe and Silver, 2002). However, viral vectors impose problems, such as a requirement for specialized laboratories, and complicated construction and preparation. Clearly, RNAi technology would benefit from the development of simple, plasmid-based vector systems that allow efficient chromosomal integration and stable expression of shRNA expression cassettes both in tissue culture and in vivo.
Transposable elements are mobile, discrete segments of DNA that can catalyze their excision from one locus and reintegrate into another. These so-called “jumping genes”, often considered junk or parasitic DNA, have been harnessed and developed into useful genetic tools (Cooley et al, 1988; Plasterk, 1996). In particular, the Sleeping Beauty (SB) element, a reconstructed, active member of the Tc1/mariner superfamily (Ivics et al, 1997), has been developed as an efficient vector for gene transfer and insertional mutagenesis in vertebrates (Clark et al, 2004; Davidson et al, 2003; Dupuy et al, 2001; Grabher et al, 2003; Horie et al, 2001; Luo et al, 1998). The application of SB as a vector for human gene therapy is also being actively explored (reviewed in Izsvák and Ivics, 2004), given its simple construction and maintenance, efficient chromosomal integration, long-term transgene expression and relative safety when compared to viral vectors. Two recent reports have shown the utility of the SB system as an shRNA vector. Heggestad et al (2004) demonstrated stable knockdown of both GFP and lamin A, whereas Chen and colleagues reported long-lasting knockdown of the human huntingtin transcript in human cells (Chen et al, 2005). The availability of multiple, transposon-based vector systems broadens the utility of these elements as genetic tools. Thus, another transposon of the Tc1/mariner superfamily, named Frog Prince (FP), was reconstructed from inactive elements found in the genome of the frog species Rana pipiens (Miskey et al, 2003). FP transposes through a “cut-and-paste” mechanism catalyzed by the transposase encoded between the two terminal inverted repeats (IRs). The transposase can be supplied in trans, allowing replacement of the transposase coding sequence with a cargo gene of choice located between the required IRs (Figure 1).
Figure 1.
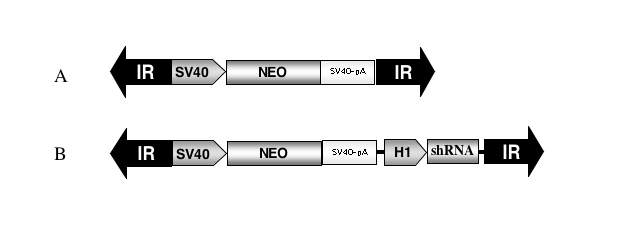
Schematic of Frog Prince-based shRNA vectors. (A) The parental FP transposon contains the neomycin resistance gene (NEO) driven by the SV40 promoter (SV40) and followed by the SV40 polyadenlyation signal (SV40-pA) between the left and right flanking terminal inverted repeats (IR). (B) The H1 promoter expression cassette from pSUPER was subcloned between the polyadenylation signal and the right IR to generate pFP/Neo-H1.
In this work, we assessed the utility of FP-based transposon vectors for efficient delivery and expression of shRNAs in cultured mammalian cells. We show that FP-based shRNA vectors can efficiently transpose in human cells. We further demonstrate efficient knockdown of expression of the exogenous marker gene EGFP, and substantial inhibition of the function of the endogenous double-strand break repair factor Ku70. The data establish the Frog Prince transposon as a useful tool for the easy and efficient generation of stable knockdown cell lines for a variety of genetic analyses.
MATERIALS AND METHODS
Plasmids
The pEGFP-C1 plasmid was obtained from Clontech (Palo Alto, CA, USA). The pSUPER plasmid was obtained from R. Agami (Brummelkamp et al, 2002). The neomycin phosphotransferase (neo) marked FP transposon, pFP/Neo, as previously described (Miskey et al, 2003), was digested with HindIII, Klenow-filled, and religated to remove the HindIII site on the vector backbone. The human H1 RNA polymerase III driven shRNA expression cassette was isolated from pSUPER with XbaI and HincII restriction digestion, Klenow-filled and ligated into pFP/Neo previously digested with BglII and Klenow-filled. The H1 shRNA cassette was subcloned between the polyadenylation signal of the neo cassette and the right IR/DR of the FP transposon. Double-stranded oligos encoding the gene-specific hairpin RNA were subcloned into the BglII/HindIII site of the H1 shRNA cassette. Due to the immediate proximity of these two restriction enzyme sites, difficulties were encountered in efficient digestion with both enzymes. Thus, a 0.3 kb spacer fragment of non-specific DNA was cloned into the HindIII site to alleviate this problem. The zeocin resistance gene (Zeo) marked SB transposon, pT/Zeo, was constructed previously as described (Izsvák et al, 2000).
The following oligos were synthesized (Biotez, Berlin, Germany), using parameters established in (Reynolds et al, 2004; Ui-Tei et al, 2004): EGFP4(482)F gatccccGCATCAAGGTGAACTTCAAttcaagagaTTGAAGTTCACCTTGATGCtttttggaaa, EGFP4(482)R agcttttccaaaaaGCATCAAGGTGAACTTCAAtctcttgaaTTGAAGTTCACCTTGATGCggg, Ku70 (1148)F gatccccGGATCATGCTGTTCACCAAttcaagagaTTGGTGAACAGCATGATCCtttttggaaa and Ku70(1148)R agcttttccaaaaaGGATCATGCTGTTCACCAAtctcttgaaTTGGTGAACAGCATGATCCggg. The numbers in parentheses refer to the start nucleotide of the 5′ sense-strand, relative to the start codon of the target transcript. Sequences in lower case are flanking and loop sequences, as recommended (Brummelkamp et al, 2002). Flanking sequences give compatible overhangs for BglII on the 5′- and HindIII on the 3′-end of the oligo. Respective forward and reverse oligos were annealed, phosphorylated, and ligated into pFP-H1 or pFP-H1/bi, previously digested with first BglII, then HindIII, and then dephosphorylated. Subclones were screened with EcoRI/HindIII restriction digestion and confirmed by sequencing.
Cell culture and transfection
Cells were cultured in DMEM (Gibco, Karlsruhe, Germany) supplemented with 10% (v/v) fetal calf serum (PAA, Pasching, Austria) at 37°C and 5% (v/v) CO2. Approximately 5 × 105 HeLa cells were plated out one day prior to transfection into 6-well plates. Purified plasmid DNA (Qiagen, Hilden, Germany) was transfected with Fugene6 (Roche, Mannheim, Germany). Two days after transfection, cells were seeded into 10 cm dishes in DMEM plus 1.4 mg/ml G418 (Gibco, Karlsruhe, Germany) and selected for two weeks. G418-resistant colonies were picked and expanded for generation of stable clones.
The HeLa EGFP clonal line H38-3 was made by cotransfection of pT/EGFP (Z. Ivics, unpublished data), pCMV-SB transposase expressing helper plasmid (Ivics et al, 1997), and pHygro (Clontech, Palo Alto, CA, USA). Hygromycin-resistant (Invitrogen, Karlsruhe, Germany; 150 μg/ml) colonies were individually picked after two weeks of selection and assayed for EGFP expression by fluorescent microscopy and flow cytometry (FACS) analysis on a FACSCalibur (Becton Dickinson, Heidelberg, Germany). An EGFP-expressing transgenic clone was subcloned by limiting dilution to obtain the clonal line H38-3.
EGFP knockdown analysis
H38-3 cells were transfected as described above with 800 ng of pFP/Neo-H1 with or without an shRNA oligo, plus the FP transposase helper plasmid pFV-FP (Miskey et al, 2003). As control, 800 ng of the pSUPER plasmid with or without an shRNA oligo was cotransfected with pFP/Neo to allow G418 selection. The transfected cells were seeded to a 10 cm dish with 1.4 mg/ml G418 and selected for two weeks. G418-resistant colonies were pooled, and EGFP expression was assayed by fluorescent microscopy and FACS analysis. The mean EGFP fluorescence was calculated by CELLQuest V3.1 (Becton Dickinson). For analysis in 96-well format, transfected H38-3 cells were diluted into DMEM containing 1.4 mg/ml G418 and plated with a multichannel pipette into black, clear bottom 96-well plates (Corning, NY, USA) two days after transfection. After two weeks of selection, EGFP expression levels were measured with a platereader (Bio-Tek Synergy HT, Bad Friedrichshall, Germany) and analyzed with KC4 V3.3 software. Colonies were then fixed, stained, and counted. EGFP values per colony were graphically displayed using Treeview 1.60 (http://rana.lbl.gov/EisenSoftware.htm).
Transient Ku70 knockdown was analyzed using western hybridization as previously described (Izsvák et al, 2004). Stable Ku70 knockdown cell lines were functionally assayed for reduced Ku70 activity by a transposition assay (Ivics et al, 1997). The SB transposon pT/Zeo was cotransfected with the pCMV-SB transposase helper plasmid into each clonal line. Cells were selected with 100 μg/ml of zeocin (Invitrogen, Karlsruhe, Germany) for two weeks before resistant colonies were stained and counted.
RESULTS
Frog Prince transposon-based shRNA vectors
Transpositional activity of FP is similar to, if not higher than, SB in diverse vertebrate cell lines (Miskey et al, 2003). Thus, it is an ideal candidate for a transposon-based shRNA vector. The H1 pol III promoter expression cassette from pSUPER (Brummelkamp et al, 2002) was subcloned into the FP transposon to drive expression of shRNAs. The H1 expression cassette was subcloned into the 3′ end of the FP transposon, between the polyadenylation signal of the neomycin-resistance (neo) expression cassette and the right terminal inverted repeat, and the resulting construct was denoted as pFP/Neo-H1 (Figure 1B). The H1 cassette insertion was recovered in both orientations relative to the direction of neo gene transcription. Both transposon constructs were tested for transpositional activity with a standard transposition assay (Ivics et al, 1997) in human HeLa cells. Both versions transposed equally well, and almost as well as the parental transposon pFP/Neo (data not shown).
Stable EGFP knockdown
EGFP was chosen as the initial target for proof-of-principle tests. Four anti-EGFP shRNA oligonucleotides were designed using web-based parameters, and subcloned into both pSUPER and pFP/Neo-H1 for transient analysis. One of these (hereafter referred to as EGFP4) resulted in over 90% reduction in mean EGFP fluorescence in transient transfection assays using both pSUPER-based and transposon-based vectors with either orientation of the H1 shRNA expression cassette (data not shown). Thus, the pFP/Neo-H1 transposon vector (Figure 1B) and the EGFP4 oligo were chosen to test long-term knockdown of EGFP expression mediated by the Frog Prince transposon. The EGFP4 shRNA oligo was subcloned into pFP/Neo-H1 to generate pFP/Neo-H1/EGFP4. EGFP knockdown was examined in a stably transgenic, HeLa-derived, EGFP-expressing clonal cell line (H38-3). To generate transposition-mediated transgenic cells, H38-3 cells were cotransfected with the transposon-based shRNA vector and the FP transposase expression plasmid pFV-FP (Miskey et al, 2003). In “no-transposition” control experiments, pS/EGFP4 was cotransfected with pFP/Neo as a selectable marker to allow colony selection. Another, “no-shRNA” control line was made by cotransfecting H38-3 cells with pFP/Neo-H1 plus pFV-FP.
Pooled colonies were assayed for EGFP fluorescence levels by FACS between 35 and 66 days post-transfection, and compared to the parental line H38-3. The mean EGFP fluorescence of each sample was compared to the mean EGFP of the parental line, which was arbitrarily set to 100% (Figure 2). Both pSUPER- and pFP-based shRNA resulted in about 85-90% reduction in mean EGFP fluorescence, respectively, relative to parental levels (Figure 2). In comparison, transgenic cells generated by the pFP/Neo-H1 transposon had a mean EGFP fluorescence approximately equal to H38-3 (Figure 2). Thus, Frog Prince-based shRNA can efficiently mediate stable silencing of an exogenously introduced gene.
Figure 2.

Stable FP-shRNA-mediated knockdown of EGFP in pooled HeLa colonies. The EGFP clonal HeLa cell line H38-3 was cotransfected with the indicated shRNA expressing plasmid plus either a transposase source or a neomycin expression plasmid. Transfected cells were selected with G418 for two weeks, and resistant colonies were harvested and pooled together. EGFP expression was analyzed by FACS. The mean EGFP expression of the H38-3 parental line was arbitrarily set to 100%. The respective overlaid FACS profiles are shown in the inset.
FP-based shRNA vectors are more efficient at colony formation and EGFP knockdown than non-transposon-based plasmid vectors
We showed that the FP-based shRNA system is capable of knocking down EGFP expression in pooled transgenic cells, but how does the efficiency of both transgenesis and knockdown compare to non-transposon systems? To answer this question, an assay was established to allow direct comparison between the numbers of antibiotic-resistant colonies generated and the level of EGFP expression per colony. H38-3 HeLa cells were transfected with either pFP/Neo-H1-EGFP4 plus pFV-FP (transposition-mediated stable transgenesis) or with pS/EGFP4 plus pFP/Neo as a selectable marker (“no-transposition” control). The cells were plated to a 96-well plate such that pFP/Neo-H1-EGFP4-transfected cells gave at least one colony in almost every well. After two weeks under G418 selection, the level of EGFP expression per well was measured using a plate reader. The cells were then fixed, stained, and the colonies counted. The value of EGFP expression per colony was calculated and expressed in a graphical format. In Figure 3, each rectangle represents a 96-well plate, with each pixel being one well. The pixels are color-coded according to the level of EGFP expression per colony - with black meaning no colonies, and increasingly lighter shades of green meaning higher EGFP expression per colony. pFP/Neo-H1-EGFP4 (Figure 3B) was clearly more efficient at forming antibiotic resistant colonies, as 95 of the 96 wells contained colonies, whereas only 52 of 96 wells contained colonies for pS/EGFP4 (Figure 3A). Furthermore, the average level of EGFP expression in wells with colonies was overall reduced in pFP/Neo-H1-EGFP4 colonies compared to pS/EGFP4. We conclude that FP transposon-based shRNA vectors are more efficient at colony formation and EGFP knockdown than non-transposon, plasmid-based vectors.
Figure 3.
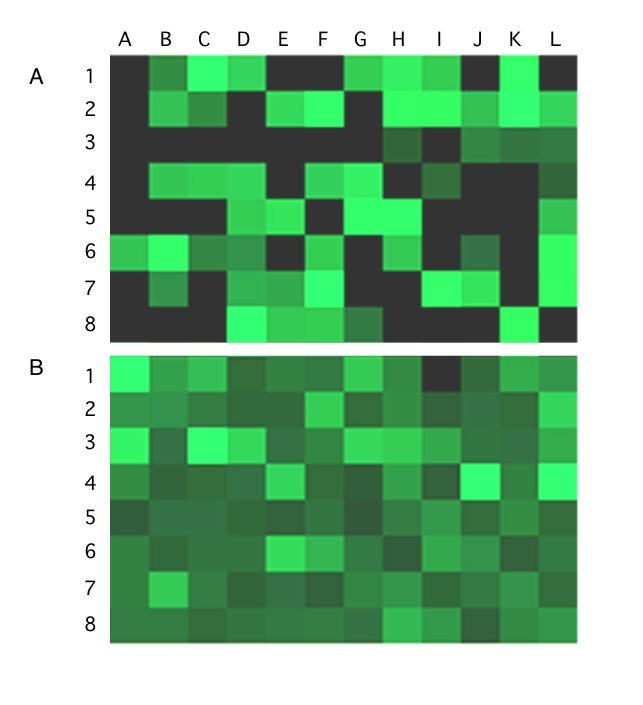
Comparison of efficiencies of colony formation and EGFP knockdown with pSUPER- versus pFP-based shRNA vectors. The EGFP clonal HeLa line H38-3 was cotransfected with (A) pSUPER/EGFP4 and pFP/Neo, or (B) pFP/Neo-H1/EGFP4 and pFV-FP. Two days after transfection, the cells were diluted and plated into 96-well plates. After two weeks under G418 selection, EGFP expression was analyzed for resistant colonies in each well with a plate reader. Colonies were then fixed, stained and counted. The EGFP expression level per colony was calculated and graphically displayed using the Treeview program. Each square pixel represents a well and is color-coded. Black represents empty wells, while the shades of green reflect the level of EGFP expression per colony, with dark meaning a low level and bright meaning a high level.
Frog Prince-based shRNA can efficiently mediate silencing of an endogenous gene
We next tested the ability of the FP-shRNA system to silence an endogenous gene. The Ku70/80 complex is a component of the nonhomologous end joining (NHEJ) pathway of DNA double strand break (DSB) repair (Durocher and Jackson, 2001). The Ku protein complex has been shown to be required for efficient SB transposition (Izsvák et al, 2004; Yant and Kay, 2003). This was studied by measuring the efficiency of SB transposition in cells with a Ku-deficient genetic background, and in wild-type cells in which Ku80 was transiently knocked down with RNAi. Thus, a colony-forming transposition assay has been shown to serve as a simple and efficient functional test of Ku knockdown (Izsvák et al, 2004).
An shRNA oligo was designed against the human Ku70 transcript and subcloned into the pSUPER and pFP/Neo-H1 vectors. The resulting shRNA-expressing constructs were transiently transfected into HeLa cells, and the ability of the shRNA to knock down endogenous Ku70 expression was assayed using Western hybridization. As shown in Figure 4A, the anti-Ku70 shRNA clearly diminished levels of Ku70 protein, whereas cellular levels of actin remained unchanged. Then, HeLa cells were transfected with pSUPER (negative control), pS/Ku70 and pFP/Neo-H1-Ku70, and individual transgenic cell clones were isolated from each transfection. pSUPER and pS/Ku70 were cotransfected with pFP/Neo as a marker to allow colony selection. The pFP/Neo-H1-Ku70 construct was transfected both in the presence and absence of pFV-FP to monitor FP transpositional efficiency. In the absence of transposase (in this experiment pCMV-Beta, encoding β-galactosidase, replaced the transposase expression plasmid, Figure 4B), transgene integration occurred randomly rather than by transposition, and served as a control. Similar to SB transposition, the shRNA-mediated knockdown of Ku70 clearly interfered with efficient FP transposition (data not shown). However, FP transposase-mediated transposition of pFP/Neo-H1-Ku70 was still higher than random plasmid integration, allowing easy establishment of stable colonies. Individual colonies were picked for each construct and expanded under selection for up to three months. Since the SB and FP transposases share only 50% amino acid identity, and they were shown not to cross-mobilize each other (Miskey et al, 2003), it was possible to test the efficacy of the Ku70 shRNA constructs using a SB transposition assay.
Figure 4.
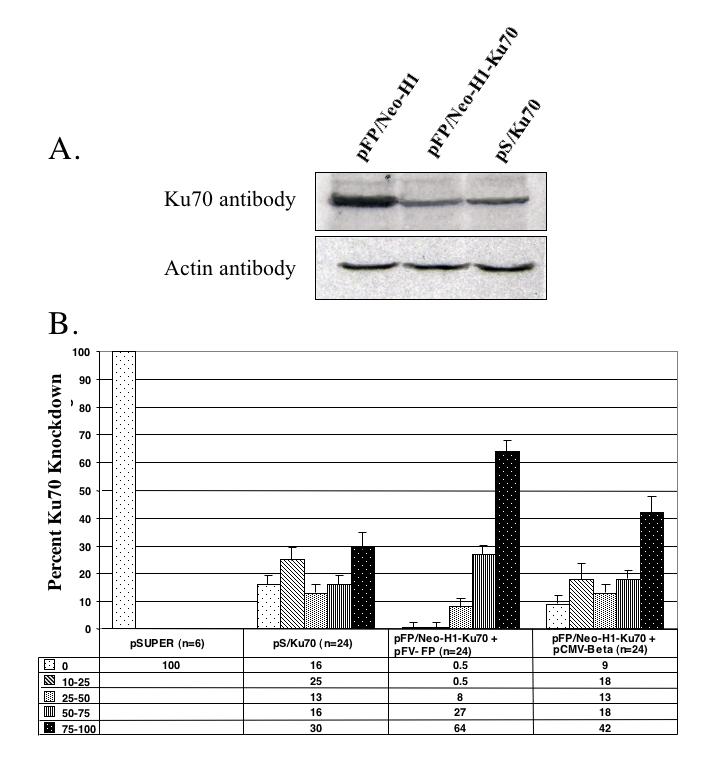
Efficiency of FP-shRNA-mediated knockdown of Ku70 in HeLa cells assayed by Western hybridization and Sleeping Beauty transposition. (A) Western hybridizations of nuclear protein extracts prepared from HeLa cells transiently transfected with the plasmid constructs shown above the lanes. Nuclear extracts from each transfection were blotted and hybridized with a Ku70 antibody (upper panel) or with an actin antibody (lower panel). (B) Efficiency of SB transposition in transgenic cell clones expressing shRNAs against Ku70. The indicated numbers of colonies (n) from each transfection were picked and expanded under selection. A transposition assay using a zeocin resistance gene-marked SB transposon and a SB transposase helper plasmid was performed in each clone. After two weeks under zeocin selection, resistant colonies were stained and counted. The efficiency of SB transposition in each picked clone was grouped into five categories: 0, 1-25, 25-50, 50-75 and 75-100% reduction, relative to the level of SB transposition in the pSUPER control clones.
Thus, to characterize the knockdown capacity of the stably integrated shRNA constructs, antibiotic-resistant colonies from each experiment were assayed for SB transposition. The level of SB transpositional efficiency is a measure of Ku70 activity, such that diminished SB activity reflects increased Ku70 knockdown. The transpositional efficiency of SB in pSUPER stable colonies was arbitrarily set to 100%. Five categories were established to characterize the efficiency of the knockdown effect: 0, 10-25, 25-50, 50-75, and 75-100% reduction in SB transposition. The results in Figure 4B show that in a sample size of 24 colonies, over 60% of the transposition-generated (pFP/Neo-H1-Ku70 plus pFV-FP) clones had a knockdown effect of 75-100%. However, this number was only about 30% for pS/Ku70 clones (Figure 4B). Furthermore, over 90% of the transposition-generated clones showed at least 50% knockdown of Ku70 activity whereas only 46% of pS/Ku70 clones were at this level (Figure 4B). In the clones generated with pFP/Neo-H1-Ku70 in the absence of transposase (a situation similar to the next generation of pSUPER-based plasmids that contain a selectable marker), the observed knockdown effect was intermediate, with about 40% of the colonies in the 75-100% category (Figure 4B). Taken together, these results show that the efficacy of the transposition-based system is greater than the plasmid-based system in reducing the function of an endogenous gene. Furthermore, these results confirm the importance of the Ku70 protein for efficient transposition of both the SB and FP transposons.
DISCUSSION
We showed that the Frog Prince-based shRNA system is capable of knocking down both exogenous as well as endogenous transcripts in human cells. In addition, it functions both transiently and in stable cell clones, either individually picked or pooled. Our results show that both the number and the knockdown effect of shRNA-expressing colonies are higher with the transposon- versus the plasmid-based approach. When the transposon vector system is used for establishing stable knockdown lines, the probability that an antibiotic-resistant clone is also a good knockdown clone was higher (Figures 3 and 4B). One explanation is that transposition-mediated genomic integration maintains the shRNA cassette and the selectable marker physically linked within the transposable element. This can enhance the stability of the gene knockdown effect, compared to non-transposase-mediated integration, where the cleavage of the plasmid DNA occurs randomly prior to chromosomal integration (Bishop, 1996), which may lead to physical separation of the selectable maker and the shRNA expression cassette. Transposition-mediated integration provided more efficient knockdown than random integration even when the selectable marker was provided in cis (Figure 4B, compare pFP/Neo-H1-Ku70 in the presence and absence of transposase), showing that the precise cut-and-paste mechanism of transposition is superior to random integration for maintaining linkage of the shRNA expression cassette and the selectable marker.
The transposon-based shRNA system also has several advantages over viral systems. Transposons, in contrast to viral vectors, are quick, simple and cost-effective to produce, and are not infectious, thus do not pose potential safety risks to the investigator. Additionally, integration of transposons into the genome is random (Liu et al, 2005; Vigdal et al, 2002; Yant et al, 2005), in contrast to retroviruses, lentiviruses (Mitchell et al, 2004) and recombinant adeno-associated viruses (Nakai et al, 2005) that tend to prefer to integrate into expressed genes. This random integration of transposons represents a reduced chance of interrupting a gene that is either essential for cell survival or that could lead to non-specific gene expression effects. In comparison to viruses and in contrast to plasmid vectors, transposons integrate through a precisely mediated mechanism that can promote stable transgene expression over a long period of time. Here we show that the FP-shRNA mediated knockdown of EGFP in a transgenic line can persist for at least six weeks (Figure 2) and that functional knockdown of Ku70 can persist for up to three months (Figure 4B). Together with previous studies demonstrating Sleeping Beauty transposon-based stable knockdown of gene expression (Heggestad et al, 2004; Chen et al, 2005), these data establish the advantages of transposon-based shRNA vectors for both stable and long-term knockdown of a target transcript. The availability of multiple transposon-based RNAi vectors increases the utility of these elements for regulating gene expression in vertebrates. We have demonstrated that the Frog Prince and the Sleeping Beauty transposon systems can be used in combination in certain applications. Specifically, the FP transposition process was used to deliver the shRNA expression cassette from a plasmid DNA source into chromosomes, and the effect of the shRNA-mediated knockdown of Ku70 was assayed using the SB transposon system. This allows the unique situation in which one transposon can be used as a tool to study the transpositional requirements (e.g., host factors) of the other.
The FP-shRNA system lends itself not only to generation of stable knockdown lines in cell culture, but also to generation of knockdown model organisms. The FP transposon system has shown activity in a wide host range of vertebrate cell types in culture (Miskey et al, 2003), as well as in zebrafish embryos (C. Miskey, unpublished data). The related transposon, SB, has been extensively used for genetic engineering of vertebrate species such as fish, frogs and mice (reviewed in Ivics and Izsvák, 2004; Miskey et al, 2005). With the inclusion of either an inducible promoter or a tissue-specific promoter (reviewed in Prawitt et al, 2004), the FP-shRNA system would allow spatial and temporal control over the silencing of developmentally important or essential genes, as well as generation of animal models of human disease through localized inhibition of specific targets.
RNA interference has also generated much interest as an alternate method for gene therapy, focusing on loss-of-function rather than gain-of-function. Current targets being tested include cancer, single gene disorders and viral infections (reviewed in Caplen, 2004). The FP-shRNA system is perfectly suited as a vector for these applications. In addition to the benefits stated above over plasmid- and viral-based systems, the FP transposon is expected to be active in many, if not all, human cell types.
ACKNOWLEDGEMENTS
We thank members of the Transposition Group at the MDC for the many helpful comments and discussions. Also thanks to Herbert Schulz and Oliver Hummel (MDC, Berlin, Germany) for their help with Treeview data analysis. The pSUPER plasmid was a kind gift from Reuven Agami (NKI, Amsterdam, Netherlands).
STATEMENT OF COMPETING INTERESTS
The authors declared no competing interests.
REFERENCES
- Abbas-Terki T, Blanco-Bose W, Deglon N, Pralong W, Aebischer P. Lentiviral-mediated RNA interference. Hum Gene Ther. 2002;13:2197–2201. doi: 10.1089/104303402320987888. [DOI] [PubMed] [Google Scholar]
- Bishop JO. Chromosomal insertion of foreign DNA. Reprod Nutr Dev. 1996;36:607–618. [PubMed] [Google Scholar]
- Brummelkamp TR, Bernards R, Agami R. A system for stable expression of short interfering RNAs in mammalian cells. Science. 2002;296:550–553. doi: 10.1126/science.1068999. [DOI] [PubMed] [Google Scholar]
- Caplen NJ. Gene therapy progress and prospects. Downregulating gene expression: the impact of RNA interference. Gene Ther. 2004;11:1241–1248. doi: 10.1038/sj.gt.3302324. [DOI] [PubMed] [Google Scholar]
- Caplen NJ, Parrish S, Imani F, Fire A, Morgan RA. Specific inhibition of gene expression by small double-stranded RNAs in invertebrate and vertebrate systems. Proc Natl Acad Sci USA. 2001;98:9742–9747. doi: 10.1073/pnas.171251798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen ZJ, Kren BT, Wong PY, Low WC, Steer CJ. Sleeping Beauty-mediated down-regulation of huntingtin expression by RNA interference. Biochem Biophys Res Commun. 2005;329:646–652. doi: 10.1016/j.bbrc.2005.02.024. [DOI] [PubMed] [Google Scholar]
- Clark KJ, Geurts AM, Bell JB, Hackett PB. Transposon vectors for gene-trap insertional mutagenesis in vertebrates. Genesis. 2004;39:225–233. doi: 10.1002/gene.20049. [DOI] [PubMed] [Google Scholar]
- Cooley L, Kelley R, Spradling A. Insertional mutagenesis of the Drosophila genome with single P elements. Science. 1988;239:1121–1128. doi: 10.1126/science.2830671. [DOI] [PubMed] [Google Scholar]
- Davidson AE, Balciunas D, Mohn D, et al. Efficient gene delivery and gene expression in zebrafish using the Sleeping Beauty transposon. Dev Biol. 2003;263:191–202. doi: 10.1016/j.ydbio.2003.07.013. [DOI] [PubMed] [Google Scholar]
- Devroe E, Silver PA. Retrovirus-delivered siRNA. BMC Biotechnol. 2002;2:15. doi: 10.1186/1472-6750-2-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dupuy AJ, Fritz S, Largaespada DA. Transposition and gene disruption in the male germline of the mouse. Genesis. 2001;30:82–88. doi: 10.1002/gene.1037. [DOI] [PubMed] [Google Scholar]
- Durocher D, Jackson SP. DNA-PK, ATM and ATR as sensors of DNA damage: variations on a theme? Curr Opin Cell Biol. 2001;13:225–231. doi: 10.1016/s0955-0674(00)00201-5. [DOI] [PubMed] [Google Scholar]
- Elbashir SM, Harborth J, Lendeckel W, Yalcin A, Weber K, Tuschl T. Duplexes of 21-nucleotide RNAs mediate RNA interference in cultured mammalian cells. Nature. 2001;411:494–498. doi: 10.1038/35078107. [DOI] [PubMed] [Google Scholar]
- Fire A. RNA-triggered gene silencing. Trends Genet. 1999;15:358–363. doi: 10.1016/s0168-9525(99)01818-1. [DOI] [PubMed] [Google Scholar]
- Gil J, Esteban M. Induction of apoptosis by the dsRNA-dependent protein kinase (PKR): mechanism of action. Apoptosis. 2000;5:107–114. doi: 10.1023/a:1009664109241. [DOI] [PubMed] [Google Scholar]
- Grabher C, Henrich T, Sasado T, Arenz A, Wittbrodt J, Furutani-Seiki M. Transposon-mediated enhancer trapping in medaka. Gene. 2003;322:57–66. doi: 10.1016/j.gene.2003.09.009. [DOI] [PubMed] [Google Scholar]
- Heggestad AD, Notterpek L, Fletcher BS. Transposon-based RNAi delivery system for generating knockdown cell lines. Biochem Biophys Res Commun. 2004;316:643–650. doi: 10.1016/j.bbrc.2004.02.090. [DOI] [PubMed] [Google Scholar]
- Horie K, Kuroiwa A, Ikawa M, et al. Efficient chromosomal transposition of a Tc1/mariner-like transposon Sleeping Beauty in mice. Proc Natl Acad Sci U S A. 2001;98:9191–9196. doi: 10.1073/pnas.161071798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ivics Z, Hackett PB, Plasterk RH, Izsvák Z. Molecular reconstruction of Sleeping Beauty, a Tc1-like transposon from fish, and its transposition in human cells. Cell. 1997;91:501–510. doi: 10.1016/s0092-8674(00)80436-5. [DOI] [PubMed] [Google Scholar]
- Ivics Z, Izsvák Z. Transposable elements for transgenesis and insertional mutagenesis in vertebrates: a contemporary review of experimental strategies. Methods Mol Biol. 2004;260:255–276. doi: 10.1385/1-59259-755-6:255. [DOI] [PubMed] [Google Scholar]
- Izsvák Z, Ivics Z. Sleeping Beauty transposition: biology and applications for molecular therapy. Mol Ther. 2004;9:147–156. doi: 10.1016/j.ymthe.2003.11.009. [DOI] [PubMed] [Google Scholar]
- Izsvák Z, Ivics Z, Plasterk RH. Sleeping Beauty, a wide host-range transposon vector for genetic transformation in vertebrates. J Mol Biol. 2000;302:93–102. doi: 10.1006/jmbi.2000.4047. [DOI] [PubMed] [Google Scholar]
- Izsvák Z, Stüwe EE, Fiedler D, Katzer A, Jeggo PA, Ivics Z. Healing the wounds inflicted by Sleeping Beauty transposition by double-strand break repair in mammalian somatic cells. Mol Cell. 2004;13:279–290. doi: 10.1016/s1097-2765(03)00524-0. [DOI] [PubMed] [Google Scholar]
- Liu G, Geurts AM, Yae K, et al. Target-site preferences of Sleeping Beauty transposons. J Mol Biol. 2005;346:161–173. doi: 10.1016/j.jmb.2004.09.086. [DOI] [PubMed] [Google Scholar]
- Luo G, Ivics Z, Izsvák Z, Bradley A. Chromosomal transposition of a Tc1/mariner-like element in mouse embryonic stem cells. Proc Natl Acad Sci U S A. 1998;95:10769–10773. doi: 10.1073/pnas.95.18.10769. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miskey C, Izsvák Z, Kawakami K, Ivics Z. DNA transposons in vertebrate functional genomics. Cell Mol Life Sci. 2005;62:629–641. doi: 10.1007/s00018-004-4232-7. [DOI] [PubMed] [Google Scholar]
- Miskey C, Izsvák Z, Plasterk RH, Ivics Z. The Frog Prince: a reconstructed transposon from Rana pipiens with high transpositional activity in vertebrate cells. Nucleic Acids Res. 2003;31:6873–6881. doi: 10.1093/nar/gkg910. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mitchell RS, Beitzel BF, Schroder AR, et al. Retroviral DNA integration: ASLV, HIV, and MLV show distinct target site preferences. PLoS Biol. 2004;2:E234. doi: 10.1371/journal.pbio.0020234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miyagishi M, Taira K. U6 promoter-driven siRNAs with four uridine 3′ overhangs efficiently suppress targeted gene expression in mammalian cells. Nat Biotechnol. 2002;20:497–500. doi: 10.1038/nbt0502-497. [DOI] [PubMed] [Google Scholar]
- Nakai H, Wu X, Fuess S, et al. Large-scale molecular characterization of adeno-associated virus vector integration in mouse liver. J Virol. 2005;79:3606–3614. doi: 10.1128/JVI.79.6.3606-3614.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paul CP, Good PD, Winer I, Engelke DR. Effective expression of small interfering RNA in human cells. Nat Biotechnol. 2002;20:505–508. doi: 10.1038/nbt0502-505. [DOI] [PubMed] [Google Scholar]
- Plasterk RH. The Tc1/mariner transposon family. Curr Top Microbiol Immunol. 1996;204:125–143. doi: 10.1007/978-3-642-79795-8_6. [DOI] [PubMed] [Google Scholar]
- Prawitt D, Brixel L, Spangenberg C, et al. RNAi knock-down mice: an emerging technology for post-genomic functional genetics. Cytogenet Genome Res. 2004;105:412–421. doi: 10.1159/000078214. [DOI] [PubMed] [Google Scholar]
- Reynolds A, Leake D, Boese Q, Scaringe S, Marshall WS, Khvorova A. Rational siRNA design for RNA interference. Nat Biotechnol. 2004;22:326–330. doi: 10.1038/nbt936. [DOI] [PubMed] [Google Scholar]
- Tran N, Cairns MJ, Dawes IW, Arndt GM. Expressing functional siRNAs in mammalian cells using convergent transcription. BMC Biotechnol. 2003;3:21. doi: 10.1186/1472-6750-3-21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ui-Tei K, Naito Y, Takahashi F, et al. Guidelines for the selection of highly effective siRNA sequences for mammalian and chick RNA interference. Nucleic Acids Res. 2004;32:936–948. doi: 10.1093/nar/gkh247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vigdal TJ, Kaufman CD, Izsvák Z, Voytas DF, Ivics Z. Common physical properties of DNA affecting target site selection of Sleeping Beauty and other Tc1/mariner transposable elements. J Mol Biol. 2002;323:441–452. doi: 10.1016/s0022-2836(02)00991-9. [DOI] [PubMed] [Google Scholar]
- Yant SR, Kay MA. Nonhomologous-end-joining factors regulate DNA repair fidelity during Sleeping Beauty element transposition in mammalian cells. Mol Cell Biol. 2003;23:8505–8518. doi: 10.1128/MCB.23.23.8505-8518.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yant SR, Wu X, Huang Y, Garrison B, Burgess SM, Kay MA. High-resolution genome-wide mapping of transposon integration in mammals. Mol Cell Biol. 2005;25:2085–2094. doi: 10.1128/MCB.25.6.2085-2094.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu JY, DeRuiter SL, Turner DL. RNA interference by expression of short-interfering RNAs and hairpin RNAs in mammalian cells. Proc Natl Acad Sci USA. 2002;99:6047–6052. doi: 10.1073/pnas.092143499. [DOI] [PMC free article] [PubMed] [Google Scholar]